

Abstract
Alzheimer's disease (AD) is the most common neurodegenerative disease among the elderly and accounts for 60-80% of all cases of dementia. Currently, the diagnosis of AD is based on cognitive tests and mental state exams, but the peptide amyloid-beta (Aβ) in cerebrospinal fluid (CSF) is increasingly used in clinical trials and settings. As for most protein and peptide biomarkers, quantification is performed using antibody-based techniques, such as enzyme-linked immunosorbent assay (ELISA). However, intra- and inter-laboratory variability in these assays hamper its use as a diagnostic marker in clinical routine.
An antibody-independent Reference Measurement Procedure (RMP) was developed based on solid-phase extraction (SPE) and liquid chromatography (LC)-tandem mass spectrometry (MS/MS), where stable, isotope-labeled Aβ peptides were used as internal standards, enabling absolute quantification. A high-resolution quadrupole-orbitrap hybrid instrument was used for the measurements. The method allows for the quantification of CSF Aβ1-42 between 150-4,000 pg/mL.
Keywords: Medicine, Issue 121, Alzheimer's disease, Amyloid Beta Peptides, Cerebrospinal Fluid, Mass Spectrometry, Liquid Chromatography, Absolute Quantification, Reference Measurement Procedure.
Introduction
Alzheimer's disease (AD) is the most common form of dementia and affects about 35 million people worldwide1. The neuropathological hallmarks of the disease widely believed to lie at the core of AD pathogenesis are intracellular neurofibrillary tangles of hyperphosphorylated tau protein2 and extracellular plaques consisting of aggregated amyloid-beta (Aβ) peptides3. In line with this, the assessment of plaque pathology in vivo by biomarkers has recently been included in the research diagnostic criteria for AD4. For CSF measurements of Aβ1-42, several immunoassays are available and are used in many clinical laboratories5. The concentration of Aβ1-42 in CSF is approximately 50% lower in AD patients than in the cognitively normal elderly, reflecting the deposition of the peptide in plaques in the brain6,7.
These biomarkers are mainly analyzed using immunoassays (i.e., antibody-based techniques), but these assays may be influenced by matrix effects8. The use of immunoassays on different technology platforms and the lack of assay standardization9,10 makes the introduction of global cut-off concentrations difficult11,12. An analytically validated RMP would permit the uniform calibration of different assay platforms, ideally resulting in better comparability across analytical platforms and in better control of factors contributing to the overall measurement variability.
The absolute quantification of Aβ1-42 using the developed LC-MS/MS method overcomes many of the issues associated with antibody-based techniques. The method, listed as an RMP by the Joint Committee for Traceability in Laboratory Medicine (JCTLM database identification number C11RMP9), will be used to determine the absolute concentration of Aβ1-42 in a Certified Reference Material (CRM) to harmonize CSF Aβ1-42 measurements across techniques and analytical platforms. The described workflow should be of relevance for the development of candidate reference methods for peptides and proteins within other areas of medicine.
Protocol
NOTE: This protocol requires aliquots of at least 50 µL, with a concentration of 50 µg/mL for each Aβ peptide, as starting material. The Aβ peptides should be dissolved in 20% acetonitrile (ACN) and 4% concentrated ammonia solution in deionized water (v/v) and stored at 80 °C.
Caution: See Table 1 for safety information.
1. Preparation of the Solutions
Prepare 100 mL of 20% ACN and 4% concentrated ammonia solution in deionized water (v/v) by diluting 20 mL of ACN and 4 mL of concentrated ammonia (~25%) in deionized water. Adjust the final volume to 100 mL with deionized water. Make fresh daily.
Prepare 50 mL of 5 M guanidine-hydrochloride by dissolving 26.08 g of guanidine-hydrochloride in deionized water to a final volume of 50 mL. Store at 20 °C and make fresh monthly.
Prepare 200 mL of 4% phosphoric acid in deionized water (v/v) by diluting 9.4 mL of concentrated phosphoric acid (~85%) in deionized water. Adjust the final volume to 200 mL with deionized water. Store in the refrigerator and make fresh weekly.
Prepare 50 mL of 75% ACN and 10% concentrated ammonia solution (v/v) in deionized water by diluting 37.5 mL of ACN and 5 mL of concentrated ammonia (~25%) in deionized water. Adjust the final volume to 50 mL with deionized water. Make fresh daily.
Thaw at least 2.5 mL of human CSF for the calibrators, obtained from de-identified leftover samples from routine clinical analysis.
Prepare artificial CSF containing 150 mM Na, 3.0 mM K, 1.4 mM Ca, 0.8 mM Mg, 1.0 mM P, and 155 mM Cl in deionized water and add bovine serum albumin to a final concentration of 4 mg/mL. Only 1 mL is needed per analysis, but prepare a large volume, aliquot it, and store for future use.
2. Preparation of the Calibrators
Prepare 0.5 mL of 4 µg/mL 15N-Aβ1-42 peptide by adding 40 µL of 50 µg/mL 15NAβ142 to 0.46 mL of 20% ACN and 4% concentrated ammonia in a 0.5-mL microcentrifuge tube. Mix on a vortex mixer for 1 min.
Prepare 2 mL of 100 ng/mL 15N-Aβ1-42 peptide by adding 50 µL of the 4 µg/mL 15NAβ142 to 1.95 mL of 20% ACN and 4% concentrated ammonia in a 2-mL microcentrifuge tube. Mix on a vortex mixer for 1 min.
Prepare six calibrator solutions (A-F) by mixing the volumes of each solution indicated in Table 2. Use 0.5, 1.5, and 2 mL microcentrifuge tubes. Mix on a vortex mixer for 1 min.
Prepare the final calibrators (in duplicate) in 0.5 mL microcentrifuge tubes by adding the corresponding calibration solutions and human CSF according to Table 3. Mix on a vortex mixer for 1 min.
3. Preparation of the Internal Standard
Prepare 2 mL of 0.8 µg/mL 13C-Aβ1-42 peptide by adding 32 µL of 50 µg/mL 13CAβ142 to 1.968 mL of 20% ACN and 4% concentrated ammonia in a 2-mL microcentrifuge tube. Mix on a vortex mixer for 1 min.
Prepare 5 mL of 16 ng/mL 13C-Aβ1-42 peptide by adding 0.1 mL of 0.8 µg/mL to 4.9 mL of 20% ACN and 4% concentrated ammonia in a 5 mL microcentrifuge tube. Mix on a vortex mixer for 1 min.
4. Preparation of the Response Factor Sample
NOTE: The response factor (RF) determination is performed to determine the concentration of the labeled peptide used for calibration (15N-Aβ1-42). This requires that the concentration of the native Aβ1-42 peptide has been determined using amino acid analysis (AAA). Thus, the volume and concentration of native Aβ1-42 peptide aliquots need to fulfill the requirements of the AAA.
Prepare 0.5 mL of 4 µg/mL native (unlabeled)Aβ1-42 by adding 40 µL of 50 µg/mL native Aβ1-42 to 0.46 mL of 20% ACN and 4% concentrated ammonia in a 0.5-mL microcentrifuge tube. Mix on a vortex mixer for 1 min.
Prepare a 2-mL 40 ng/mL mix of native and 15N-Aβ1-42 by adding 20 µL of 4 µg/mL native Aβ1-42 and 20 µL of 4µg/mL 15N-Aβ1-42 to 1.96 mL of 20% ACN and 4% concentrated ammonia in a 2 mL microcentrifuge tube. Mix on a vortex mixer for 1 min.
Add 20 µL of the 40 ng/mL mix to 0.38 mL of artificial CSF in a 0.5 mL microcentrifuge tube. Prepare duplicates and mix on a vortex mixer for 1 min.
5. Sample Preparation
NOTE: Thaw the samples to be measured at room temperature on a roller.
Add 0.18 mL of each calibrator, response factor, and unknown sample (including quality control [QC] samples, if used) to a 1 mL protein 96-deep-well plate, according to Figure 1 (assuming a full plate is used). Make sure to add the samples in, or close to, the bottom of the wells.
Add 20 µL of internal standard to each well (i.e., calibrators, response factors, QCs, and unknowns); it is crucial to release the drop on the side of the well close to the surface of the sample without submerging the pipette tip.
Add 0.2 mL of 5 M guanidine-hydrochloride to each well.
Place the sample plate on a microplate shaker and mix the samples for 45 min at 1,100 rpm. The optimal frequency might differ depending on instrumentation. Set the frequency and amplitude of the mixer so that the solutions are thoroughly mixed and no drops of internal standard or CSF are left unmixed on the side of the wells.
Add 0.2 mL of 4% phosphoric acid to each well. Vortex mix briefly.
6. Solid Phase Extraction
NOTE: In all washing, loading, and elution steps, apply the lowest possible vacuum after adding the solution and gradually increase as needed to load or elute the solution. Disable the vacuum between each loading and elution step.
Put a reservoir tray for waste under a mixed-mode, cation-exchange, 96-well solid phase extraction (SPE) plate in the extraction plate manifold chamber.
Condition the SPE sorbent by adding 0.2 mL of methanol to each well.
Equilibrate the sorbent by adding 0.2 mL of 4% phosphoric acid to each well.
Transfer all samples (about 0.62 mL in each well) from the deep-well plate to the SPE plate. Use an eight-channel pipette when transferring the samples from the deep-well plate to the SPE plate; it is not crucial to transfer the entire or equal volumes of all samples, since the samples contain an internal standard that will compensate for variations.
Wash the sorbent after the samples have passed through by adding 0.2 mL of 4% phosphoric acid to each well.
After the washing solvent has eluted from the sorbent, replace the reservoir tray with a collection plate or tubes.
- Elute the sample from the sorbent by twice adding 50 µL of 75% ACN/10% concentrated ammonia, and note that this solution requires a very low vacuum to pass through the sorbent. Remember to disable the vacuum between each addition.
- OPTIONAL: Seal the collection plate or tubes and freeze them at -80 °C. Remove the seal from the collection plate or tubes before proceeding to step 6.8.2.
- Dry the eluates by using vacuum centrifugation (without applying heat); this can take from one to several hours, depending on the vacuum centrifuge.
- Seal the containers and freeze them at -80 °C.
7. Liquid Chromatography
- Prepare mobile phase A (5% ACN and 0.3% concentrated ammonia in deionized water [v/v]), B (4% deionized water and 0.1% concentrated ammonia in ACN [v/v]), and needle wash (50% ACN and 4% concentrated ammonia in deionized water [v/v]).
- For 500 mL of mobile phase A, add 25 mL of ACN and 1.5 mL of concentrated ammonia to deionized water. Adjust the final volume to 500 mL with deionized water.
- For 500 mL of mobile phase B, add 500 µL of concentrated ammonia and 25 mL of deionized water to ACN. Adjust the final volume to 500 mL with ACN.
- Prepare 250 mL of needle wash by adding 120 mL of ACN and 10 mL of concentrated ammonia to deionized water. Adjust the final volume to 250 mL with deionized water.
- Put the mobile phases A and B and needle wash bottles open in a sonication bath for 20 min before use with the LC system
Dissolve each sample with 25 µL of 20% ACN and 4% concentrated ammonia solution and place them on a shaker for 20 min. Centrifuge down the samples and place them in the autosampler (keep at 7 °C).
- Inject 20 µL of sample on a 1 x 250 mm2 polystyrene-divinylbenzene (reversed-phase) monolithic column maintained at 50 °C.
- Use the LC gradient shown in Table 4 with a flow rate of 0.3 mL/min. Divert the first two and the last five min to waste (post-column) using a divert valve to reduce the contamination of the mass spectrometer.
8. Mass Spectrometric Analysis
NOTE: These parameters were used for a quadrupole-orbitrap hybrid mass spectrometer equipped with aheated electrospray ionization source.
Set the parameters for the ion source according to Table 5.
Set the MS instrument to isolate the 4+ charge states of native Aβ1-42 (1,129.48 mass-to-charge ratio [m/z]), 15N-Aβ1-42 (1,143.00 m/z), and 13C-Aβ1-42 (1,179.50 m/z) in the quadrupole mass analyzer with an isolation width of 2.5 m/z.
Fragment the isolated peptides in the collision cell with a normalized collision energy (NCE) of 17.0. This might need to be tuned for each instrument, even of the same type (and especially if using other types of instruments, such as a triple quadrupole MS).
Record the fragment spectra with a resolution of 17.500, with an automatic gain control target of 2 x 105 charges and a maximum injection time of 250 ms.
9. Data Processing
Use the sum of the product ions (with a mass tolerance of ± 250 milli mass units [mmu]) in Table 6 to calculate the chromatographic areas for each peptide. Note that the ion types and numbers are only shown for native Aβ1-42 product ions, since they are the same for both 15N-Aβ1-42 and 13C-Aβ1-42.
Determine the average response factor of the two response factor samples by dividing the area under the curve (chromatographic peak) of 15N-Aβ1-42 with the area under the curve of native Aβ1-42.
Adjust the concentration of the 15N-Aβ1-42 used for calibration by multiplying it with the response factor calculated in step 9.2.
Construct a calibration curve by plotting the area ratios of 15N-Aβ1-42 to 13C-Aβ1-42 from the two sets of calibrators against the concentration.
Calculate the slope and intercept of the calibration curve using linear regression.
Calculate the area ratio of native Aβ1-42 to the internal standard (13C-Aβ1-42) for unknown samples.
Extrapolate the concentration of unknown samples from the calibration curve using the slope and intercept obtained in step 9.5.
Representative Results
The plate setup in Figure 1 is used for a full plate of samples. If fewer unknown samples are to be analyzed, the second calibrator, RF, and QC sets should be placed after the first half of the unknown samples.
As seen in Figure 2, the calibrators are close to the regression line, with low standard deviations. This method has a lower level of quantification of 150 pg/mL and an upper level of quantification of 4,000 pg/mL. The residual standard deviation of the calibration should of course be as low as possible. If calibration is non-linear, the run should be discarded (depending on severity), and the deviation is most likely due to incorrect pipetting technique and/or errors in the dilution of calibrators. The coefficient of variation (CV) of replicates should be below 20%, but preferably below 10%.
The native 15N- and 13C-Aβ1-42 elute from the LC column simultaneously (since they only differ on the isotopic level), with close to symmetrical peaks and without significant tailing (Figure 3). At least ten measurements should be performed for each chromatographic peak and can be adjusted with the maximum injection time in the instrument method. All three peptides can be measured simultaneously for all measurements (calibration, RF, and unknowns) during the MS analysis. However, if the sensitivity of the method is suboptimal, only peptides of interest should be measured for each injection (i.e., only measure 15N- and 13C-Aβ1-42 for calibrators, native and 15N-Aβ1-42 for RF samples, and native and 13C-Aβ1-42 for unknown samples).
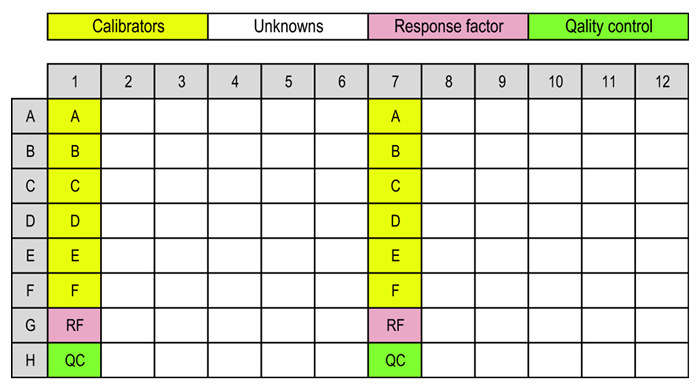 Figure 1:Layouts of SPE and deep-well plates. Typical layout of calibrators (A-F), response factor sample (RF), quality control samples (QC), and unknowns. Please click here to view a larger version of this figure.
Figure 1:Layouts of SPE and deep-well plates. Typical layout of calibrators (A-F), response factor sample (RF), quality control samples (QC), and unknowns. Please click here to view a larger version of this figure.
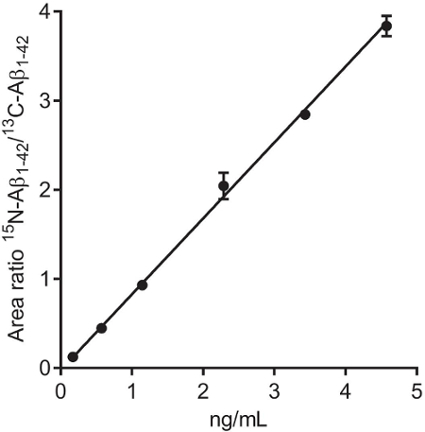 Figure 2:Calibration curve. Calibration curve constructed using 15N-Aβ1-42 at 172, 572, 1,144, 2,287, 3,431, and 4,574 pg/mL (adjusted using the response factor) and 13C-Aβ1-42 as the internal standard in human CSF (n = 2). The area ratio of 15N-Aβ1-42/13C-Aβ1-42 is plotted (Y-axis) against the concentration (X-axis). Please click here to view a larger version of this figure.
Figure 2:Calibration curve. Calibration curve constructed using 15N-Aβ1-42 at 172, 572, 1,144, 2,287, 3,431, and 4,574 pg/mL (adjusted using the response factor) and 13C-Aβ1-42 as the internal standard in human CSF (n = 2). The area ratio of 15N-Aβ1-42/13C-Aβ1-42 is plotted (Y-axis) against the concentration (X-axis). Please click here to view a larger version of this figure.
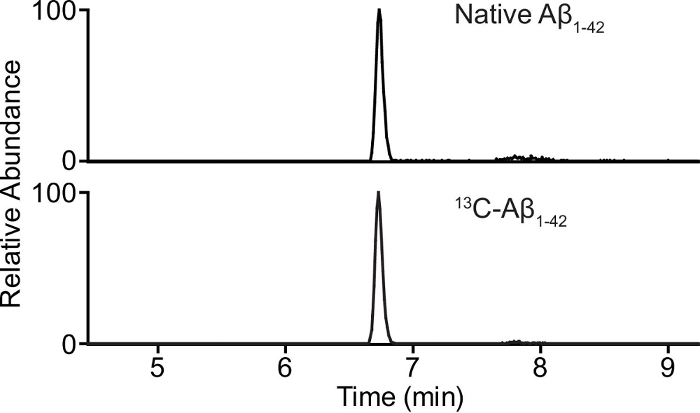 Figure 3:Chromatogram. Chromatogram of 0.500 ng/mL native (endogenous) Aβ1-42 (top panel) and 1.6 ng/mL 13C-Aβ1-42 (bottom panel) in human CSF. Please click here to view a larger version of this figure.
Figure 3:Chromatogram. Chromatogram of 0.500 ng/mL native (endogenous) Aβ1-42 (top panel) and 1.6 ng/mL 13C-Aβ1-42 (bottom panel) in human CSF. Please click here to view a larger version of this figure.
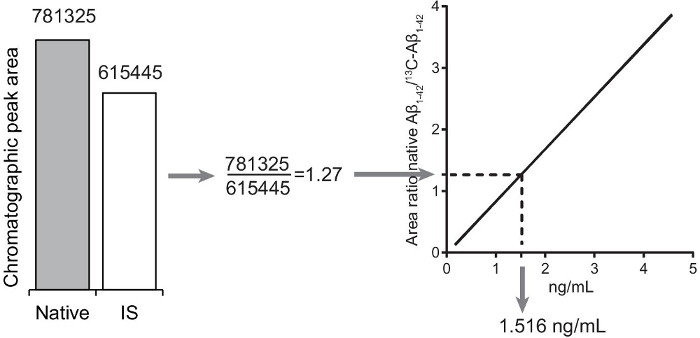 Figure 4:Quantification of unknown Aβ1-42 in unknown samples. The peak area ratio is calculated by dividing the native Aβ1-42 chromatographic peak area with the internal standard (13C-Aβ1-42) chromatographic peak area. The concentration of native Aβ1-42 in the sample is extrapolated from the calibration curve. Please click here to view a larger version of this figure.
Figure 4:Quantification of unknown Aβ1-42 in unknown samples. The peak area ratio is calculated by dividing the native Aβ1-42 chromatographic peak area with the internal standard (13C-Aβ1-42) chromatographic peak area. The concentration of native Aβ1-42 in the sample is extrapolated from the calibration curve. Please click here to view a larger version of this figure.
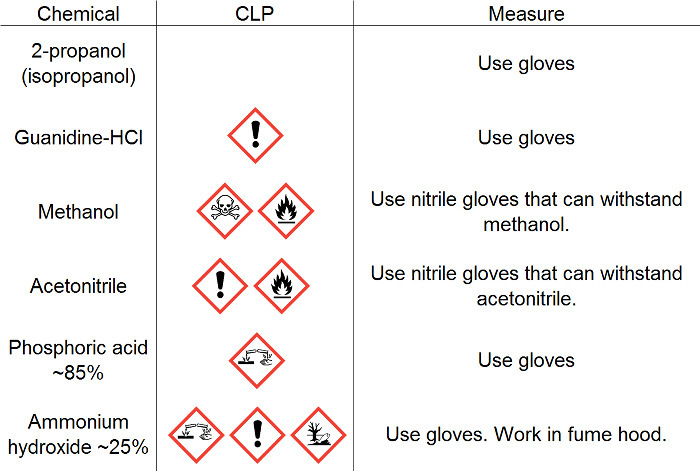 Table 1: Safety information. Safety information for chemicals used for this protocol.
Table 1: Safety information. Safety information for chemicals used for this protocol.
| Calibrator solution | Volume of 100 ng/mL 15N-Aβ1-42 solution (mL) | Volume of 20% ACN & 4% ammonia (mL) | Final volume (mL) | Final 15N-Aβ1-42 concentration (ng/mL) |
| A | 0.20 | 0.30 | 0.50 | 40.00 |
| B | 0.15 | 0.35 | 0.50 | 30.00 |
| C | 0.20 | 0.80 | 1.00 | 20.00 |
| D | 0.10 | 0.90 | 1.00 | 10.00 |
| E | 0.05 | 0.95 | 1.00 | 5.00 |
| F | 0.03 | 1.97 | 2.00 | 1.50 |
Table 2: Calibrator solutions. Calibrator solutions prepared in 20% ACN and 4% concentrated ammonia used for spiking CSF calibrators.
| Calibrator | Volume of corresponding calibrator solution (mL) | Volume of human CSF (mL) | Final volume (mL) | Final 15N-Aβ1-42 concentration (ng/mL) |
| A | 0.02 (A) | 0.18 | 0.20 | 4.00 |
| B | 0.02 (B) | 0.18 | 0.20 | 3.00 |
| C | 0.02 (C) | 0.18 | 0.20 | 2.00 |
| D | 0.02 (D) | 0.18 | 0.20 | 1.00 |
| E | 0.02 (E) | 0.18 | 0.20 | 0.50 |
| F | 0.02 (F) | 0.18 | 0.20 | 0.15 |
Table 3: Calibrators. Calibrators prepared in human CSF.
| Time (min) | % mobile phase B |
| 0 | 5 |
| 1 | 5 |
| 6 | 20 |
| 7 | 90 |
| 9 | 90 |
| 10 | 5 |
| 15 | 5 |
Table 4: LC gradient. The LC gradient used with a constant flow rate of 300 µL/min.
| Parameter | Value |
| Sheath gas | 50 |
| Auxiliary gas | 6 |
| Spray voltage | 4.4 kV |
| S-lens RF | 61 |
| Heater temperature | +190 °C |
| Capillary temperature | +350 °C |
Table 5: Ion source settings. Parameters for the ion source to be set in the instrument tune software.
| Precursor ion | Product ions |
| Native Aβ1-42 (m/z 1129.58, 4+) | 915.19 (b334+), 943.21 (b344+), 975.98 (b354+), 1000.74 (b364+), 1029.51 (b384+), 1054.03 (b394+), 1078.79 (b404+), 1107.06 (b414+), 1163.23 (b313+), 1200.25 (b323+), 1257.29 (b343+), 1300.96 (b353+), 1333.66 (b363+), 1372.00 (b383+), 1405.02 (b393+) |
| 15N-Aβ1-42 (m/z 1143.00, 4+) | 926.41, 954.68, 987.95, 1012.71, 1041.22, 1066.99, 1091.75, 1120.28, 1177.18, 1215.55, 1272.58, 1316.92, 1349.94, 1388.63, 1422.31 |
| 13C-Aβ1-42 (m/z 1179.50, 4+) | 955.33, 985.11, 1019.37, 1045.14, 1074.65, 1100.67, 1126.69, 1156.40, 1253.43, 1313.14, 1358.50, 1393.19, 1432.21, 1466.90 |
Table 6: Ions used for quantification. The 4+ charge states of the precursor ions are isolated in the quadrupole mass analyzer, with an isolation width of 2.5 m/z. The product ions (with a mass tolerance of ± 250 mmu) are used to calculate the chromatographic areas for each peptide. Ion types and numbers are only shown for native Aβ1-42 product ions, since they are the same for both 15N-Aβ1-42 and 13C-Aβ1-42.
Discussion
For the described method, instead of using a surrogate matrix, we used the surrogate analyte approach13,14,15,16, which enables calibration in human CSF. The surrogate analyte approach involves two different isotopically labeled standards. One (15NAβ142) is used to generate the calibration curve in human CSF, while another (13CAβ142) is used as the internal standard. Unknown endogenous Aβ142 concentrations are then extrapolated from the calibration curve constructed using the 15NAβ142/13CAβ142 ratio by the calculated endogenous Aβ142/13CAβ142 ratio. The surrogate analyte approach was used since there is no analyte-free CSF available, and low Aβ142 recovery was observed when using native Aβ142 in a surrogate matrix during method development.
Since 15NAβ142 and native Aβ142 may give different responses in the MS, the concentration of 15NAβ142 is adjusted by measuring an RF sample-an artificial CSF sample containing equal concentrations of 15NAβ142 and native Aβ142, with a known concentration determined by AAA.The response factor might differ between different mass spectrometers due to possible variations in the isotopic purity of the 15N-labeled peptide between batches. Thus, the response factor should be determined for each measurement day.
The most critical steps in this protocol are the preparation of the calibrators and the RF samples. Aβ peptides, especially Aβ1-42, are very hydrophobic and easily stick to pipette tips and tubes surfaces8,17,18. To minimize the loss of Aβ peptides during pipetting, it is extremely important to saturate the pipette tips prior to delivery. Preferably, three volumes of peptide solution should be discarded prior to delivery to a new tube containing solution. Depending on the volume and concentration of the stock solution, this is not always possible. The second-best approach is of course to pipette the peptide solution up and down three times prior to delivery. For the same reason, it is important to use appropriate sizes for tubes, avoiding large void volumes.
Previously published data for the method show that recovery was within 100% (15%)15. The relative errors for the back-calculated calibrators were below 15% of the whole range defined by the calibrator curve19.
One obvious limitation of this technique is that it is low-throughput compared to automated immunoassays. However, the purpose of the described method is high accuracy and not throughput. This method can also be expanded to include Aβ1-38 and Aβ1-40, which are shorter19. Another limitation of this method is that the operator will need extensive mass spectrometry training before running the analysis on the instrument.
Quantification using immunoassays is dependent upon the interaction between the antibody and the antigen. This interaction could be affected by the presence of sample components that may interfere or compete with the interaction. In addition, the interaction may also be affected by the conformation of the antigen. These effects are difficult to control and are believed to be the main reason why it has been difficult to harmonize results between immunoassay platforms and between laboratories. Because quantification with MS is based on directly counting the target molecules relative to a stable, isotope-labeled standard, quantification is absolute and generally unaffected by such matrix effects. In addition, diagnostic protein measurements by immunoassays should be supported by an unbroken chain of higher-order measurement procedures and materials, from validated LC-MS/MS and stable, isotope-labeled internal standards to RMPs and a CRM, thus improving the comparability and reliability of results20,21.
In conclusion, the described RMP for the measurement of Aβ142 in CSF is an important step in developing a CRM that will help to establish general cut-off concentration for Aβ142 in CSF. Exact cut-offs are highly important for accurately diagnosing AD early and are of the utmost importance when new types of disease-modifying drugs reach the clinic.
Disclosures
JP and EP reports no disclosures. HZ has served on the advisory boards of Roche Diagnostics, Eli Lilly, and Pharmasum Therapeutics. KB has served as a consultant or on the advisory boards for Fujirebio Europe, IBL International, and Roche Diagnostics. HZ and KB are co-founders of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg.
Acknowledgments
This work was performed on behalf of the International Federation of Clinical Chemistry Working Group on CSF Proteins, which has the following composition: Kaj Blennow (Chair) and Henrik Zetterberg, Gothenburg University, Sweden; Les Shaw and Magdalena Korecka, University of Philadelphia, PA, USA; Ingrid Zegers, Institute for Reference Materials and Measurements, Geel, Belgium; Piotr Lewczuk, Universitätsklinikum Erlangen, Germany; and Rand Jenkins, PPD Bioanalytical Laboratory, Richmond, VA, USA; Randall Bateman, Washington University, MO, USA; and H Vanderstichele, Innogenetics NV, Ghent, Belgium. This work was also part of the Global Consortium for Biomarker Standardization CSF Reference Methods Working Group, which is led by Maria C. Carrillo, Ph.D., Senior Director, Medical & Scientific Relations, Alzheimer's Association. The study was supported by The Swedish Research Council (grants #14002, #521-2011-4709, and #2013-2546); the Knut and Alice Wallenberg Foundation; Demensförbundet; and Emil and Wera Cornell's, Aina Wallström and Mary-Ann Sjöblom's, Gun and Bertil Stohne's, Magnus Bergvall's, and Gamla Tjänarinnor's Foundations.
References
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368(9533):387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Dubois B, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014;13(6):614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- Buchhave P, et al. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69(1):98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- Olsson B, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. The Lancet Neurology. 2016;15(7):673–684. doi: 10.1016/S1474-4422(16)00070-3. [DOI] [PubMed] [Google Scholar]
- Bjerke M, et al. Confounding factors influencing amyloid Beta concentration in cerebrospinal fluid. Int J Alzheimers Dis. 2010. [DOI] [PMC free article] [PubMed]
- Dumurgier J, et al. Intersite variability of CSF Alzheimer's disease biomarkers in clinical setting. Alzheimers Dement. 2013;9(4):406–413. doi: 10.1016/j.jalz.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Mattsson N, et al. CSF biomarker variability in the Alzheimer's Association quality control program. Alzheimers Dement. 2013;9(3):251–261. doi: 10.1016/j.jalz.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JH, Korecka M, Toledo JB, Trojanowski JQ, Shaw LM. Clinical Utility and Analytical Challenges in Measurement of Cerebrospinal Fluid Amyloid-beta1-42 and tau Proteins as Alzheimer Disease Biomarkers. Clin Chem. 2013;59(6):903–916. doi: 10.1373/clinchem.2013.202937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, et al. Reference measurement procedures for Alzheimer's disease cerebrospinal fluid biomarkers: definitions and approaches with focus on amyloid beta42. Biomark Med. 2012;6(4):409–417. doi: 10.2217/bmm.12.39. [DOI] [PubMed] [Google Scholar]
- Ahmadkhaniha R, Shafiee A, Rastkari N, Kobarfard F. Accurate quantification of endogenous androgenic steroids in cattle's meat by gas chromatography mass spectrometry using a surrogate analyte approach. Anal Chim Acta. 2009;631(1):80–86. doi: 10.1016/j.aca.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Jemal M, Schuster A, Whigan DB. Liquid chromatography/tandem mass spectrometry methods for quantitation of mevalonic acid in human plasma and urine: method validation, demonstration of using a surrogate analyte, and demonstration of unacceptable matrix effect in spite of use of a stable isotope analog internal standard. Rapid Commun Mass Spectrom. 2003;17(15):1723–1734. doi: 10.1002/rcm.1112. [DOI] [PubMed] [Google Scholar]
- Leinenbach A, et al. Mass spectrometry-based candidate reference measurement procedure for quantification of amyloid-beta in cerebrospinal fluid. Clin Chem. 2014;60(7):987–994. doi: 10.1373/clinchem.2013.220392. [DOI] [PubMed] [Google Scholar]
- Li W, Cohen LH. Quantitation of endogenous analytes in biofluid without a true blank matrix. Anal Chem. 2003;75(21):5854–5859. doi: 10.1021/ac034505u. [DOI] [PubMed] [Google Scholar]
- Perret-Liaudet A, et al. Cerebrospinal fluid collection tubes: a critical issue for Alzheimer disease diagnosis. Clin Chem. 2012;58(4):787–789. doi: 10.1373/clinchem.2011.178368. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, et al. Effect of sample collection tubes on cerebrospinal fluid concentrations of tau proteins and amyloid beta peptides. Clin Chem. 2006;52(2):332–334. doi: 10.1373/clinchem.2005.058776. [DOI] [PubMed] [Google Scholar]
- Pannee J, et al. Reference measurement procedure for CSF amyloid beta (Aβ)1-42 and the CSF Aβ1-42 /Aβ1-40 ratio - a cross-validation study against amyloid PET. J Neurochem. 2016;139(4):651–658. doi: 10.1111/jnc.13838. [DOI] [PubMed] [Google Scholar]
- Thienpont LM, Van Houcke SK. Traceability to a common standard for protein measurements by immunoassay for in-vitro diagnostic purposes. Clin Chim Acta. 2010;411(23-24):2058–2061. doi: 10.1016/j.cca.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Vesper HW, Thienpont LM. Traceability in laboratory medicine. Clin Chem. 2009;55(6):1067–1075. doi: 10.1373/clinchem.2008.107052. [DOI] [PubMed] [Google Scholar]