

Abstract
The identification and exploration of the detailed organization of organs or of the whole body at the cellular level are fundamental challenges in biology. Transitional methods require a substantial amount of time and effort to obtain a 3D image and including sectioning the intact tissue, immunolabeling, and imaging serially-sectioned tissue, which produces a loss of information at each step of the process. In recently developed approaches for high-resolution imaging within intact tissue, molecular characterization has been restricted to the labeling of proteins. However, currently available protocols for organ clearing require a considerably long process time, making it difficult to implement tissue clearing techniques in the lab. We recently established a rapid and highly-reproducible protocol termed ACT-PRESTO (active clarity technique–pressure related efficient and stable transfer of macromolecules into organs), which allows for tissue clearance within several hours. Moreover, ACT-PRESTO enables rapid immunolabeling with conventional methods and accelerates antibody penetration into the deep layer of densely-formed, thick specimens by applying pressure or convection flow. We describe how to prepare tissues, how to clear by lipid removal using electrophoresis, and how to immuno-stain by a pressure-assisted delivery. The rapidity and consistency of the protocol will expedite the performance of 3D histological research and volume-based diagnoses.
Keywords: Bioengineering, Issue 118, tissue clearing, immunolabeling, 3D imaging, tissue-hydrogel, tissue engineering, neuroscience, ACT-PRESTO, X-CLARITY
Introduction
One of the challenges in neuroscience is the visualization of neuronal circuit wiring and individual cells within intact brain tissue. Until recently, demonstrating the connections between neurons in this way required 1) serial sectioning of tissues; 2) molecular labeling of specific targets, such as axons or proteins; and 3) visualization by 3D reconstruction of the whole brain via computational registering or alignment of 2D serial images1. These steps are laborious, require a great deal of time, and are liable to lose information during sectioning and labeling, making neuronal network mapping exceedingly difficult. However, many methods that allow for the visualization of intact tissue without sectioning have been developed. Biological tissues can be made optically transparent by tissue-clearing techniques2-10. One of the major methods is to reduce refractive index differences between the intact tissue and the immersion solution in order to reduce light scattering in the intact tissue, thus rendering the tissue transparent and enabling the observation of deep structures. Some types of immersion solutions have hydrophobic properties, which result in fast fluorescent quenching during the dehydration procedure. Therefore, those methods are not compatible with fluorescent imaging over a long period of time11,12. Instead of hydrophobic reagents, other methods use hydrophilic reagents for tissue clearing, such as SeeDB4 and Scale6, which maintain the structural information and fluorescence of the biological tissue4,6,8. However, macromolecules, including antibodies, cannot reach the core of intact tissue by diffusion alone. Therefore, the pre-labeling of target molecules in deep areas of densely-packed tissues is technically challenging.
In a recently-developed tissue-clearing method, CLARITY3, a hydrogel-embedded biological tissue is formed with acrylamide, and lipids are removed by electrophoresis under sodium dodecyl sulfate (SDS)-containing solution. The sample is then immersed in a solution with a matching reflective index to reduce light scattering3. The lipid removal system was later modified in advanced CLARITY13 and PACT (passive clarity technique)10. After polymerization, acrylamide chains crosslink with proteins, forming hydrogel-tissue. Lipid components cannot crosslink with acrylamide; thus, lipids can be eliminated by electrophoresis under the SDS-containing buffer. Through the active elimination of lipids, brain tissue markedly increases in transparency9. However, these methods do not address the impossibility of deep-labeling by free diffusion. To overcome this limitation, techniques for the active transport of reagents into the deepest parts of thick tissues are required.
Although CLARITY allows tissue clearing and deep-tissue visualization, it is not an easy or fast procedure. It can take several weeks to clear a whole mouse brain7,14. Rapid clearing of tissues is essential for the application of such methods to basic or clinical laboratory settings for life science research or volume diagnosis. The current protocol provides a simplified process for biological tissue clearance and subsequent protein detection by pressure-assisted delivery immuno-staining. It is suitable for high-resolution volume imaging of large filed and 3D data processing systems through combination with imaging methods.
Protocol
Animal experiments must conform to all relevant governmental and institutional regulations.
1. Preparation of Reagents
- For the hydrogel monomer solution (A4P0): Add 20 g of acrylamide to 450 mL of 0.1x phosphate-buffered saline (PBS) and adjust the volume to 500 mL with 0.1x PBS. CAUTION: Acrylamide monomers are toxic. Perform all procedures in a fume hood with personal protective equipment, including a face shield, a laboratory coat, gloves, and closed-toe shoes.
- Immediately prior to use, add 100 mg of 2,2'-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride to 40 mL of hydrogel monomer solution (A4P0) in a 50 mL conical tube under a fume hood.
For the electrophoretic tissue clearing (ETC) buffer: Add 40 g of sodium dodecyl sulfate (SDS) and 12.37 g of boric acid to 800 mL of deionized H2O (dH2O). Adjust the pH to 8.5 with NaOH and adjust the volume to 1 L with additional dH2O. CAUTION: SDS is a harmful chemical; therefore, handle it with care.
For the CUBIC-mount solution: Add 250 g of sucrose, 125 g of urea, and 125 g of N,N,N',N'-tetrakis(2-hydroxypropyl)ethylenediamine to 150 mL of dH2O and bring the volume to 500 mL with dH2O.
2. Sample Preparation & Fixation
- Euthanasia of the mouse.
- Prepare the alfaxalone (1 mg/kg) and xylazine (0.5 mg/kg) in the same syringe.
- Intraperitoneally inject the mixture of alfaxalone and xylazine and allow 3 min for the mouse to become unconscious.
- Wait until the anaesthetized mouse no longer responds to painful stimuli, such as a tail pinch, before proceeding. CAUTION: Follow appropriate institutional guidelines for handling animals.
Transcardially perfuse the mouse with 100 mL of 0.9% NaCl (pH 7.4-7.5) solution containing heparin (100 U/mL) to exsanguinate, followed by 100 mL of 4% paraformaldehyde (PFA) in 1x PBS (pH 7.4)14. CAUTION: PFA solution is toxic. The preparation of PFA solution and all subsequent handling must be conducted in a fume hood and with personal protective equipment.
Cut off the mouse head, open up the skin, and break the skull between the eyes using small scissors.
Remove pieces of the skull using small, curved forceps.
Take out the brain or desired organs.
Incubate the brain and organs in post-fix 4% PFA solution at 4 °C overnight. NOTE: For specific-region tissue clearing, dissect the specific region or trim the tissue after tissue fixation. CAUTION: PFA solution is toxic. The transcardial perfusion and all subsequent mouse handling must be conducted in a fume hood and with personal protective equipment.
3. Hydrogel Monomer Infusion and Polymerization
Incubate the fixed organs in A4P0 hydrogel monomer solution at 4 °C for 12-24 h with gentle shaking.
- Hydrogel monomer-infused tissue polymerization.
- Transfer the sample to a 10 mL round-bottom tube with 5 mL of hydrogel monomer solution. Wrap the top of the round-bottom tube with Parafilm.
- Remove the oxygen from the round-bottom tube containing the hydrogel-infused whole mouse brain sample by bubbling nitrogen through the liquid for 1 min. Quickly and tightly close the sample-containing tube.
Transfer the tube to a water-bath (37 °C) for 2 h.
Wash the polymerized sample briefly with 0.1x PBS to remove excess hydrogel. CAUTION: Hydrogel monomers are toxic. To avoid skin contact with hydrogel monomer, perform all procedures in a fume hood and with personal protective equipment, including a face shield, a laboratory coat, and gloves. NOTE: Polymerized tissues can be stored in 0.1x PBS containing sodium azide for more than 2 weeks at 4 °C. CAUTION: Sodium azide is highly and acutely toxic. Perform all procedures in a fume hood and with personal protective equipment, including a face shield, a laboratory coat, and gloves.
4. Electrophoretic Tissue Clearing (ETC)
Transfer the polymerized sample to a tissue container and place the tissue container in the ETC chamber.
Fill the ETC chamber with ETC buffer using peristatic pump.
- Set the ETC conditions and run the ETC. Use the following settings.
- For ETC settings for whole organs, use the following settings: (1.5 amp), temperature (37 °C), running time (6.0 h), pump speed (30 rpm).
- For ETC settings for 1- to 2-mm thick mouse brain slices, use the following settings: current (1.5 amp), temperature (37 °C), running time (2.0 h), pump speed (30 rpm).
Transfer the cleared sample to a 50-mL conical tube with 45 mL of 0.1x PBS. Wash the cleared sample several times with 0.1x PBS until no bubbles are seen when the tube is briefly shaken (to confirm the complete removal of SDS).
5. Immunolabeling of ACT-processed Tissues
- For mouse whole brain: Incubate the sample in a 3-mL volume of antibody solution containing 1x PBS, 6% bovine serum albumin(BSA), 0.1% Triton X-100, and 0.01% sodium azide (antibody dilution solution) with a dilution factor of 1/500 for tyrosine hydroxylase (TH) antibody for 4 days at 37 °C with mild shaking. Replace the antibody solution at the end of day 2.
- Wash the sample in a 50-mL conical tube with 45 mL of 0.1x PBS for 3 - 5 h and change the buffer every hour.
- Incubate the sample in a 3-mL volume of antibody dilution solution with a dilution factor of 1/500 for donkey anti-rabbit 488 secondary antibody for 4 days at 37 °C with mild shaking. Replace the diluted antibody solutions on day 2.
- For 1- to 2-mm thick sliced brain tissue: Incubate the sample overnight or for up to 1 day in a 0.5- to 1-mL volume of antibody dilution solution with a dilution factor of 1/500 for collagen type IV antibody at 37 °C with mild shaking.
- Wash the sample in a 15-mL conical tube with a 12-mL volume of 0.1x PBS for 1-2 h. Change the buffer every hour.
- Incubate the sample in a 0.5- to 1-mL volume of antibody dilution solution with a dilution factor of 1/500 for Cy3-conjugated anti-rabbit secondary antibody overnight or for up to 1 day at 37 °C with mild shaking.
Wash the sample in 0.1x PBS for 3 - 5 h; change the buffer every hour.
Prior to imaging, incubate the sample in an appropriate amount of CUBIC-mount solution for 1 h at room temperature with gentle shaking. Replace with fresh CUBIC-mount solution and incubate for an additional 1 h. NOTE: 76 type of antibodies worked well in mouse ACT-processed tissue, including 31 monoclonal antibodies14. Pre-labeled tissue can be combined with the immunolabeling of two more fluorochromes in ACT-processed tissue.
6. Immunolabeling of Dense Tissues (PRESTO)
- c-PRESTO NOTE: c-PRESTO is suitable for small-sized samples, such as whole testis, whole kidney, or small sections of larger organs.
- Transfer the sample into a 1.5-mL tube, add 500 µL of antibody dilution solution with a dilution factor of 1/500 for collagen type IV antibody, and centrifuge the tube at 600 × g for 2 h.
- Wash the stained sample with 0.1x PBS by centrifugation at 600 × g for 30 min.
- Add 500 µL of antibody dilution solution with a dilution factor of 1/500 for Cy3-conjugated anti-rabbit secondary antibody and centrifuge at 600 × g for 2 h.
- Wash the stained sample with 0.1x PBS by centrifugation at 600 × g for 30 min.
- s-PRESTO NOTE: s-PRESTO is suitable for larger tissues.
- Prepare a syringe (30-mL volume) and connect it to a 3-way valve (Figure 1A). Glue the valve with a glue gun for the high-pressure conditions (Figure 1B).
- Transfer the sample into the 3-way valve-connected syringe in the closed-valve position.
- Add 5 - 7 mL of antibody dilution solution with a dilution factor of 1/500 for collagen type IV antibody. Release the antibody solution into the sample by opening the valve.
- Set the syringe piston to the 17-mL position to provide sufficient room for infusion/withdrawal movement. This can be done in the open-valve position. Close the 3-way valve after the syringe setting is finished.
- Put the syringe containing the sample on a syringe pump. Set the syringe pump conditions to an infusion/withdrawal volume of 10 mL/min and a 4-min pause time on continuous cycle mode (Figure 1C). NOTE: The pump infuses until it reaches the target volume (10 mL), and then the direction of flow changes after a brief pause (4 min).
- Run the syringe pump for 3 - 24 h at room temperature (Figure 1F).
- Open the 3-way valve and replace the solution with 0.1x PBS.
- Wash the stained sample twice with 0.1x PBS for 1 h each time using the syringe pump.
- Change with antibody dilution solution containing Cy3-conjugated anti-rabbit secondary antibody (dilution factor of 1/500) and run the syringe pump for 3 - 24 h.
- Open the 3-way valve and replace the solution with 0.1x PBS.
Prior to imaging, incubate the stained sample in an appropriate amount of CUBIC-mount solution for 1 h at room temperature with gentle shaking. Replace with fresh CUBIC-mount solution and incubate for an additional 1 h.
7. Imaging
Place the labeled tissue in a confocal dish and add CUBIC-mount solution until the tissue is covered. Place a coverslip on the tissue. Use a confocal microscope to image the tissue14 under the 10X objective.
Representative Results
ACT-tissue Clearing
One important factor affecting tissue clearing is tissue fixation. PFA fixation and acrylamide infusion are separate steps in this protocol. After the tissue-hydrogel polymerization step, the free A4P0 solution is not polymerized, although tissue-infused hydrogels are cross-linked with endogenous macromolecules. Therefore, no gel should form outside of the tissues (Figure 2A). The ACT procedure results in less cross-linking between proteins and acrylamide compared to the CLARITY protocol. Accordingly, the tissue-hydrogel sample is more porous. This feature enables the fast extraction of lipids and the diffusion of macromolecules. Sections of mouse brain 1- to 2-mm thick were sufficiently cleared within 1 - 2 h (Figure 2B), and 5 - 6 h of ETC was sufficient for clearance of whole mouse brains (Figure 2C). Hydrogel-mediated tissue clearing induced the expansion of the tissue after the ETC step, but the tissue returned to its original size in reflective index-matched solution. The tissue-hydrogel sample formed in the ACT procedure is highly porous, and thus, small compounds and macromolecules could penetrate efficiently and diffuse easily (Figure 3). After a 2-h incubation of 1-mm brain slices with tyrosine hydroxylase (TH) and collagen type IV antibody, a subsequent 2-h incubation with a secondary antibody was sufficient to label dopaminergic neurons at a 500-µm depth (Figure 3).
PRESTO-tissue Immunolabeling
Dense organs generally have high concentrations of ECM fibers, which affects the penetration of antibodies into the tissues. Thus, antibodies penetrated to a depth of only 20-30 µm in dense tissues, such as kidney tissue, after 12 h of incubation. To overcome this limitation, we designed active macromolecule penetration methods that enable the delivery of reagents into deep tissues, namely PRESTO (pressure related efficient and stable transfer of macromolecules into the organs) (Figure 4). Compared to the static incubation of ACT-processed kidney tissue with antibody solutions, the application of centrifugal forces (600 x g) with a table-top centrifuge (centrifugal PRESTO, c-PRESTO) greatly improved antibody delivery into deep tissue (Figure 5). When we applied the c-PRESTO procedure to dense tissues, 3 h of incubation was sufficient to label 250- to 300-µm deep structures. To improve the tissue distortion without significant tissue damage, we also designed a machine that provides convection flow with a syringe pump (syringe PRESTO, s-PRESTO). This process similarly improved antibody penetration compared to passive diffusion (Figure 5).
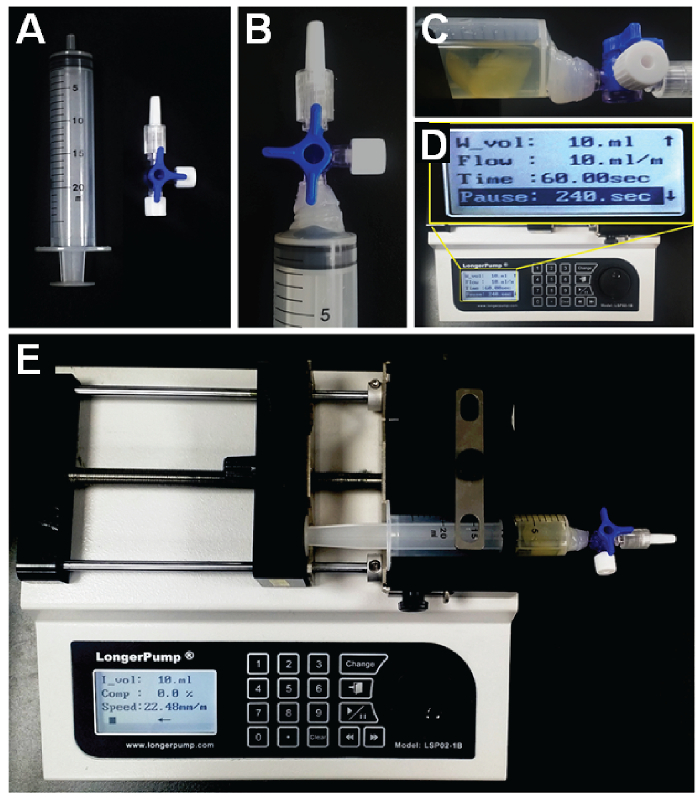 Figure 1. Preparation of the s-PRESTO apparatus. A. A syringe (30 mL) and three-way stopcock. B. The three-way stopcock connected and glued to the syringe. C. The syringe containing the sample and antibody solution, and the syringe placed on the syringe pump. D. The syringe pump and working condition setup. E. The pump infuses a solution containing the labeling reagents into the specimen. When the designated volume is reached, the pumping direction changes and withdraws the solution after a designated period of time. Please click here to view a larger version of this figure.
Figure 1. Preparation of the s-PRESTO apparatus. A. A syringe (30 mL) and three-way stopcock. B. The three-way stopcock connected and glued to the syringe. C. The syringe containing the sample and antibody solution, and the syringe placed on the syringe pump. D. The syringe pump and working condition setup. E. The pump infuses a solution containing the labeling reagents into the specimen. When the designated volume is reached, the pumping direction changes and withdraws the solution after a designated period of time. Please click here to view a larger version of this figure.
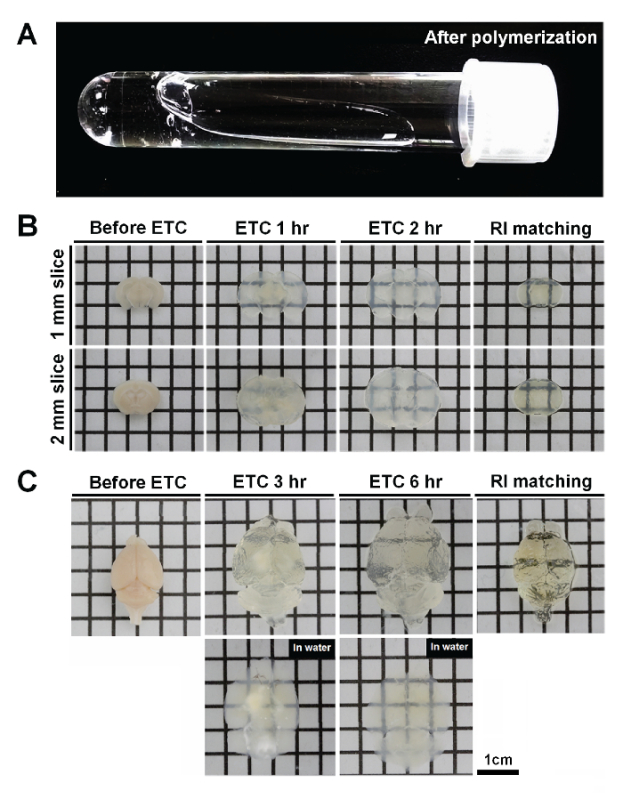 Figure 2. ACT tissue clearing of mouse brain. A. After polymerization, free A4P0 solutions are not polymerized; tissue-infused hydrogels are gelated by cross-linking with endogenous macromolecules. B. 1- to 2-mm thick brain slices were cleared for 1 - 2 h, and the extent of expansion and size recovery in reflective index (RI)-matching solution were recorded. C. The thickness of the whole mouse brain is about 0.8 cm, and 5 - 6 h of ETC was sufficient to completely clear the brain. After 3 h of ETC, there was a significant amount of non-cleared tissue. By 6 h of ETC, however, clearing of the entire brain had occurred. The color of tissues after ACT in the ETC buffer is white or opaque. By RI adjustment, tissues become transparent. Please click here to view a larger version of this figure.
Figure 2. ACT tissue clearing of mouse brain. A. After polymerization, free A4P0 solutions are not polymerized; tissue-infused hydrogels are gelated by cross-linking with endogenous macromolecules. B. 1- to 2-mm thick brain slices were cleared for 1 - 2 h, and the extent of expansion and size recovery in reflective index (RI)-matching solution were recorded. C. The thickness of the whole mouse brain is about 0.8 cm, and 5 - 6 h of ETC was sufficient to completely clear the brain. After 3 h of ETC, there was a significant amount of non-cleared tissue. By 6 h of ETC, however, clearing of the entire brain had occurred. The color of tissues after ACT in the ETC buffer is white or opaque. By RI adjustment, tissues become transparent. Please click here to view a larger version of this figure.
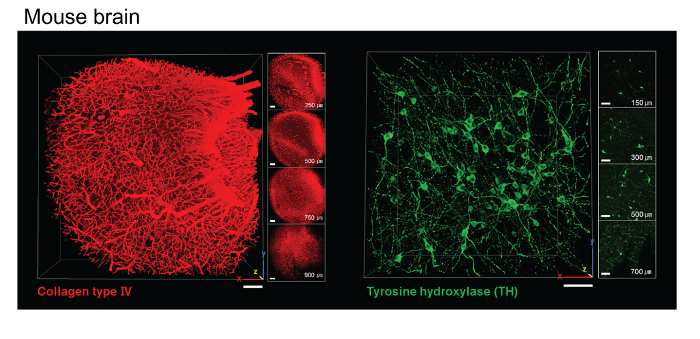 Figure 3. Immunolabeling with ACT-cleared mouse brain tissue. Images of ACT-processed 1-mm mouse brain slices showing mouse brain cortex stained with an antibody to collagen type IV and part of the midbrain stained with an antibody to tyrosine hydroxylase (TH). Scale bar, 100 µm. Please click here to view a larger version of this figure.
Figure 3. Immunolabeling with ACT-cleared mouse brain tissue. Images of ACT-processed 1-mm mouse brain slices showing mouse brain cortex stained with an antibody to collagen type IV and part of the midbrain stained with an antibody to tyrosine hydroxylase (TH). Scale bar, 100 µm. Please click here to view a larger version of this figure.
 Figure 4. PRESTO immunolabeling methods. Antibodies cannot penetrate into dense tissues by diffusion. PRESTO immunolabeling methods were designed to actively infuse macromolecules and to deliver reagents into dense tissues. The application of centrifugal forces using a standard table-top centrifuge (centrifugal PRESTO, c-PRESTO) markedly facilitated the delivery of antibodies. Appling convection flow with a syringe pump (syringe PRESTO, s-PRESTO) improved antibody penetration. Please click here to view a larger version of this figure.
Figure 4. PRESTO immunolabeling methods. Antibodies cannot penetrate into dense tissues by diffusion. PRESTO immunolabeling methods were designed to actively infuse macromolecules and to deliver reagents into dense tissues. The application of centrifugal forces using a standard table-top centrifuge (centrifugal PRESTO, c-PRESTO) markedly facilitated the delivery of antibodies. Appling convection flow with a syringe pump (syringe PRESTO, s-PRESTO) improved antibody penetration. Please click here to view a larger version of this figure.
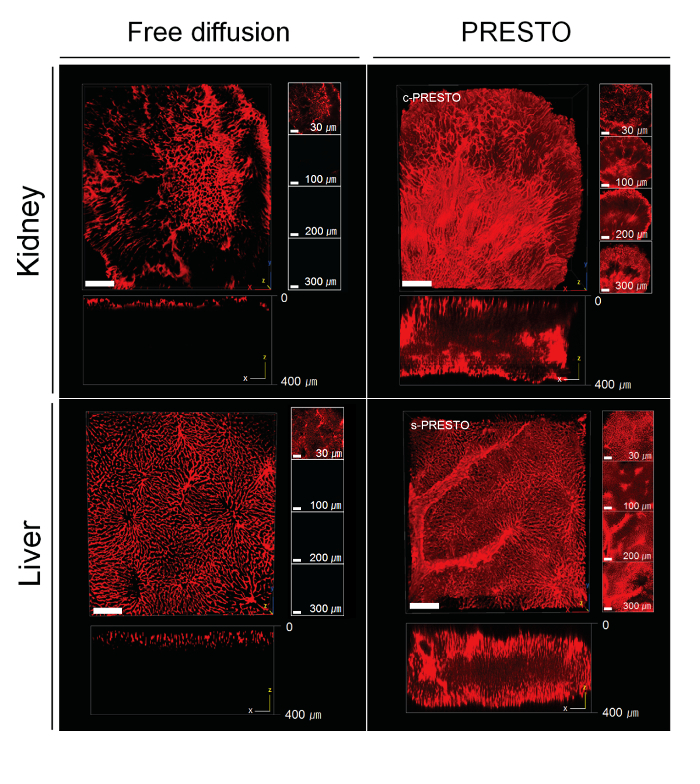 Figure 5. PRESTO immunolabeling of ACT-cleared dense tissue. For c-PRESTO, tissues were centrifuged at 600 x g for 3 h using a standard table-top centrifuge in order to accelerate the penetration of primary and secondary antibodies. For s-PRESTO, a syringe pump was used to deliver the antibodies. Mouse organs, such as the kidneys and liver, were labeled with collagen type IV. Compared to conventional methods, 3 h of c- or s-PRESTO markedly enhances the labeling depth. In the kidney and liver, the depth of the Z-axis reaches 300 - 350 µm or deeper in PRESTO samples (right), while control samples are labeled at a depth of only 40 - 50 µm (left). A 3D reconstructed image was obtained with a confocal microscope. Scale bar, 100 µm. Please click here to view a larger version of this figure.
Figure 5. PRESTO immunolabeling of ACT-cleared dense tissue. For c-PRESTO, tissues were centrifuged at 600 x g for 3 h using a standard table-top centrifuge in order to accelerate the penetration of primary and secondary antibodies. For s-PRESTO, a syringe pump was used to deliver the antibodies. Mouse organs, such as the kidneys and liver, were labeled with collagen type IV. Compared to conventional methods, 3 h of c- or s-PRESTO markedly enhances the labeling depth. In the kidney and liver, the depth of the Z-axis reaches 300 - 350 µm or deeper in PRESTO samples (right), while control samples are labeled at a depth of only 40 - 50 µm (left). A 3D reconstructed image was obtained with a confocal microscope. Scale bar, 100 µm. Please click here to view a larger version of this figure.
Discussion
Poor fixation or hydrogel immersion into tissues can cause the loss of proteins and the distortion of tissue during the tissue clearing process. The whole adult mouse brain should be incubated in 4% paraformaldehyde overnight, followed by immersion in a minimum volume of 20 mL of hydrogel monomer solution for 12 - 18 h with gentle shaking. For large tissues, including human tissue such as brain slices and spinal cord, extended soaking time in the hydrogel monomer solution is required. The ACT protocol is easily applicable to the clearing of fixed tissues because the tissue fixation and polymer infusion steps are separated. After tissue clearing, human tissues were well-stained with several antibodies. Note that the ACT clearing technique is not compatible with alcohol fixatives, such as ethanol and methanol.
The tissue-hydrogel polymerization step is also critical to produce good-quality tissue following the clearing process. Because oxygen inhibits the polymerization of acrylamide, it should be removed by degassing the tissue-containing solution under a nitrogen gas infusion system. Alternatively, de-oxygenation can be executed using a vacuum chamber with a heat block for 23 h.
The clearing rate is dependent upon numerous factors, including the organ size, the contents of lipids or ECM fibrous proteins, the condition of fixation, etc. Most adult mouse tissues can be cleared by ETC overnight. However, there is a risk of unnecessary over-clearing and tissue swelling; thus, the differences in clearing time are of important consideration. Tissue-clearing conditions should be empirically determined. Importantly, the contents of the ECM may affect the appearance of the cleared tissue. The color of the tissues remains white or opaque, even after the ACT in ETC buffer, because ACT does not clear dense protein fibers. However, when these organs were immersed in RI matching solution, they became transparent.
The rate of tissue swelling appears to be dependent upon the ECM contents of the tissues, and soft organs, such as the brain, exhibit a greater swelling ratio than dense organs. Because increased tissue swelling will help the tissue-clearing procedure, ACT routinely utilizes distilled water-based buffer in most cases. In some cases, when tissue swelling or transient deformity is not desirable, such as in embryos, buffer containing 0.1x PBS can be applied for high-resolution imaging. It should be also noted that higher salt concentrations in the buffer could cause the shrinkage of tissues.
The ACT method can clarify whole organs and even the whole body of a mouse14. However, deep-tissue imaging of transparent tissue requires special microscopes and objectives. Thus, brain tissue 1- to 2-mm thick is most effective for imaging with a conventional confocal microscope. In our hands, removal of the labeled antibodies from ACT-processed tissues is antibody-dependent, and rounds of different antibody labeling are not recommended. Several antibodies, such as TH and GFAP, worked especially well for whole-brain immunolabeling14. On the other hand, some antibodies, such as Tuj1 or Map2, often labeled only the surface of the tissues. This might be improved by other methods, such as SWITCH15. Another potential limitation is that it might be difficult to preserve fine protein structures in ACT-processed tissue because of the tissue swelling and shrinkage during the tissue-clearing step.
The PRESTO technique is applicable to a wide variety of tissue-labeling techniques. For example, in tissue-transparent methods using pre-labeled tissue, such as SeeDB4 and iDISCO7, immunolabeling of dense tissue requires incubation periods longer than several days to weeks. However, PRESTO can shorten the incubation time to several hours, enabling the completion of the entire process within a day with 1-mm thick dense tissue. PRESTO can enhance the efficacy of deep-labeling thick, dense tissues, or even un-cleared thick tissues. Because PRESTO uses a table-top centrifuge or syringe pump and does not require any special equipment, it is relatively easy to implement this technique with routine lab procedures.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This research was supported by the Brain Research Program through the National Research Foundation (NRF) funded by the Korean Ministry of Science, ICT, and Future Planning (NRF-2015M3C7A1028790).
References
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt H-U, et al. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat Methods. 2007;4:331–336. doi: 10.1038/nmeth1036. [DOI] [PubMed] [Google Scholar]
- Chung K, et al. Structural and molecular interrogation of intact biological systems. NATURE. 2013;497:332–337. doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M-T, Fujimoto S, Imai T. SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat Neurosci. 2013;16:1154–1161. doi: 10.1038/nn.3447. [DOI] [PubMed] [Google Scholar]
- Ertürk A, et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat Protoc. 2012;7:1983–1995. doi: 10.1038/nprot.2012.119. [DOI] [PubMed] [Google Scholar]
- Hama H, et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- Renier N, et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell. 2014;159:896–910. doi: 10.1016/j.cell.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Susaki EA, et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell. 2014;157:726–739. doi: 10.1016/j.cell.2014.03.042. [DOI] [PubMed] [Google Scholar]
- Tainaka K, et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 2014;159:911–924. doi: 10.1016/j.cell.2014.10.034. [DOI] [PubMed] [Google Scholar]
- Yang B, et al. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell. 2014;158:945–958. doi: 10.1016/j.cell.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-Y, Chung K, Deisseroth K. Light microscopy mapping of connections in the intact brain. Trends Cogn Sci. 2013;17:596–599. doi: 10.1016/j.tics.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Yushchenko DA, Schultz C. Tissue clearing for optical anatomy. Angew Chem Int Edit. 2013;52:10949–10951. doi: 10.1002/anie.201306039. [DOI] [PubMed] [Google Scholar]
- Tomer R, Ye L, Hsueh B, Deisseroth K. Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nat Protoc. 2014;9:1682–1697. doi: 10.1038/nprot.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, et al. ACT-PRESTO: Rapid and consistent tissue clearing and labeling method for 3-dimensional (3D) imaging. Sci Rep. 2016;6:18631. doi: 10.1038/srep18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray E, et al. Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems. Cell. 2015;163:1500–1514. doi: 10.1016/j.cell.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]