

Abstract
Biological small angle X-ray scattering (BioSAXS) is a powerful technique in molecular and structural biology used to determine solution structure, particle size and shape, and surface-to-volume ratio of macromolecules. The technique is applicable to a very wide variety of solution conditions spanning a broad range of concentrations, pH values, ionic strengths, temperatures, additives, etc., but the sample is required to be monodisperse. This caveat led to the implementation of liquid chromatography systems on SAXS beamlines. Here, we describe the upstream integration of size-exclusion (SEC) and ion-exchange chromatography (IEC) on a beamline, different methods for optimal background subtraction, and data reduction. As an example, we describe how we use SEC- and IEC-SAXS on a fragment of the essential vaccinia virus protein D5, consisting of a D5N helicase domain. We determine its overall shape and molecular weight, showing the hexameric structure of the protein.
Keywords: Biochemistry, Issue 119, Bio-Small Angle X-ray Scattering (BioSAXS), online size-exclusion chromatography (SEC), online ion-exchange chromatography (IEC), background subtraction, D5, vaccinia
Introduction
BioSAXS is a powerful tool to determine the shape of nano-sized objects1-4. The scattering of X-rays by a solution containing macromolecules, sized in the nm range, is recorded at very low angles. This angular range contains information about global parameters: the radius of gyration; the largest intraparticle distance; the particle shape; and the degree of folding, denaturation, or disorder. The technique does not require crystals, and the macromolecule stays in solution and thus can be kept in conditions mimicking certain important parameters of the cell, such as ionic strength, pH, etc. The knowledge of these factors might help to determine, for example, the physiologically relevant oligomeric state of a protein of interest or to validate a proposed model of a complex. The characterization of protein-protein interactions in different buffer conditions, the creation of models of missing domains, the refinement of homology models, and the determination of discrete folded and unfolded states can be performed quickly and easily5.
As with any technique, BioSAXS has intrinsic weaknesses: aggregated or denatured samples, mixtures of particles, heterogeneous samples, radiation damage, and buffer mismatches may result in un-interpretable data. For many analysis methods, it is implicitly assumed that the sample is monodisperse, a requirement that is often difficult to obtain in practice. In many cases, the degradation of the sample is subtle and cannot be detected in the data on its own, and any attempt to interpret the data gives inaccurate or even misleading results. To overcome these obstacles, the combination of size-exclusion chromatography (SEC) and SAXS was implemented on many beamlines to ensure data quality and to make this technique more accessible for increasingly difficult samples6-11. Recently, we added a new method to the repertoire by developing online ion-exchange chromatography (IEC)-coupled SAXS12. Both techniques are opening SAXS to a wide range of biological particles formerly impossible to analyze. The choice of which method to use depends on the biophysical properties of the particles of interest.
SEC separates macromolecules by their size, whereby at least a 10% difference in apparent molecular mass is needed for the separation. Physical limitations of the column and physiological properties of the samples, like hydrophobic surfaces, flexibility, and lack of stability, also complicate data collection, analysis, and interpretation.
Ion-exchange chromatography, which separates molecules based on their charge and, hence, their binding affinity to the IEC column, can be used instead of or in addition to SEC. The total charge can be readily manipulated by changing the pH, or varying the salt concentration of the buffer, providing a relatively simple method for the controlled elution of the molecules from the IEC column. By using the charge, the separation of similar types and sizes of molecules, which would otherwise be difficult to separate, can be performed routinely with IEC. Additionally, IEC has the advantage of being able to deal with diluted samples, allowing one to avoid the concentration steps, which carry the potential risk of denaturing the protein. Unfortunately, as the charge distribution is highly sample-dependent, IEC requires optimization regarding the pH and the salt concentrations13,14.
For many proteins that are difficult to express, purify, or both, only low quantities of sample are available to study. It is important to be efficient and to minimize the number of purification steps and, therefore, the losses. For this reason, the last purification step is online directly prior to SAXS data acquisition, in order to increase the likelihood of collecting a good data set.
Here, we present and compare online SEC-SAXS and IEC-SAXS. Both techniques were implemented on the BioSAXS beamline BM29 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France15. As a test case, we used the D5N and helicase domain of the vaccinia virus protein D5, which was rather difficult to analyze structurally using other methods. The vaccinia virus is a member of the Poxviridae family and is 98% identical to the variola virus, the cause of small pox. Using the vaccinia system, we study the replication machinery, focusing here on the essential helicase-primase D5.
D5 is a 95-kDa protein with an N-terminal archeo-eukaryotic primase (AEP) domain16 followed by a cysteine cluster region (res. 240-345)16. Further towards the C-terminus comes a D5N domain (res. 340-460), which is always associated with D5-type helicases, and finally a superfamily 3 (SF3) helicase domain (res. 460-785) 17 (Figure 1A). The helicase domain of D5 builds a hexameric ring structure that is needed for tight binding to DNA. Thanks to recent SAXS and EM studies, the low-resolution structures of the primase and the helicase domains are now known18.
Here, we show how to use the implemented online chromatography techniques on the BioSAXS beamline BM29 at ESRF to gain insights into the structure of the C-terminal fragment (residue 323-785) of D5.
Protocol
1. Description of the Offline Preparation and Sample Generation of a D5 Deletion Protein
NOTE: The D5323-785 construct (Figure 1A) was cloned, expressed, and purified as described18.
- Clone the construct into the pProEx HTb vector
- Perform a polymerase chain reaction (PCR) on D5R using the 5'-GCGCCATGGGTAATAAACTGTTTAATATTGCAC-3' and 5'-ATGCAAGCTTTTACGGAGATGAAATATCCTCTATGA-3' primers and a PCR reaction mix with polymerase, following the manufacturer's advice.
- Purify the PCR fragments via spin columns, following the manufacturer's advice.
- Digest the PCR fragments and the plasmid with the NcoI and HindIII restriction enzymes, following the manufacturer's recommendations for buffer composition and incubation time.
- Run the fragments on a 1% agarose gel in 0.5x TAE buffer (20 mM Tris, 10 mM acetic acid, and 0.5 mM EDTA) and stain the gel with a DNA dye.
- Expose the gel to UV light. Caution: Wear personal safety equipment! Cut out the bands of the digested PCR fragments and the digested plasmid and purify the DNA using a gel purification spin column kit, following the manufacturer's recommendations.
- Ligate the purified PCR fragments into the plasmid using a fast ligation kit, following the manufacturer's recommendations.
- Transform via heat shock the product from the ligation reaction into competent bacteria optimized for plasmid amplification.
- Add half of the ligation product to a vial of bacteria.
- Incubate the reaction tube on ice for 20 min.
- Incubate the tube at 42 °C for 30 s.
- Place the tube on ice for 2 min.
- Add 1 mL of Luria Bertani (LB) Broth (Miller) without antibiotics and let the cells recover at 37 °C for 1 h.
- Spin down the cells at 16,100 x g for 3 s in a tabletop microtube centrifuge and remove most of the medium.
- Spread the cells onto an LB agar plate with ampicillin (50 µg/mL final) and let the colonies grow at 37 °C overnight.
- Put a colony into 3 mL of LB with ampicillin (50 µg/mL final) and grow the culture at 37 °C, shaking at 160 rpm overnight.
- Follow the instruction of the miniprep kit to extract the plasmid.
- Send a sample of the plasmid for sequencing and analyze the sequence for the absence of mutations and for the correct insertion.
- Transform the pProEx HTb D5323-785 construct into bacteria optimized for protein expression (use 1 µL and follow the steps in step 1.1.7). Spread the bacteria onto an LB agar plate with ampicillin (50 µg/mL final).
- To create a starting culture, put one colony in 300 mL of LB with ampicillin (50 µg/mL final) and grow the bacteria at 37 °C, shaking at 160 rpm overnight.
Inoculate 12 x 1 L of LB in the presence of ampicillin (50 µg/mL final), each with 20 mL of the pProEx HTb D5323-785 bacteria starter culture at 37 °C until an OD600 = 0.3 is reached. Reduce the temperature to 18 °C and induce expression at an OD600 = 0.5 with 1 mM IPTG. Incubate the cultures, shaking them at 160 rpm and 18 °C overnight.
Harvest the bacteria by centrifugation at 50,000 x g and at 4 °C for 30 min. Resuspend the bacterial pellets in approximately 150 mL of lysis buffer (50 mM Tris-HCl [pH 7], 150 mM NaCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, and 10% glycerol) with 1x protease inhibitor and 25 units (U)/10 mL DNase. Lyse the bacteria via sonication on ice (1-inch probe, 50% power, 0.5-s pulse, 0.5-s pause, 3x 5 min).
Load the supernatant onto a nickel affinity column (Ni-column, 1.5-mL bed volume), and wash it with 10 column volumes (CV) of binding buffer (50 mM Tris-HCl [pH 7], 150 mM NaCl, 10 mM β-mercaptoethanol, and 10% glycerol), 10 CV of washing buffer (50 mM Tris-HCl [pH 7], 1 M NaCl, 10 mM β-mercaptoethanol, and 10% glycerol), and 10 CV of imidazole wash (50 mM Tris-HCl [pH 7], 150 mM NaCl, 10 mM β-mercaptoethanol, 10% glycerol, and 30 mM imidazole). Elute the D5323-785 (20 mM Tris-HCl [pH 7], 150 mM NaCl, 10 mM β-mercaptoethanol, 10% glycerol, and 200 mM imidazole).
- Inspect the different purification steps via sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE).
- Load 2-20 µL of each purification step flow-through mixed with 1x loading dye (2x: 4% SDS, 20% glycerol, 120 mM Tris-HCl [pH 6.8], and 0.02% weight/volume bromophenol blue) on a 10% SDS gel and run them at 200 V in 1x Laemmli running buffer (25 mM Tris, 0.192 M glycine, and 0.1% SDS).
- Stain the gel with a ready-to-use Coomassie blue solution.
Exchange the buffer of the eluted protein with binding buffer using a desalting column according to the manufacturer's manual.
Cleave the His-tag by adding Tobacco Etch Virus nuclear-inclusion-a endopeptidase (TEV, 1 mg/100 mg of protein) to the protein solution. Incubate at room temperature overnight.
Check the digestion by performing SDS-PAGE (see step 1.5)
Equilibrate the Ni-column in binding buffer. Let the protein solution pass through and wash the beads with 2 CV of imidazole washing buffer while collecting the flow-through.
Concentrate the protein using a centrifugal concentrator to a final volume of 0.5 mL.
Inject the concentrated protein onto a size-exclusion column equilibrated with gel filtration buffer (20 mM Tris-HCl [pH 7], 150 mM NaCl, 10% glycerol, and 1 mM dithiothreitol [DTT]).
Collect the flow-through in 0.5-mL fractions.
Check the presence and purity of the protein in the peak samples by performing SDS-PAGE (see step 1.5).
Combine the fractions of the eluted peak of D5323-785. Dilute the sample to 25 mM NaCl and 5% glycerol while keeping the Tris and DTT concentrations constant (for 5 mL of protein solution in 20 mM Tris-HCl [pH 7], 150 mM NaCl, 10% glycerol, and 1 mM DTT, first add 5 mL of 20 mM Tris-HCl [pH 7] and 1 mM DTT, and then add 20 mL of 20 mM Tris-HCl [pH 7], 5% glycerol, and 1 mM DTT).
Load the sample onto an ion-exchange column. Use buffer A (20 mM Tris [pH 7], 25 mM NaCl, 5% glycerol, and 1 mM DTT) and buffer B (20 mM Tris [pH 7], 1 M NaCl, 5% glycerol, and 1 mM DTT) to form a salt gradient from 25 mM to 1 M.
Collect the eluted protein in 1.5-mL fractions.
Check the presence and purity of the protein in the peak samples by performing SDS-PAGE (see step 1.5 and Figure 1B)
Reconcentrate the protein solution in a centrifugal concentrator to a final volume of 0.5 mL.
Rerun the concentrated protein on a size-exclusion column in gel filtration buffer (20 mM Tris-HCl [pH 7], 150 mM NaCl, 5% glycerol, and 1 mM DTT; see step 1.11).
2. Data Collection
NOTE: Request beam time as early as possible. For ESRF, guidelines on available access types and on how to submit an application can be found at: http://www.esrf.eu/UsersAndScience/UserGuide/Applying. After the acceptance of the proposal and an invitation for the experiment, all participants have to complete a safety training. After validation of the training, fill in the “A-form” (via the ESRF user portal) to declare the researchers visiting the beamline for the experiment, along with the required safety information on the samples. Contact the local contact person to discuss the experiment.
- SEC-SAXS data collection NOTE: For online purification, use the high-performance liquid chromatography (HPLC) system installed at BM29. The system consists of an in-line degasser, a binary pump, a valve for buffer selection and gradients, an autosampler, a UV-VIS array photospectrometer, and a conductometer. It is directly attached to the flow-through capillary of the SAXS exposure unit19.
- Put 1 mL of the gel filtration buffer aside. Connect the buffer bottle to the SEC system (the default is Port A).
- Choose the flow rate depending on the column in use. Note: For the size-exclusion column used here, the flow rate is 0.1 mL/min. Define the maximum pressure for the column, take note of the back-pressure, and set the acquisition time to at least 1.2 CV.
- Flush the pumps and the upstream tubes via the "auto-purge" function.
- Wait until the system is filled with the buffer and connect the column. Equilibrate the column by passing at least 1.5 CV.
- Interlock the hutch and measure the buffer set aside in step 2.1.1, using the sample changer to control for variation of the signal due to radiation damage. Only continue if there is no radiation damage to the buffer.
- Switch the beamline control system to HPLC mode. Make a test run of buffer, collecting 200 frames with 1 s per frame. Write down the number of counts given by the detector in the summed intensity plot. NOTE: The signal should match the previously measured buffer, and the summed intensity over time should remain constant. If the signal does not match or is not constant, a longer equilibration of the system is required.
- Spin down the sample at 13,000 x g in a tabletop microtube centrifuge for at least 10 min.
- Fill the sample into an HPLC-compatible glass vial (provided) and put it into the auto-injector. Note the well position.
- Interlock the experimental hutch using standard procedure, as explained in the User Safety Training.
- Log into and create a folder for data storage through the beamline control software.
- Program the HPLC system using the "quick batch" feature. Set the storage location for the UV data to the folder created in step 2.1.10. Make sure to activate the automatic ASCII conversion of the UV data, storing the ASCII file in the same folder.
- Choose the injection volume and well position. Add the measurement to queue by pressing the "Start" button. The software will ask to save the batch. Prepare for saving, but do not press the "Save" button, as this automatically starts the measurement.
- Set up data collection parameters in the beamline control software. Choose the number of frames so that the total data collection time is in slight excess of the acquisition time defined in step 2.1.2.
- Open the safety shutter and start SAXS data collection using the beamline control software. Verify that the data are correctly acquired. Compare the newly collected data to the data collected in step 2.1.6 and check that the number of counts in the summed intensity remains constant for at least 100 frames and matches the value from step 2.1.6.
- Start the SEC run by pressing the "Save" button, as prepared in step 2.1.11, and note the frame number displayed in the camserver software when the injection is completed and UV data acquisition starts.
- After the data collection, open the ISPyB database20 at Open the "data acquisition" tab and press the "Go" button to access the data set and the results of the automatic analysis21.
- IEC-SAXS data collection NOTE: Instead of the continuous linear gradient commonly applied in IEC experiments, use a step-wise gradient in which the amount of buffer B is increased in pre-set discrete steps.
- Create the HPLC program for a sample-free buffer run. Program a step-wise gradient starting from 0% buffer B and increasing by 5 percentage points after two CV until 100% of buffer B is reached.
- Put some of each buffer aside. Connect the bottle with the low-salt buffer A to port A and the one with the high-salt buffer B to port B.
- Choose the flow rate and stay below the pressure limit of the column (in this example, 1 mL/min).
- As in step 2.1.3, flush the pumps and upstream tubing by using the "auto-purge" function.
- Equilibrate the system in low-salt buffer A (see step 1.15) and connect the ion-exchange column. Wait until the column is completely equilibrated (at least 1.5 CV).
- Use the buffer set aside in step 2.2.2 to examine buffers A and B for radiation damage using the sample changer, as in step 2.1.5.
- Check the buffer coming out of the column, as in step 2.1.6. Compare the signal to the signal of buffer A recorded in step 2.2.6. Note again the number of counts in the summed intensity plot.
- Create a folder for data storage for the buffer run.
- Set up data collection in HPLC system mode. Choose the number of frames so that the total data collection time is in excess of the total time of the IEC run (see step 2.2.1).
- Open the safety shutter and start the SAXS data collection. Verify that it proceeds correctly and double-check that the summed intensity remains constant at the value noted in step 2.2.7 for at least 100 frames.
- Use the "Single Run" feature of the HPLC software and start the step-wise gradient programmed in step 2.2.1. For the injection, set the vial number to -1 in order to inject no sample in this step. Write down the number of frames acquired as the program starts.
- Create the HPLC program for the sample run. Program a step-wise gradient starting from 0% of buffer B. Estimate the percentage of buffer B at each peak measured offline in step 1.1.5 (Figure 1B). Set at least 3 steps exactly 1 percentage point below and 1.5 points above the peaks of interest, each for 2.5 CV (Figure 1C). Add a final step at 100% buffer B for 2.5 CV.
- Spin down the sample at 13,000 x g in a tabletop microtube centrifuge for at least 10 min.
- Dilute D5323-785 into the salt concentration of buffer A (25 mM) by mixing it with 20 mM Tris-HCl [pH 7], 10% glycerol, and 1 mM DTT.
- Load the sample manually onto the column. Put the buffer A input tube into the diluted sample after pausing the pumps to avoid the formation of air bubbles. Disconnect the column from the detectors to directly collect the flow-through from the column.
- When the sample container is almost empty, return the input tube into buffer A (again pausing the pumps while moving the tube) and restart the pumping with buffer A to transfer the sample from the tubing to the column. Once the entire sample has been loaded, pass 2 CV of buffer A, and then reconnect the column to the detectors.
- For the sample run, create a folder for data storage.
- Open the safety shutter and start the SAXS data collection. Verify that the data collection proceeds correctly and double-check that the summed intensity remains constant at the value noted in step 2.2.7 for at least 100 frames.
- Use the "Single Run" feature of the HPLC software to start the step-wise gradient programmed in step 2.2.12. For the injection, set the vial number to -1 in order to inject no sample in this step. Write down the number of frames acquired as the program starts.
- Open "data acquisition" in ISPyB20 and press "Go" to look at the data set and the automatic analysis21.
- Export the UV data from the HPLC system to ASCII format through the "Postrun Analysis" software.
3. SEC-SAXS Data Reduction & Analysis
Open the data in data acquisition in ISPyB by pressing the "Go" button. Determine in the overview which frames of the SAXS data collection correspond to the elution peak. Verify that the radius of gyration is stable throughout the peak.
Confirm that the automated buffer correction was performed correctly. Load frames from the central part of the peak into a program that performs manipulations with experimental SAXS data files22 and average them. Then, load the automatically generated buffer file and check whether the high q regions of the averaged frames and the buffer frames match.
- Compare the frames acquired throughout the peak quantitatively using the CORMAP test23.
- Load the automatically created subtractions (*_sub.dat files) of frames covering the region of interest.
- Scale them to each other by pressing the "Scale" button.
- Compare them using the "Data Comparison" feature. There should be no systematic changes between the frames throughout the peak.
Open all *-_sub.dat frames of interest, merge them using the "Merge" button, and save the merged file in .dat format.
Determine the radius of gyration with the "Radius of Gyration" tool in the program, noting the first data point in the Guinier range for later cropping of the data at low resolution.
Determine the pair-distance distribution function with the "Distance Distribution" tool in the program and save the resulting .out file as gnom.out.
4. Ab Initio Modeling
On a Unix machine, run a 40 x ab initio model reconstruction program without symmetry restrictions. Create a new folder and copy the gnom.out file to it. Run the program as follows: for (( i = 1; i <= 40; i++ )); do dammif gnom.out -m s -p dammifp1_$i; done
Analogously, run an ab initio model reconstruction program with symmetry restrictions for six-fold symmetry in a second folder: for (( i = 1; i <= 40; i++ )); do dammif gnom.out -m s -s P6 -p dammifp6_$i; done
Align all ab initio model reconstruction program output files (*-1.pdb). In the two folders from steps 4.1 and 4.2, run damaver -a *-1.pdb.
Open the union of all models (damaver.pdb) as well as the model filtered to the correct volume (damfilt.pdb) in a suitable molecular visualization software26 and compare them.
Open the damsel.log file in a text editor. The model listed as the "reference" at the bottom of the file is the most representative model.
5. IEC-SAXS Data Reduction, Analysis, and Modeling
In the program that performs the manipulation with experimental SAXS data files22, load about 50 SAXS frames from each mixing step of the buffer run, press the "AVERAGE" button, and save the results.
Based on the auto-processing results available via ISPyB, identify which frames of the experiment correspond to the elution of the protein and verify that the (preliminary) radius of gyration is stable throughout the peak.
Load the frames into the experimental SAXS data files manipulation program22 and average them, as done for the buffer frames (see step 5.1). Then, load the buffer files generated in step 5.1 and compare the high q regions (above 4.2 nm-1) to find a buffer that matches the average from the peak. NOTE: There should be no systematic offset between the average from the peak and the buffer. If the sample curve seems to fall between two buffer curves, interpolate their curves by calculating the average.
Subtract the buffer identified as the best match from the average scattering curve of the peak fraction and save the resulting subtracted file. In addition, create subtracted curves using the average buffer curves from the preceding and following mixing steps to estimate the error margin of the subtraction.
Repeat steps 5.3 and 5.4 individually for the left and right halves of the peak and compare the results to those from step 5.4 to check for stability of the signal throughout the peak.
Follow steps 3.5 and 3.6 for all three subtracted curves from step 5.4.
Compare the results for all three subtracted curves. Note: Only results that remain robust within the error margin of subtraction can be trusted.
Perform modeling as in steps 4.1 and 4.2 for the best subtraction identified in step 5.3.
Follow step 4.3 to 4.5 to align the models and to identify the most representative one.
6. Comparison of the Results
Run a program to superimpose 3D structures12 on the representative models of the SEC (step 4.5) and IEC-SAXS data (step 5.9) with P6 symmetry: supcomb model_sec.pdb model_iec.pdb
Import the model_sec.pdb and the model_iec-r.pdb into a molecular visualization software.
Open the .fir files of the modelsec.pdb and the modeliec-r.pdb in a spreadsheet and create a 2D graph of experimental and calculated scattering curves.
Representative Results
The results of the model-free invariant analysis are listed in Table 1. The analysis of the D5323-785 SEC-SAXS data showed a molecular mass estimate (from a Porod analysis) of 345 kDa versus 338 kDa, observed using IEC-SAXS. Both are in agreement with the expected mass of 6 times 53.5 kDa (321 kDa) for a hexamer. The ab initio modeling of both data sets was undertaken with no imposed symmetry (SEC-SAXS: χ2 = 0.88, IEC-SAXS: χ2 =3.1) and using C6 symmetry (SEC-SAXS: χ2 = 1.0, IEC-SAXS: χ2 = 4). As the overall fits to the scattering data for both reconstructions are comparable in both cases (Figure 1D and E), C6 symmetry can be assumed. Thus, the model corresponds to a hexagonal cone-like structure with a central channel, which appears partially obstructed (SEC: Figure 1F; IEC: Figure 1G, overlay Figure 1H). An examination of individual models before averaging shows that this obstruction is likely to be an artifact of the averaging process.
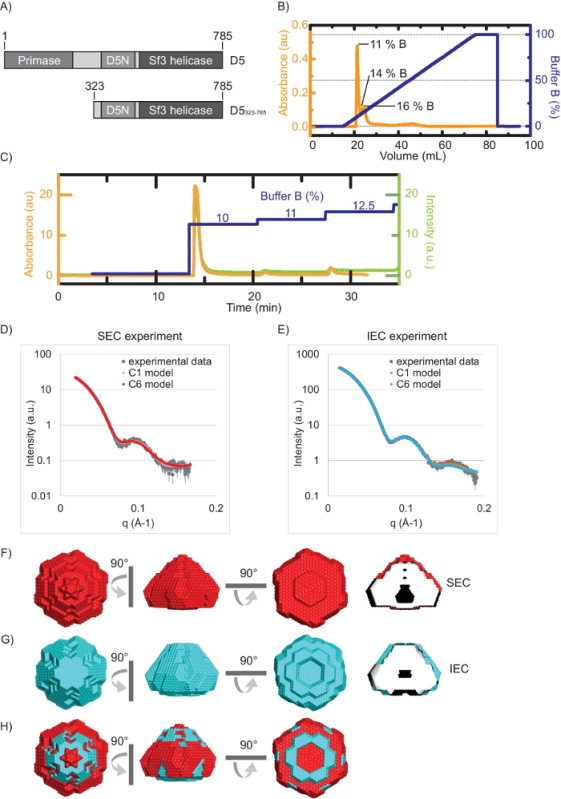 Figure 1: SAXS data analysis and comparison of D5323-785 (figure adapted from References 12 and 18). A) Schematic domain structure of D5 protein and D5323-785. B) Offline ion-exchange chromatogram with the indication of the three peaks. Orange curve: Absorbance at 280 nm (au), blue curve: percentage of buffer B. Percentages of buffer B at the peaks are indicated. C) Online ion-exchange chromatogram. Color key as in B. Additionally, the measured intensity is indicated in green. The experimental scattering curve and calculated curve of the model obtained by SEC-SAXS D) and IEC E) data analysis of D5323-785. F) Bead model of D5323-785 based on SEC data and G) IEC data. H) Overlay of the SEC and IEC models. The panels in F) to H) were created using a molecular visualization software24. Please click here to view a larger version of this figure.
Figure 1: SAXS data analysis and comparison of D5323-785 (figure adapted from References 12 and 18). A) Schematic domain structure of D5 protein and D5323-785. B) Offline ion-exchange chromatogram with the indication of the three peaks. Orange curve: Absorbance at 280 nm (au), blue curve: percentage of buffer B. Percentages of buffer B at the peaks are indicated. C) Online ion-exchange chromatogram. Color key as in B. Additionally, the measured intensity is indicated in green. The experimental scattering curve and calculated curve of the model obtained by SEC-SAXS D) and IEC E) data analysis of D5323-785. F) Bead model of D5323-785 based on SEC data and G) IEC data. H) Overlay of the SEC and IEC models. The panels in F) to H) were created using a molecular visualization software24. Please click here to view a larger version of this figure.
| Data-collection parameters | |
| Instrument: | ESRF BM29 |
| Wavelength (Å) | 0.99 |
| q-range (Å-1) | 0.0032 – 0.49 |
| Sample-to-detector distance | 2.864 m |
| Exposure time (sec) | 1 per frame |
| Concentration range | n.a. |
| Temperature (K) | 293 |
| Detector | Pilatus 1M (Dectris) |
| Flux (photons/s) | 1 × 1012 |
| Beam size (µm2) | 700 × 700 |
| Structural parameters for D5323-785, SEC | |
| I0 (cm-1) [from Guinier] | 0.0237 |
| Rg (Å) [from Guinier] | 48 |
| qminRg – qmaxRg used for Guinier | 0.77 - 1.24 |
| Dmax (Å) [from p(r) ] | 145 |
| q-range used for p(r) (Å-1) | 0.02 - 0.17 |
| Porod volume Vp (Å3) [from Scatter] | (570 ± 5) × 103 |
| Molecular mass Mr (kDa) [from Vp] | 345 |
| Calculated monomeric Mr from sequence (kDa) | 321 |
| Structural parameters for D5323-785, IEC | |
| I0 (cm-1) [from Guinier] | 0.386 |
| Rg (Å) [from Guinier] | 46.5 ± 0.1 |
| qminRg – qmaxRg used for Guinier | 0.44 - 1.29 |
| Dmax (Å) [from p(r) ] | 120 |
| q-range used for p(r) (Å-1) | 0.03 - 0.18 |
| Porod volume Vp (Å3) [from Scatter] | (577 ± 5) × 103 |
| Molecular mass Mr (kDa) [from Vp] | 339 |
| Calculated hexameric Mr from sequence (kDa) | 321 |
Table 1: SAXS data parameters. The table summarizes the parameters of SAXS data acquisition and analysis.
Discussion
For many macromolecules, a final purification step using chromatography is required prior to SAXS data collection to obtain a good quality data set. However, not all samples remain stable; they may be prone to aggregation or re-equilibration to a mixture of oligomerization states. Therefore, a final online purification step on the beamline is required to minimize the time between purification and data collection in order to obtain the best-quality SAXS data. Depending on the biophysical properties of the protein of interest, SEC-SAXS or IEC-SAXS might be chosen to obtain optimal sample quality. Here, on a protein construct derived from the helicase/primase D5, both techniques are explained and discussed.
Acquisition of SEC-SAXS data is becoming more and more standardized and is available on many BioSAXS beamlines. Data analysis, especially background subtraction, is relatively straightforward and easy. However, a stable buffer signal and the sufficient separation of the macromolecular species remains essential. Therefore, it is critical to reserve enough time to equilibrate the column thoroughly. Failure of this method can be due to persistent contaminants of similar size to the protein of interest, low concentrations, and radiation-sensitive buffers.
In practice, initially, SEC-SAXS is likely to be used as the method of choice for most macromolecular samples. Still, many purification protocols require a prior IEC step due to the presence of contaminants or aggregation. Given that each concentration and chromatography step is associated with losses of sample (estimated at 30-50%) and time, direct IEC-SAXS is advantageous. For samples that cannot be purified by SEC, be it due to the presence of similarly sized "contaminants" or because they severely aggregate at the necessary concentrations, IEC-SAXS would always be the better-suited approach. Also, the higher flow rates supported by many IEC columns can help to reduce the transit time between purification and measurement. In the example presented here, IEC was used with a step elution, which allows for the separation of the close peaks of D5323-785 from contaminants by carefully choosing the salt concentration steps. In principle, the number of steps is unlimited, but practically, at least 1 step per peak is required, and not too many should be chosen. For the background subtraction method described above, it is crucial to measure a relatively high number of different buffer compositions in order to find the matching one.
A shared downside of both techniques is the lack of precise protein concentration information. Due to this, precise mass determination based on forward scattering is not possible. For globular proteins such as D5323-785, the Porod volume provides an alternative, albeit less precise, mass estimate, but for highly flexible or disordered proteins, this approach would not be valid.
A variation of the step-wise gradient IEC-SAXS method presented here is the use of a linear gradient instead. While it is possible to work with as many steps as desired to isolate sub-peaks using a step-wise elution, in the linear gradient approach, it is required to optimize the gradient conditions carefully in order to separate the peaks entirely before starting the SAXS experiment. Background subtraction in this approach could be done frame-wise and could be verified by the comparison of the individual frames, but it requires more advanced data handling, and a dedicated software does not exist yet.
The choice of a suitable column is critical for both techniques, as it determines the separation of the macromolecular species. Size-exclusion columns differ in loading capacity, the size range of separable macromolecules, and resolution, while ion-exchange columns vary in the kind and the density of their immobilized charges.
While the protocol presented here is specific to the ESRF beamline BM29, adaptation to any other SAXS beamline is, in principle, straightforward. The main requirements are a sufficiently high X-ray flux and a suitable detector (ideally single-photon-counting), to acquire reasonable signal-to-noise data in the range of seconds or less, and an online liquid chromatography system capable of creating gradients. The exact implementation would, of course, depend on the local beamline environment.
The results obtained on D5323-785 using the two methods differ slightly. The radius of gyration is slightly smaller for the IEC data than for the SEC data, and the local minima of the scattering curve are shifted to slightly larger scattering vectors. This means that the D5323-785 measured with IEC-SAXS is slightly more compact than the D5323-785 measured with SEC-SAXS. This might be due to differences in the sample preparation, in the time between purification and measurement (IEC is faster), or, less likely, to a contaminant in the SEC-purified sample. The bead models obtained completely independently with both methods are comparable (Figure 1H). D5323-785 shows the expected hollow, hexameric structure18.
In conclusion, online ion-exchange and online size-exclusion chromatography are important biochemical purification methods that can be coupled directly to SAXS6,7,9-12,25,26. The background subtraction of IEC-SAXS data is slightly more difficult and ambiguous than for SEC-SAXS, but it is nevertheless possible. Depending on the biophysical properties of the protein of interest, both SEC and IEC-SAXS allow for the optimization of species separation with inherent advantages. Providing that the validation steps (as described) are correctly observed, the resulting data can be analyzed with confidence, and models can be determined using the standard tools available within the community. Together, both techniques allow online separation for a broad range of biological macromolecules, yielding data not accessible via standard static measurements.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We acknowledge financial support for the project from the French grant REPLIPOX ANR-13-BSV8-0014 and by research grants from the Service de Santé des Armées and the Délégation Générale pour l'Armement. We are thankful to the ESRF for the SAXS beam time. We thank Andrew McCarthy, Gordon Leonard, Wim Burmeister, and Guy Schoehn for financial and scientific support.
References
- Graewert MA, Svergun DI. Impact and progress in small and wide angle X-ray scattering (SAXS and WAXS) Curr. Opin. Struct. Biol. 2013;23:748–754. doi: 10.1016/j.sbi.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Jacques DA, Trewhella J. Small-angle scattering for structural biology-Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010;19:642–657. doi: 10.1002/pro.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikhney AG, Svergun DI. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015;589:2570–2577. doi: 10.1016/j.febslet.2015.08.027. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Vestergaard B. Analysis of biostructural changes, dynamics, and interactions - Small-angle X-ray scattering to the rescue. Arch. Biochem. Biophys. 2016. [DOI] [PubMed]
- David G, Pérez J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 2009;42:892–900. [Google Scholar]
- Graewert MA, et al. Automated Pipeline for Purification, Biophysical and X-Ray Analysis of Biomacromolecular Solutions. Sci. Rep. 2015;5 doi: 10.1038/srep10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambright D, et al. Complementary techniques enhance the quality and scope of information obtained from SAXS. ACA Trans. 2013. pp. 1–12.
- Mathew E, Mirza A, Menhart N. Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. J. Synchrotron Radiat. 2004;11:314–318. doi: 10.1107/S0909049504014086. [DOI] [PubMed] [Google Scholar]
- Round A, et al. Determination of the GH3. 12 protein conformation through HPLC-integrated SAXS measurements combined with X-ray crystallography. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2013;69:2072–2080. doi: 10.1107/S0907444913019276. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Inoko Y. Size-exclusion chromatography combined with small-angle X-ray scattering optics. J. Chromatogr. 2009;1216:7461–7465. doi: 10.1016/j.chroma.2009.02.053. [DOI] [PubMed] [Google Scholar]
- Hutin S, Brennich ME, Maillot B, Round A. Online ion-exchange chromatography for small angle X-ray scattering. Acta Cryst. 2016. [DOI] [PMC free article] [PubMed]
- Selkirk C. Ion-exchange chromatography. Protein Purification Protocols. 2004. pp. 125–131. [DOI] [PubMed]
- Yigzaw Y, Hinckley P, Hewig A, Vedantham G. Ion exchange chromatography of proteins and clearance of aggregates. Curr. Pharm. Biotechnol. 2009;10:421–426. doi: 10.2174/138920109788488842. [DOI] [PubMed] [Google Scholar]
- Pernot P, et al. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 2013;20:660–664. doi: 10.1107/S0909049513010431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Koonin EV, Leipe DD, Aravind L. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 2005;33:3875–3896. doi: 10.1093/nar/gki702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- Hutin S, et al. Domain organization of vaccinia virus helicase-primase D5. J. Virol. 2016. pp. 4604–4613. [DOI] [PMC free article] [PubMed]
- Round A, et al. BioSAXS Sample Changer: a robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2015;71:67–75. doi: 10.1107/S1399004714026959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maria Antolinos A, et al. ISPyB for BioSAXS, the gateway to user autonomy in solution scattering experiments. Acta Crystallographica Section D. 2015;71:76–85. doi: 10.1107/S1399004714019609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennich ME, et al. Online data analysis at the ESRF bioSAXS beamline, BM29. J. Appl. Crystallogr. 2016;49 [Google Scholar]
- Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1-towards automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 2007;40:223–228. [Google Scholar]
- Franke D, Jeffries CM, Svergun DI. Correlation Map, a goodness-of-fit test for one-dimensional X-ray scattering spectra. Nat. Methods. 2015;12:419–422. doi: 10.1038/nmeth.3358. [DOI] [PubMed] [Google Scholar]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.8. 2015.
- Jensen MH, et al. Time-resolved SAXS measurements facilitated by online HPLC buffer exchange. J. Synchrotron Radiat. 2010;17:769–773. doi: 10.1107/S0909049510030372. [DOI] [PubMed] [Google Scholar]
- Hynson RM, Duff AP, Kirby N, Mudie S, Lee L. Differential ultracentrifugation coupled to small-angle X-ray scattering on macromolecular complexes. J. Appl. Crystallogr. 2015;48:769–775. [Google Scholar]