

Abstract
Nitric Oxide (NO•) is a small radical, which mediates multiple important cellular functions in mammals, bacteria and plants. Despite the existence of a large number of methods for detecting NO• in vivo and in vitro, the real-time monitoring of NO• at the single-cell level is very challenging. The physiological or pathological effects of NO• are determined by the actual concentration and dwell time of this radical. Accordingly, methods that allow the single-cell detection of NO• are highly desirable. Recently, we expanded the pallet of NO• indicators by introducing single fluorescent protein-based genetically encoded nitric oxide (NO•) probes (geNOps) that directly respond to cellular NO• fluctuations and, hence, addresses this need. Here we demonstrate the usage of geNOps to assess intracellular NO• signals in response to two different chemical NO•-liberating molecules. Our results also confirm that freshly prepared 3-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-propanamine (NOC-7) has a much higher potential to evoke change in intracellular NO• levels as compared with the inorganic NO• donor sodium nitroprusside (SNP). Furthermore, dual-color live-cell imaging using the green geNOps (G-geNOp) and the chemical Ca2+ indicator fura-2 was performed to visualize the tight regulation of Ca2+-dependent NO• formation in single endothelial cells. These representative experiments demonstrate that geNOps are suitable tools to investigate the real-time generation and degradation of single-cell NO• signals in diverse experimental setups.
Keywords: Molecular Biology, Issue 121, Ca2+, eNOS, Fluorescence Microscopy, Fura-2, Genetically Encoded Probes, Live-Cell Imaging, Multichannel Imaging, Nitric Oxide, NO•-Donors, NOC-7, Single Cell Analysis, SNP
Introduction
We have recently developed a novel class of genetically encoded fluorescent NO• probes, called the geNOps1. These sensors consist of a simply built, bacteria-derived, NO• binding domain2, which is conjugated to a distinct fluorescent protein (FP) variant3 (cyan, green or orange FP). NO• binding to the non-heme iron(II) center4 within the geNOps instantly reduces the fluorescence intensity1. Importantly, the geNOps fluorescence recovers rapidly and fully when intracellular NO• levels decline1. Accordingly, geNOps allow real-time imaging of (sub)cellular NO• fluctuations. Although the NO• sensing mechanism of geNOps remain unclear so far, they have proven to be excellent NO• reporters, and thus have the potency to open up a new era of polychromatic, quantitative NO• bioimaging with high spatial and temporal resolutions1,5. Other available fluorescent NO• probes are based on small chemical compounds, which need to be loaded into cells, and are irreversibly modified by NO•6. Additional disadvantages of NO• sensitive small fluorophores are their potential cytotoxicity and relatively low specificity which make it difficult to use them in a reliable, analytical and conclusive manner7,8,9. Although the effective usage of genetically encoded fluorescent probes requires efficient gene transfer techniques, FP-based genetically encoded sensors have emerged as indispensable tools that have revolutionized our understanding of the inner functioning of cells10,11. Before the development of the single FP-based geNOps, a Förster resonance energy transfer (FRET)-based NO• sensor, referred to as NOA-112, was constructed. Sato et al. designed this sophisticated probe that consists of two subunits of the NO•-sensitive soluble guanylate cyclase (sGC), both conjugated to FRET-based sensors reporting cyclic guanosine monophosphate (cGMP) levels12. As this probe responds to cGMP, it only indirectly senses intracellular NO• fluctuations12. Although NOA-1 responds to NO• elevations in the nano-molar range, this tool has not been used frequently so far, probably due to limitations regarding the availability and practicability of this bulky bipartite sensor.
Versatile functions of NO•, which impact fundamental biological processes have been well characterized13,14. Many studies proved that the NO• concentration within cells and subdomains determines cell fate in health and diseases14,15,16. In mammalians, NO• is mainly generated enzymatically in various cell types by the well characterized nitric oxide synthase (NOS) family17. So far, three isoforms of NOS have been described18,19,20; these are the Ca2+/calmodulin-dependent endothelial NOS (eNOS or NOS-3)18 and neuronal NOS (nNOS or NOS-1)19, and the Ca2+/calmodulin-independent constitutively active inducible NOS (iNOS or NOS-2)20. Moreover, the existence of mitochondrial NOS (mtNOS) has also been suggested21. However, mtNOS is considered as a splicing variant of nNOS, and is therefore not separately classified as an isoform21. Another isoform, apart from those in mammalian cells, is the so-called bacterial NOS (bNOS), mainly found in gram-positive bacteria22. The enzymatic production of NO• is highly controlled and depends on the availability of several cofactors such as nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), tetrahydrobiopterin (BH4), molecular oxygen and L-arginine17. The cationic amino acid L-arginine is the substrate that is converted to L-citrulline upon NO• production17. In addition to the highly regulated enzymatic generation of NO•, it has been postulated that the radical can be reduced non-enzymatically from nitrite pools by mitochondria under hypoxic conditions23. Once NO• is produced within a cell, it can freely diffuse through biomembranes14,15. However, the very short half-life of this radical is mainly determined by the environmental conditions, and various pathways and chemical reactions efficiently degrade NO• levels24. Eventually, the formation, diffusion, and degradation of NO• depends on diverse environmental parameters which determine the effective concentration of the highly biologically active molecule24.
The geNOps technology allows the direct detection of (sub)cellular NO• fluctuation1 and is therefore suitable to reinvestigate, and newly discover the mechanisms responsible for the buildup and decomposition of cellular NO• signals. Here, we provide simple protocols and representative results for the usage of geNOps to visualize exogenously evoked and endogenously generated NO• profiles, on the level of individual cells. Moreover, the geNOps technology can be adapted for application in other cell model systems to study the complex patterns of NO• formation, diffusion, and degradation in response to diverse cellular stimuli and stresses.
Protocol
1. Preparation of Chemical Buffers and Solutions
Prepare a storage buffer containing 135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 2.6 mM NaHCO3, 0.44 mM KH2PO4, 0.34 mM Na2HPO4, 10 mM D-glucose, 2 mM L-glutamine, 1x MEM vitamins, 1x MEM amino acids, 1% pen strep, and 1% amphotericin B. Dissolve all components in distilled water and stir the buffer for 20 min using a magnetic stirrer at room temperature. Adjust the pH to 7.44 using 1 M NaOH. Upon the dropwise addition of NaOH, measure pH using a pH meter while continuously stirring.
Prepare a physiological Ca2+-containing buffer which consists of 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM D-glucose, and 10 mM HEPES. Adjust the pH to 7.4 using NaOH as described in step 1.1.
Prepare a Ca2+-free buffer which consists of the same ingredients as listed in step 1.2. Use 1 mM EGTA instead of 2 mM.
Solubilize Fura-2AM in DMSO to obtain a 1 mM stock solution. Aliquot the stock solution in tightly sealed vials and store at -20 °C for up to one month. If frozen, allow the stock solution to equilibrate at room temperature for at least 1 hr protected from light. Dilute the Fura-2AM stock solution into 1 ml storage buffer (step 1.1) to obtain a final concentration of 3.3 µM.
Prepare 1 ml of a 100 mM histamine stock solution in distilled water (pH 7.0). Dilute the 100 mM histamine stock solution in 100 ml Ca2+-containing physiological buffer to a final concentration of 100 µM histamine.
Solubilize Nω-Nitro-L-arginine (L-NNA) in 100 ml of calcium-containing physiological buffer to obtain a final concentration of 300 µM. Store the solutions at 37 °C water bath for at least 1 hr until the L-NNA is completely dissolved.
Solubilize 10 mg NOC-7 in distilled water (pH 7.0) to obtain a 10 mM stock solution. Aliquot the stock solution in small aliquots in tightly sealed vials and store immediately at -70 °C. Dilute the NOC-7 stock solution in a physiological buffer to obtain a final concentration of 10 µM NOC-7. NOTE: NOC-7 is a small chemical compound which spontaneously releases NO with a short half-life. Always prepare the working buffers that consists NOC-7 just prior to each experiment.
Make a 1 mM sodium nitroprusside (SNP) solution in a physiological calcium buffer and prepare a 10 µM dilution. NOTE: Always prepare small amounts (~10 ml) of NO-donor solutions because of the rapid degradation rate.
Prepare a phosphate buffered solution (PBS) consisting of 137 mM NaCl, 2.7 mM KCl, 9.2 mM Na2HPO4, and 1.5 mM KH2PO4. Adjust the pH value to 7.44 with NaOH/HCl.
2. Cell Preparation
NOTE: In order to achieve uniformity in the performance of measuring nitric oxide (NO•) in single cells using geNOps, cells need to be pre-incubated with an iron(II)/vitamin C containing buffer prior to imaging experiments in order to restore Fe2+ within the NO•-binding domain of the NO•-probes. In case of EA.hy926 and HEK293 cells, incubation for 20 min with the iron(II) booster solution leads to full activation of the NO• sensors.
Seed 5.5 x 105 EA.hy926 or HEK293 cells for next day or 3.5 x 105 cells for the day after next day, on 30-mm microscope cover glasses into a well of a 6-well plate. Incubate cells at 37 °C in a humidified environment with 5% CO2. NOTE: Detailed information regarding adenoviral infection of mammalian cells and virus construction has been described by Zhang et al.25. This step does not apply for the HEK 293 cells stably expressing G-geNOp.
Take fetal calf serum (FCS) and antibiotic-free Dulbecco's eagle modified medium (DMEM) and add adeno-associated virus type 5 (AAV5) vector carrying the gene encoding for G-geNOp (MOI: 500 for EA.hy926 cells yield almost 100% positive cells; MOI: 1 is efficient for HEK293 cells). NOTE: Use of adeno-associated viral vectors is assigned to risk group 2. Biosafety level 2 containment is generally required for work with this vector. If necessary, virus infection can also be performed in FCS containing medium. Alternatively, cells can be transiently transfected using lipid-based carriers1.
Remove the culture medium and wash cells with pre-warmed (37 °C) PBS. Add 1 ml of DMEM/AAV5 medium on each well for 1 hr. This step does not apply for the HEK 293 cells stably expressing G-geNOp.
Add 1 ml of 20% FCS-containing DMEM on each well to a final concentration of 10% FCS. Do not remove the AAV5-containing media from the cells. Gently seesaw the plate to homogenize the medium within the wells. Incubate cells for 48 hr at 37 °C in a humidified environment with 5% CO2. This step does not apply to the HEK 293 cells stably expressing G-geNOp.
After 48 hr, wash cells with pre-warmed PBS. Subsequently add 2 ml pre-warmed storage buffer (section 1.1) to each well and incubate cells at room RT for at least 1 hr protected from light radiation.
Replace the storage buffer with 1 ml/well of the iron(II) booster solution at room temperature (RT). Incubate cells for exactly 20 min in darkness. NOTE: Do not exceed or reduce the optimal incubation time as this might affect the responsiveness of the NO• probes.
Wash cells once with storage buffer and incubate each well with 2 ml storage buffer for at least 2 hr at RT in order to allow the cells to equilibrate.
Replace the storage buffer with 3.3 µM Fura-2AM in 1 ml storage buffer for 45 min at RT, protected from light.
Wash cells twice with storage buffer and incubate, once again for at least 30 min in order to allow cells to equilibrate.
3. Live Cell Imaging of NO• and Ca2+ Signals in Individual Cells
Fix a 30 mm cover slip coated with EAhy.926 or HEK293 cells (from step 2.1) in a metal perfusion chamber and place it on the microscope. Connect the influx tube to the buffer reservoirs and the efflux to a vacuum pump. Ensure a consistent flow and avoid draining of the cover slip.
Start the gravity-driven perfusion with physiological calcium buffer (from step 1.2) using a semi-automatic perfusion system. NOTE: Such a system consists of buffer reservoirs, respective tubing, magnetic valves that are electronically controlled and a vacuum pump (see Figure 1B). The flow rate can range from 1 to 3 ml/min, dependent on the height of the perfusion reservoirs. For consistent local drug application, the flow rate of all used reservoirs should be approximately equal. This should be tested prior to imaging experiments. Consider that endothelial cells respond to increased shear stress which can be induced by rigorous perfusion.
Switch on the imaging system and allow warm up of all devices for 30 min.
Define imaging settings using respective software. Select excitation wavelength 340 nm and 380 nm for fura-2 imaging and 480 nm for exciting G-geNOp. To minimize fluorescence bleaching increase the camera binning to 4 and reduce excitation intensities and exposure times. See also steps 3.6-3.8. NOTE: Imaging settings and parameters depend on the devices used, fura-2 loading efficiency and G-geNOp expression levels.
Select the imaging region by moving the xyz-table of the microscope until several fluorescent cells are in the focus. Then define regions of interest (ROIs) via the respective software tool. Draw regions covering several whole single fluorescent cells per imaging field manually. In addition, define a background region of similar size. NOTE: Once images have been acquired and stored ROIs can also be defined newly after the imaging process for further analysis using respective imaging analysis software (see Materials List).
Start collecting data on an inverted and advanced fluorescent microscope with a motorized sample stage, and a monochromatic light source. Alternately excite at 340 nm and 380 nm for Fura-2AM and 480 nm for G-geNOp, respectively. Set respective exposure times so that for all channels, a clear fluorescence signal is detectable over time. See also step 3.4. NOTE: This depends on the intensity of the excitation light and the camera binning. For example, use 15% intensity of the excitation light, a camera binning of 4 and 150 ms for 340 nm, 50 ms for 380 nm, and 300 ms for 480 nm. See also step 3.4.
Collect the emitted light at 510 nm for fura-2/am, 520 nm for G-geNOp using a charge-coupled device (CCD) camera with appropriate filter set consisting of the 500 nm exciter, a 495 nm dichroic and a 510-520 nm emitter. Record one total frame every 3 sec.
Record the first minutes (if necessary up to 3 min) in physiological Ca2+ buffer (1.2) to obtain the baseline of respective fluorescence signals over time.
Once a stable baseline fluorescence is observed, switch to 100 µM histamine or ATP (1 µM or 100 µM) containing physiological Ca2+ buffer to stimulate cells for 3 min. NOTE: EA.hy926 cells that respond to the agonists show a sharp increase of the fura-2 ratio (fluorescence excited at 340 nm divided through fluorescence excited at 380 nm) and a clear decrease of the G-geNOp fluorescence signal.
Switch back to the physiological Ca2+ buffer without histamine or ATP and L-NNA for 5 min to remove the compounds from the cells. This step might be prolonged until the fluorescence changes are recovered completely.
Administrate 10 µM NOC-7 in physiological Ca2+ buffer for 2 min using the perfusion system. The NO donor strongly affects G-geNOp fluorescence which usually decreases by >20% in response to 10 µM NOC-7. In EA.hy926 cells the NOC-7 effect is approximately 3 fold stronger compared to the agonist induced G-geNOp fluorescence quench.
Wash out the NO•-releasing compound for approximately 10 min with physiological Ca2+ buffer and stop recording once basal fluorescence is recovered.
4. Data Analysis
Export acquired average fluorescence intensity data of single cells over time, to the data analysis software.
Subtract respective background values and calculate the ratio of 340 nm by 380 nm of the respective fura-2 signals of each single cell over time.
Subtract the background values of the G-geNOp channel to obtain the actual fluorescence intensity of the NO probe (F) over time using any calculation software.
Take baseline fluorescence values as F0 (F0 is the fluorescence of the NO probe over time without stimulation). See also step 4.5 and Figure 1C.
Calculate a function over time for the fluorescence bleaching effects using the following equation: F0 = Finital•exp(-K•Time) + Fplateau. Finital: maximal fluorescence signal once imaging is started; K: rate constant of fluorescence bleaching over time; Fplateau: fluorescence minimum reached by bleaching over time; See also steps 4.3- 4.4 and Figure 1C. NOTE: For the approximation, all fluorescence values over time prior to and after cell stimulation might be used. Further details are specified by Bentley et al.27.
To normalize the G-geNOp signal over time, calculate 1-F/F0 (Steps 4.3-4.5). See Figure 1C and 1D.
Representative Results
Visualization of Single-cell NO• Profiles in Response to Transiently Applied NO Donors
We used an HEK cell clone which stably expresses G-geNOp (Figure 1A), in order to visualize NO• signals on the single cell-level in response to two different NO-liberating small chemical compounds, NOC-7 and SNP. The NO• donors were consecutively applied to and removed from cells during imaging using a gravity-based perfusion system that ensured continuous flow (Figure 1B). All cells expressing G-geNOp with different intensities showed a clear reduction of fluorescence in response to NOC-7 and SNP (Figure 1C), indicating fast NO• accumulation within cells upon the addition of the NO• donors. Normalized fluorescence signals (1-F/F0) demonstrated that both NO• donors evoked homogeneous cellular NO• elevations that completely recovered upon washout of the NO•-releasing compounds (Figure 1D). However, 10 µM SNP induced only 50% of the cellular NO• signal (9.63 ± 1.05%, n = 3/38) that was reached by 10 µM NOC-7 (18.10 ± 1.20%, n = 3/38, p < 0.0001). In order to achieve equal intracellular NO• levels with both NO• donors, a concentration of 1 mM of SNP was required (Figures 1C, 1D).
Next, we tested the capacity of freshly prepared versus expired NOC-7 to elevate intracellular NO• levels in HEK cells. For this purpose, we prepared four experimental buffers containing 5 µM NOC-7. The NO• donor was either added freshly just prior to measurement, or kept within reservoirs for 1 hr, 2 hr, and 3 hr at room temperature prior to measurement. The different buffers were consecutively applied to and removed from the G-geNOp expressing cells using a perfusion system (Figure 2B). This approach unveiled the stability of aqueous NOC-7 solutions which, as expected, showed decreased capacities over time to elevate intracellular NO• levels (Figures 2A, 2C). Interestingly, NO• signals recovered significantly faster upon the removal of expired buffers, as compared to the intracellular NO• response that was evoked by fresh NOC-7 (Figure 2C), perhaps indicating adhesion of the intact NO•-liberating molecules at cellular components.
Simultaneous Visualization of Ca2+ and NO• Signals in Single Endothelial Cells
In order to study Ca2+-triggered NO• formation in endothelial cells, the commonly used endothelial immortalized cell-surrogate, EA.hy926 cell line, was transiently transfected with G-geNOp and loaded with Fura-2/am (see Protocol 2.8). Transfection yielded approximately 10% of G-geNOp positive endothelial cells (n = 6, Figure 3A), which is sufficient to record Ca2+-evoked NO• production on the level of single endothelial cells. However, we achieved nearly 100% G-geNOp positive EA.hy926 cells using an adeno-associated viral vector (n = 6; see Protocol 2.2). Prior to multichannel measurements, cells were incubated for 20 min at room temperature in storage buffer consisting L-NNA, a potent irreversible NOS-inhibitor26. Control cells were kept in the same storage buffer without L-NNA (see Protocol 1.1) (Figure 3B). Treatment of control cells with histamine, a potent inositol 1,4,5-trisphosphate (IP3)-generating agonist, instantly elevated cytosolic Ca2+ levels followed by a gradual increase of intracellular NO• until the agonist was removed (Figures 3C, 3D). Cells pre-treated with the NOS-inhibitor showed similar cytosolic Ca2+ signals, whereas the intracellular NO• level remained almost unaffected in response to histamine (Figure 3E). EA.hy926 cells expressing G-geNOp were also treated with 1 µM and 100 µM of the IP3-generating agonist ATP in order to test whether or not geNOps are suitable to monitor NO• signals in response to both low physiological and supra-physiological concentrations of an agonist (Figure 3F). In the endothelial cell line 1 µM ATP evoked a clear cytosolic NO• signal, which was approximately half of the signal obtained by 100 µM ATP (Figure 3F).
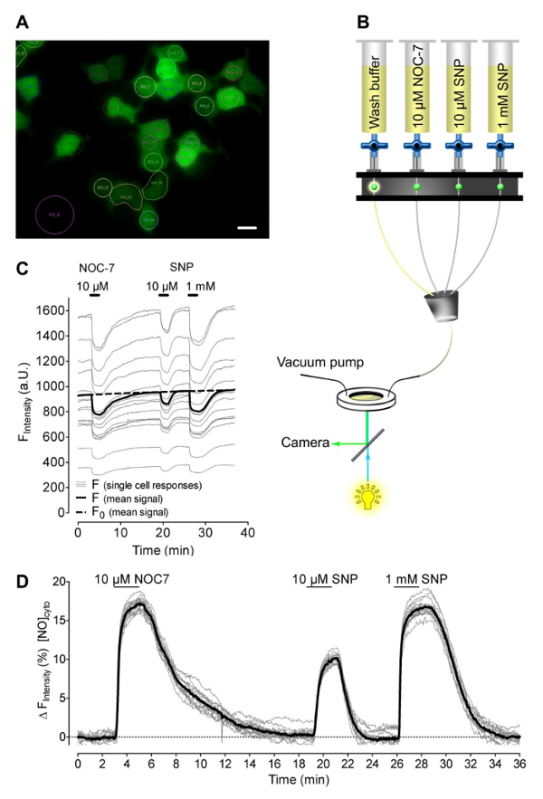 Figure 1: Intracellular NO profiles in response to different NO-liberating molecules. (A) Wide field images of HEK cells stably expressing cytosolic G-geNOp. Scale bar = 20 µm. (B) Schematic illustration of a gravity based half-automatic perfusion system for the controlled application and removal of NOC-7 and SNP. (C) Representative (out of 3 independent experiments) non-normalized single-cell fluorescence intensity traces in arbitrary units versus time of HEK cells stably expressing cytosolic G-geNOp in response to 10 µM NOC-7, 10 µM SNP, and 1 mM SNP. Black bold curve represents the average curve of 26 single-cell traces (light grey curves). Dotted black curve represents F0, which was used for normalization. (D) Normalized and inverted single traces (1-F/F0, light grey curves), and mean curve (black bold curve) over time in response to 10 µM NOC-7, 10 µM SNP, and 1 mM SNP extracted from panel C. Please click here to view a larger version of this figure.
Figure 1: Intracellular NO profiles in response to different NO-liberating molecules. (A) Wide field images of HEK cells stably expressing cytosolic G-geNOp. Scale bar = 20 µm. (B) Schematic illustration of a gravity based half-automatic perfusion system for the controlled application and removal of NOC-7 and SNP. (C) Representative (out of 3 independent experiments) non-normalized single-cell fluorescence intensity traces in arbitrary units versus time of HEK cells stably expressing cytosolic G-geNOp in response to 10 µM NOC-7, 10 µM SNP, and 1 mM SNP. Black bold curve represents the average curve of 26 single-cell traces (light grey curves). Dotted black curve represents F0, which was used for normalization. (D) Normalized and inverted single traces (1-F/F0, light grey curves), and mean curve (black bold curve) over time in response to 10 µM NOC-7, 10 µM SNP, and 1 mM SNP extracted from panel C. Please click here to view a larger version of this figure.
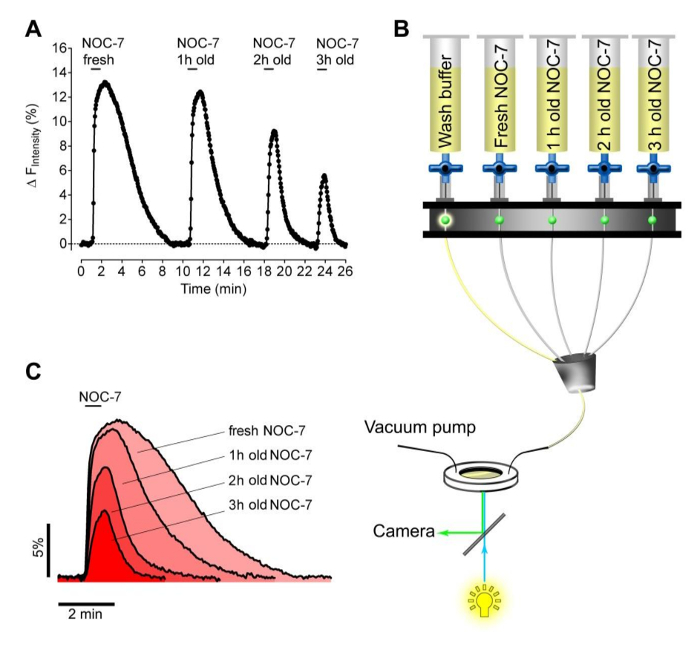 Figure 2: Stability test of NOC-7 using stably G-geNOp expressing HEK cells. (A) Representative NO• concentration response curve over time of stably G-GeNOps expressing HEK cells upon application of fresh and old NOC-7 buffer solutions. All NOC-7 containing buffers were prepared initially with a final concentration of 5 µM using the same stock solution (50 mM). The elapse time of the respective solutions after preparation until imaging amounts to 1 hr, 2 hr, and 3 hr as indicated. (B) Schematic illustration of a gravity based half-automatic perfusion system to consecutively add and remove NOC-7 containing solutions during imaging. (C) Temporal correlations of cellular NO• signals in response to freshly prepared and old NOC-7 buffers. Please click here to view a larger version of this figure.
Figure 2: Stability test of NOC-7 using stably G-geNOp expressing HEK cells. (A) Representative NO• concentration response curve over time of stably G-GeNOps expressing HEK cells upon application of fresh and old NOC-7 buffer solutions. All NOC-7 containing buffers were prepared initially with a final concentration of 5 µM using the same stock solution (50 mM). The elapse time of the respective solutions after preparation until imaging amounts to 1 hr, 2 hr, and 3 hr as indicated. (B) Schematic illustration of a gravity based half-automatic perfusion system to consecutively add and remove NOC-7 containing solutions during imaging. (C) Temporal correlations of cellular NO• signals in response to freshly prepared and old NOC-7 buffers. Please click here to view a larger version of this figure.
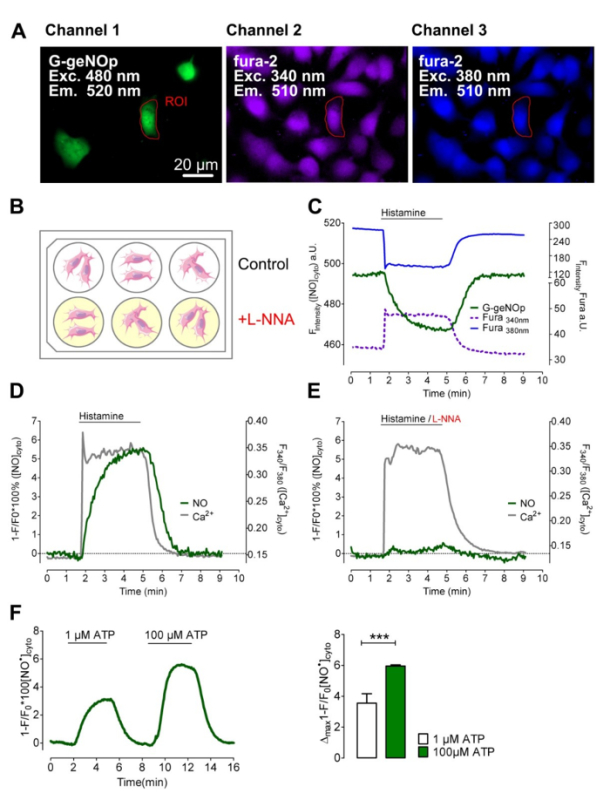 Figure 3: Simultaneous multichannel imaging of NO• and Ca2+ signals in single EA.hy926 cells. (A) Representative wide-field fluorescence images of EAhy.926 cells expressing G-geNOp (left panel) that are loaded with fura-2/am (middle and right panels). Scale bar = 20 µm. (B) Schematic illustration of the treatment of EAhy.926 cells with 500 µM Nω-nitro-L-arginine (L-NNA) for 20 min within a six-well plate prior to measurement. (C) Representative time course of a simultaneous record of fura-2 (excitation: 340 nm/380 nm emission: 510 nm) and G-geNOp signals (excitation: 480 nm; emission: 520 nm) in response to 100 µM histamine. As indicated histamine was removed after 3 min using a perfusion system. (D) Curves represent simultaneous recordings of cytosolic Ca2+ (fura-2 ratio signals, F340/F380 is grey solid line) and NO• (normalized and inverted curve, 1-F/F0 is green solid line) signals over time of a single fura-2/am loaded endothelial cell transiently expressing G-geNOp. Cells were stimulated with 100 µM histamine for 3 min in a Ca2+ (2 mM CaCl2) containing buffer (n = 8/3). (E) Representative simultaneously recorded Ca2+ (grey solid line) and NO• (green solid line) signals over time of an EA.hy926 cell that was pretreated with 500 µM Nω-Nitro-L-arginine (L-NNA) prior to measurement (n = 12/3). Cells were treated with 100 µM histamine in the presence of 2 mM Ca2+. (F) Average curves representing cytosolic NO• signals in response to 1 µM ATP followed by a second cell stimulation with 100 µM ATP. Bars represent mean values ± SD of maximal G-geNOp signals in response to 1 µM ATP (white bar) and 100 µM ATP (green bar) of 4 independent experiments; p < 0.001 vs. 1 µM ATP. P-value was calculated using an unpaired t-test. Please click here to view a larger version of this figure.
Figure 3: Simultaneous multichannel imaging of NO• and Ca2+ signals in single EA.hy926 cells. (A) Representative wide-field fluorescence images of EAhy.926 cells expressing G-geNOp (left panel) that are loaded with fura-2/am (middle and right panels). Scale bar = 20 µm. (B) Schematic illustration of the treatment of EAhy.926 cells with 500 µM Nω-nitro-L-arginine (L-NNA) for 20 min within a six-well plate prior to measurement. (C) Representative time course of a simultaneous record of fura-2 (excitation: 340 nm/380 nm emission: 510 nm) and G-geNOp signals (excitation: 480 nm; emission: 520 nm) in response to 100 µM histamine. As indicated histamine was removed after 3 min using a perfusion system. (D) Curves represent simultaneous recordings of cytosolic Ca2+ (fura-2 ratio signals, F340/F380 is grey solid line) and NO• (normalized and inverted curve, 1-F/F0 is green solid line) signals over time of a single fura-2/am loaded endothelial cell transiently expressing G-geNOp. Cells were stimulated with 100 µM histamine for 3 min in a Ca2+ (2 mM CaCl2) containing buffer (n = 8/3). (E) Representative simultaneously recorded Ca2+ (grey solid line) and NO• (green solid line) signals over time of an EA.hy926 cell that was pretreated with 500 µM Nω-Nitro-L-arginine (L-NNA) prior to measurement (n = 12/3). Cells were treated with 100 µM histamine in the presence of 2 mM Ca2+. (F) Average curves representing cytosolic NO• signals in response to 1 µM ATP followed by a second cell stimulation with 100 µM ATP. Bars represent mean values ± SD of maximal G-geNOp signals in response to 1 µM ATP (white bar) and 100 µM ATP (green bar) of 4 independent experiments; p < 0.001 vs. 1 µM ATP. P-value was calculated using an unpaired t-test. Please click here to view a larger version of this figure.
Discussion
Since the discovery of NO• as an important signaling molecule in biology28, the specific real-time measurement of the radical in single cells, tissues and whole animals with high resolution in a feasible and reliable manner has been aspired. Here we report the application of recently developed genetically encoded fluorescent NO• probes (geNOps) that enable exact live-cell imaging of NO• signals using wide-field fluorescence microscopy1.
To circumvent elaborate and invasive transfection procedures HEK cell clone that stably expresses green fluorescent G-geNOp was used to quantify exogenously generated single-cell NO• profiles. As HEK cells normally do not produce NO• endogenously29, this cell type is suitable for the generation of a geNOp-based sensor cell line, which might be useful for many other applications in co-culture conditions with NO• producing primary cells or even in living animals30. However, in this study we demonstrate the capacity of various NO•-liberating compounds, of different concentrations and stabilities, to evoke intracellular NO• signals using the G-geNOp expressing HEK cell model. Our data revealed that the NO•-donor concentration, quality and method of application eventually determine the patterns of intracellular NO• profiles. Such information is indispensable for the in situ pharmacokinetic characterization of different NO• donors, which are indicative of several diseases. Notably, geNOps have been shown to stably respond to multiple repetitive applications of NO•-donor pulses over a very long time1. Accordingly, the experiments using NO•-liberating compounds presented herein, allow semi-quantitative conclusions regarding the various amplitudes and kinetics of respective cellular NO• signals (Figures 1 and 2).
Although the stably expressing HEK cell clone probably originates from a single cell, a broad heterogeneity of G-geNOps expression levels was observed (Figure 1). This is a common feature of stable cell clones as the transcription of the (genome-integrated) gene of interest is under the control of many factors such as diverse environmental stresses31 that influence cell growth rates32, and the cell cycle status33. The single FP-based geNOps are non-ratiometric probes and, hence, the NO•-induced loss of fluorescence intensity increases with the geNOp expression level1. Accordingly, normalization of the geNOps signals is essential for quantifying cellular NO• signals particularly in case of a comparative analysis. As shown in our recent study, a strict linear correlation between the basal fluorescence intensity of geNOps and the strength of the NO•-induced fluorescence quenching over a broad range of fluorescence intensities has been found1. This is an important feature of geNOps for absolute quantification of cellular NO• signals. As shown in Figure 1, normalization of the G-geNOps signals in response to NOC-7 and SNP revealed homogeneous NO• signals in different HEK cells from the same plate, indicating that HEK cells are not diverse with regards to their capacity to take up and degrade the NO• radical that originates from the NO• donor. In contrast, using geNOps in HeLa cells demonstrated clear heterogeneities of cellular NO• signals among different cells in response to NOC-7. These differences point to cell type-specific NO• metabolism and decomposition rates, which might have multiple implications in cell physiology and pathology, and can be uncovered using the geNOps technology.
Nevertheless, two important features of geNOps have to be considered carefully for the correct usage of the sensors and data interpretations: i) geNOps require adequate iron(II) to fully respond to NO•1 and ii) depending on the FP variant, geNOps can be pH sensitive1. Here we describe a protocol that has been found to be suitable for the nontoxic iron(II) supplementation of geNOps, which are expressed in either HEK, HeLa or EA.hy926 cells (see Protocol 2.6). While it has been demonstrated that cell treatment with iron(II)/vitamin C did not affect cell morphology, cell viability and metabolic activity of cells1, it might be essential to optimize this important step for other cell types and tissues. However, in some experimental conditions, the requirement of iron(II) loading might limit the applicability of geNOps. Notably, it has been shown that ascorbate can reduce NO•35 and ascorbate-iron(II) complexes are able to scavenge NO•36,37. Moreover, excess iron(II) and ascorbate can induce inflammatory responses39 and uncouple eNOS41. Such effects need to be considered when using the geNOps technology. It has been shown that under certain experimental conditions, the intracellular pH is affected significantly34, which has the potential to influence the geNOps fluorescence1. Notably, the cyan and green geNOps variants are relatively pH sensitive showing a decrease of fluorescence upon acidification1. Hence, acute changes of the (sub)cellular pH might simulate false NO• signals when using the pH sensitive geNOps. As suggested in our previous work, the parallel usage of NO• insensitive geNOps (geNOpsmut) as negative controls is recommended to dissect real cellular NO• signal from pH changes1. In addition cellular pH changes can be inspected using pH probes such as SypHer34.
Further, we visualized the endogenous enzymatic NO• formation in response to a physiological Ca2+-mobilizing agonist in the endothelial cell surrogate EA.hy926. The EA.hy926 cell line is a frequently used model system consistently expressing eNOS38. Use of geNOps transiently expressed in EA.hy926 cells, confirmed that IP3-mediated Ca2+ signals evoke profound NO• formation in this cell type, which was almost completely blocked by L-NNA. In order to temporally correlate Ca2+ with NO• signals, G-geNOp-expressing cells were loaded with the UV-excitable chemical Ca2+-indicator fura-2/am. The spectral separation of the Ca2+ bound and unbound fura-2 fluorescence from the G-geNOp signal can be easily achieved with commercially available filter sets40. Imaging both probes unveiled that the Ca2+-triggered enzymatic NO• formation occurs much slower compared to the cytosolic Ca2+ rise in this endothelial cell type. Similar kinetics of single-cell NO• signals in endothelial cells from bovine pulmonary artery upon cell treatment with the IP3-generating agonist bradykinin, as well as shear stresses have been reported using NOA-1, an indirect highly NO•-sensitive sensor12. Accordingly, these data emphasize that the Ca2+-evoked eNOS-derived NO• formation requires a certain starting time until the full enzymatic activity is reached. Although the kinetics of cellular NO• formation, diffusion and degradation can be extracted from other data, e.g. tension-based measurements of NO•-induced vessel relaxation26, the great benefit of fluorescent NO• probes is that they directly convert cellular NO• fluctuations into visible signals in real-time. Hence, imaging cellular NO• signals with geNOps provides high spatial and temporal resolution, and offers unique possibilities in (re)investigating the (sub)cellular NO• homeostasis. For instance, imaging eNOS shuttling42 in combination with the geNOps technology in single endothelial cells might be suitable to correlate NO• formation with the subcellular localization and translocation of the NO•-producing enzyme or other relevant proteins such as calmodulin and caveolin43.
Here we describe the practicable application of G-geNOp expressing HEK and EA.hy926 cells to visualize exogenously and endogenously generated cellular NO• signals on the single-cell level and in real-time on a conventional wide field fluorescence microscope. Our data imply that geNOps are suitable to specifically track (sub)cellular NO• dynamics under various experimental conditions using all kinds of interesting cell types.
Disclosures
E.E, M.W., R.M. and W.F.G., staff members of the Medical University of Graz, have filed a U.K. patent application (patent application number WO2015EP74877 20151027, priority number GB20140019073 20141027) that describe parts of the research in this manuscript. Licenses related to this patent are provided to Next Generation Fluorescence Imaging (NGFI) GmbH (http://www.ngfi.eu/), a spin-off company of the Medical University of Graz.
Acknowledgments
The authors acknowledge C.J. Edgell, Pathology Department, University of North Carolina at Chapel Hill, NC, USA for providing the EA.hy926 cells. Author E.E. is supported by Nikon Austria within the Nikon-Center of Excellence, Graz and is a fellow of the Ph.D. program in Molecular Medicine at the Medical University of Graz. The researchers are also supported by the Ph.D. program Metabolic and Cardiovascular Disease (DK-W1226) of the Medical University of Graz. This work was also funded by the FWF project P 28529-B27. Microscopic equipment is part of the Nikon-Center of Excellence, Graz that is supported by the Austrian infrastructure program 2013/2014, Nikon Austria Inc., and BioTechMed, Graz.
References
- Eroglu E, et al. Development of novel FP-based probes for live-cell imaging of nitric oxide dynamics. Nat Commun. 2016;7:10623. doi: 10.1038/ncomms10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush M, et al. The structural basis for enhancer-dependent assembly and activation of the AAA transcriptional activator NorR. Mo. Microbiol. 2015;95(1):17–30. doi: 10.1111/mmi.12844. [DOI] [PubMed] [Google Scholar]
- Cranfill PJ, et al. Quantitative assessment of fluorescent proteins. Nat. Methods. 2016;13(7):557–562. doi: 10.1038/nmeth.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Autréaux B, Tucker N, Spiro S, Dixon R. Characterization of the Nitric Oxide-Reactive Transcriptional Activator NorR. In: Poole ed.Poole,R.K.,(ed).,, editor. Globins and Other Nitric Oxide-Reactive Proteins, Part B. 1st edition. Vol. 437. Elsevier textbooks, s.l; 2008. pp. 235–251. [DOI] [PubMed] [Google Scholar]
- Strack R. Sensors and probes: Yes to genetically encoded NO• sensors. Nat Methods. 2016;13(4):288. doi: 10.1038/nmeth.3827. [DOI] [PubMed] [Google Scholar]
- Auten RL. Response to 'The use of diaminofluorescein for nitric oxide detection: Conceptual and methodological distinction between NO and nitrosation. Free Radic. Biol. Med. 2011;50(12):1812. doi: 10.1016/j.freeradbiomed.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Sivaraman G, Anand T, Chellappa D. A Fluorescence Switch for the Detection of Nitric Oxide and Histidine and Its Application in Live Cell Imaging. ChemPlusChem. 2014;79(12):1761–1766. [Google Scholar]
- Ye X, Rubakhin SS, Sweedler JV. Detection of nitric oxide in single cells. Analyst. 2008;133(4):423–433. doi: 10.1039/b716174c. [DOI] [PubMed] [Google Scholar]
- Thyagarajan B, Malli R, Schmidt K, Graier WF, Groschner K. Nitric oxide inhibits capacitative Ca2+ entry by suppression of mitochondrial Ca2+ handling. Br J Pharmacol. 2002;137(6):821–830. doi: 10.1038/sj.bjp.0704949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germond A, Fujita H, Ichimura T, Watanabe TM. Design and development of genetically encoded fluorescent sensors to monitor intracellular chemical and physical parameters. Biophy. Rev. 2016;8:121–138. doi: 10.1007/s12551-016-0195-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malli R, Eroglu E, Waldeck-Weiermair M, Graier WF. Filling a GAP-An Optimized Probe for ER Ca2+ Imaging In Vivo. Cell Chem Biol. 2016;23(6):641–643. doi: 10.1016/j.chembiol.2016.06.003. [DOI] [PubMed] [Google Scholar]
- Sato M, Hida N, Umezawa Y. Imaging the nanomolar range of nitric oxide with an amplifier-coupled fluorescent indicator in living cells. Proc Natl Acad Sci USA. 2005;102(41):14515–14520. doi: 10.1073/pnas.0505136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidinger A, Kozlov AV. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules. 2015;5(2):472–484. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolo S. Encyclopedia of life sciences. Chichester: Wiley; 2005. Nitric Oxide in Human Health and Disease. [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonafe F, Guarnieri C, Muscari C. Nitric oxide regulates multiple functions and fate of adult progenitor and stem cells. J Physiol Biochem. 2015;71(1):141–153. doi: 10.1007/s13105-014-0373-9. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012;33(7):829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- Zhou L, Zhu D-Y. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20(4):223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75(6):639–653. doi: 10.1016/j.lfs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26(4):190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Crane BR, Sudhamsu J, Patel BA. Bacterial nitric oxide synthases. Annu Rev Biochem. 2010;79:445–470. doi: 10.1146/annurev-biochem-062608-103436. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7(2):156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- Kelm M. Nitric oxide metabolism and breakdown. Biochim Biophys Acta. 1999;1411(2-3):273–289. doi: 10.1016/s0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. Estrogen-related receptors stimulate pyruvate dehydrogenase kinase isoform 4 gene expression. J Biol Chem. 2006;281(52):39897–39906. doi: 10.1074/jbc.M608657200. [DOI] [PubMed] [Google Scholar]
- Holzmann S, Kukovetz WR, Windischhofer W, Paschke E, Graier WF. Pharmacologic differentiation between endothelium-dependent relaxations sensitive and resistant to nitro-L-arginine in coronary arteries. J Cardiovasc Pharmacol. 1994;23(5):747–756. doi: 10.1097/00005344-199405000-00009. [DOI] [PubMed] [Google Scholar]
- Bentley M, et al. Vesicular calcium regulates coat retention, fusogenicity, and size of pre-Golgi intermediates. Mol Biol Cell. 2010;21(6):1033–1046. doi: 10.1091/mbc.E09-10-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ. Nitric oxide: a unique endogenous signaling molecule in vascular biology. Biosci Rep. 1999;19(2):51–71. doi: 10.1023/a:1020150124721. [DOI] [PubMed] [Google Scholar]
- Upreti M, Kumar S, Rath PC. Replacement of 198MQMDII203 of mouse IRF-1 by 197IPVEVV202 of human IRF-1 abrogates induction of IFN-β, iNOS, and COX-2 gene expression by IRF-1. Biochem Biophys Res Com. 2004;314(3):737–744. doi: 10.1016/j.bbrc.2003.12.156. [DOI] [PubMed] [Google Scholar]
- Lacin E, Muller A, Fernando M, Kleinfeld D, Slesinger PA. Construction of Cell-based Neurotransmitter Fluorescent Engineered Reporters (CNiFERs) for Optical Detection of Neurotransmitters In Vivo. J Vis Exp. 2016. [DOI] [PMC free article] [PubMed]
- de Nadal E, Ammerer G, Posas F. Controlling gene expression in response to stress. Na. Rev Genet. 2011;12(12):833–845. doi: 10.1038/nrg3055. [DOI] [PubMed] [Google Scholar]
- Latchman DS. Encyclopedia of life sciences. Chichester: Wiley; 2005. Transcriptional Gene Regulation in Eukaryotes. [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RAM. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14(8):518–528. doi: 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poburko D, Santo-Domingo J, Demaurex N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J Biol Chem. 2011;286(13):11672–11684. doi: 10.1074/jbc.M110.159962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez SA, et al. Nitric oxide is reduced to HNO by proton-coupled nucleophilic attack by ascorbate, tyrosine, and other alcohols. A new route to HNO in biological media. J Am Chem Soc. 2015;137(14):4720–4727. doi: 10.1021/ja512343w. [DOI] [PubMed] [Google Scholar]
- Kuropteva ZV, Kudryavtsev ME. Ferrous-ascorbate complexes as carriers of nitric oxide. Gen Physiol Biophys. 1997;16(1):91–96. [PubMed] [Google Scholar]
- Vanin AF, Huisman A, Stroes ES, Ruijter-Heijstek FC, Rabelink TJ, van Faassen EE. Antioxidant capacity of mononitrosyl-iron-dithiocarbamate complexes: implications for NO trapping. Free Radic Biol Med. 2001;30(8):813–824. doi: 10.1016/s0891-5849(01)00466-x. [DOI] [PubMed] [Google Scholar]
- Lindberg RA, Dewhirst MW, Buckley BJ, Hughes CS, Whorton AR. Ca2+-dependent nitric oxide release in endothelial but not R3230Ac rat mammary adenocarcinoma cells. Am J Physiol. 1996;271(1):332–337. doi: 10.1152/ajpcell.1996.271.1.C332. [DOI] [PubMed] [Google Scholar]
- Campo GM, et al. The SOD mimic MnTM-2-PyP(5+) reduces hyaluronan degradation-induced inflammation in mouse articular chondrocytes stimulated with Fe (II) plus ascorbate. Int J Biochem Cell Biol. 2013;45(8):1610–1619. doi: 10.1016/j.biocel.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Waldeck-Weiermair M, et al. Spatiotemporal correlations between cytosolic and mitochondrial Ca2+ signals using a novel red-shifted mitochondrial targeted cameleon. PLOS ONE. 2012;7(9):45917. doi: 10.1371/journal.pone.0045917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: implications for uncoupling endothelial nitric oxide synthase. Biochem Pharmacol. 2005;70(3):343–354. doi: 10.1016/j.bcp.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Liu J, Hughes TE, Sessa WC. The first 35 amino acids and fatty acylation sites determine the molecular targeting of endothelial nitric oxide synthase into the Golgi region of cells: a green fluorescent protein study. J Cell Biol. 1997;137(7):1525–1535. doi: 10.1083/jcb.137.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feron O. The Endothelial Nitric-oxide Synthase-Caveolin Regulatory Cycle. J Biol Chem. 1998;273(6):3125–3128. doi: 10.1074/jbc.273.6.3125. [DOI] [PubMed] [Google Scholar]