

Abstract
Proteomics is the large-scale analysis of proteins. Proteomic techniques, such as liquid chromatography tandem mass spectroscopy (LC-MS/MS), can characterize thousands of proteins at a time. These powerful techniques allow us to have a systemic understanding of cellular changes, especially when cells are subjected to various stimuli, such as infections, stresses, and specific test conditions. Even with recent developments, analyzing the exosomal proteome is time-consuming and often involves complex methodologies. In addition, the resultant large dataset often needs robust and streamlined analysis in order for researchers to perform further downstream studies. Here, we describe a SILAC-based protocol for characterizing the exosomal proteome when cells are infected with HIV-1. The method is based on simple isotope labeling, isolation of exosomes from differentially labeled cells, and mass spectrometry analysis. This is followed by detailed data mining and bioinformatics analysis of the proteomic hits. The resultant datasets and candidates are easy to understand and often offer a wealth of information that is useful for downstream analysis. This protocol is applicable to other subcellular compartments and a wide range of test conditions.
Keywords: Infection, Issue 121, Exosomes, Extracellular Vehicles, HIV-1, Proteomics, Mass spectrometry, SILAC, Bioinformatics
Introduction
Many human diseases, including viral infections, are often associated with distinctive cellular processes that take place in and around the affected cells. Proteins, often acting as the ultimate cellular effectors, mediate these processes. Analysis of the proteins often can provide invaluable information as to the local environment of affected cells and help us to understand the underlying mechanism of disease pathogenesis. Among various protein analysis techniques, proteomics holds particularly great promise. As a powerful, large-scale tool, proteomics can provide a systemic understanding of cellular processes, particularly in the area of the function and interaction of proteins. Analyzing specific proteins is made simpler through the development of labeling techniques, which allow investigators to monitor the expression of cellular components, particularly proteins, in the site of investigation. Although many proteomic analyses have been performed at cellular proteome scale, proteomic characterizations on subcellular compartments have proved to be particularly informative1. This is exemplified well in the studies of HIV-1 infection.
Exosomes, 30-100 nm membrane vesicles secreted by a wide range of cell types2,3, are critical components of intercellular communication and molecular transport. They were previously discovered to play important roles in the HIV-1 budding process4,5. By combining proteomic analysis with functional dissection, we found that exosomes released from HIV-1 infected cells are composed of a unique and quantitatively different protein signature and harbor regulatory molecules that impact cellular properties on neighboring receptive cells, including cellular apoptosis and proliferation6. The methods are described in this protocol, namely SILAC (stable isotope labeling by amino acids in cell culture)7 based proteomic characterization of exosomes from HIV-1 infected cells. Similar approaches can be applied to better understand other subcellular compartments during pathogenesis by adjusting the experimental stress to the specific compartment or fraction of interest and making necessary changes to the described procedures.
Given the recent development of quantitative proteomic methods, there are many to choose from when selecting the most efficient method for a particular experiment. Among these are the chemical-based iTRAQ (isobaric tags for relative and absolute quantification)8 and the label-free MRM (multiple reaction monitoring)9 techniques. Both methods are powerful tools and are good choices for specific settings. For a typical laboratory mainly working with cell lines, however, these two methods have relatively higher costs and are more time-consuming when compared to the SILAC based method. SILAC is a metabolic based labeling technique that incorporates nonradioactive isotopic forms of amino acids from the culture media into cellular proteins. Typically, SILAC experiments start with two cell populations, for example, infected and uninfected. Each is differentially labeled in its specific isotopic environment until full labeling is achieved. The labeled exosomes of these cells are then subjected to protein extraction. Once extracted, the labeled exosomal proteins are analyzed using liquid chromatography tandem mass spectroscopy10. Finally, the mass spectrometry results and significantly labeled proteins are subjected to statistical and bioinformatics analyses as well as rigorous biochemical verification. Our previous investigative reports suggest that the SILAC/exosome procedures are more appropriate for cell lines than primary cells, as cell lines are usually in an active proliferating state for efficient isotopic labeling,
Protocol
1. Cell Culture and HIV-1 Infection
NOTE: Before starting experiments, it is recommended to check the cells' viability through Trypan Blue staining11 and their proliferation through a MTT assay12. It is also critical to use newly prepared SILAC medium. Various cell lines can be used, as long as they are in an actively proliferative stage, and are susceptible to HIV-1 infection, or the test condition of choice. In this protocol, use H9 cell line as the example.
Seed 2 x 106 H9 cells into each cell culture flask. Grow one group in 10 ml labeled RPMI 1640 medium containing 10% dialyzed fetal bovine serum (FBS), 100 mg/L 13C6 L-lysine and 100 mg/L 13C615N4 L-arginine. Grow the other group in 10 ml unlabeled RPMI 1640 medium, with 10% dialyzed FBS, 100 mg/L L-Lysine and 100 mg/L L-Arginine.
Grow the cells for six doublings at which point the proteins of the cells in the labeled medium are practically completely labeled (>99%) with heavy amino acids. Add fresh media or change media regularly (every 1-3 days depending on the type of cell. For H9 cell, change medium every 3 days). At the end of labeling, increase culture volume (e.g. ~30 ml) to accommodate the growth of more cells. NOTE: Determine exact cell doubling time at the beginning of the experiment via the Trypan Blue staining and cell counting.
Infect the labeled cells with HIV-1NL4-3 using standard HIV-1 infection protocol13 for 48 hr. Incubate the cells with appropriate amounts of virus with a multiplicity of infection (MOI) around 0.3. Check HIV-1 infection by p24 quantification of the culture supernatants using an HIV-1 p24 antigen ELISA kit. Perform an immunofluorescence assay14 to confirm successful infection of cells. Keep growing the unlabeled cells in unlabeled medium without HIV-1 infection.
Harvest the supernatants of both groups at the end of infection (step 2.1).
2. Exosome Isolation
NOTE: Through a series of ultracentrifugation steps, exosomal fractions from culture supernatants are enriched15. Perform all the following steps at 4 °C, with an ultracentrifuge rotor that can reach a speed of at least 100,000 x g.
Collect the supernatants of the cultures in 50 ml conical tubes, avoiding cells. Centrifuge them for 10 min at 300 x g to remove remaining cells.
Collect the supernatants in new 50 ml conical tubes and centrifuge them for 10 min at 2,000 x g to remove dead cells. Transfer resulting supernatants to commercial rotor-compatible tubes that are able to withstand ultracentrifugation.
Be sure to balance the ultracentrifuge tubes. Centrifuge the tubes for 30 min at 10,000 x g to remove cell debris.
Collect the supernatants in ultracentrifuge tubes and centrifuge for 70 min at 100,000 x g. Discard the supernatants.
Resuspend the exosome-rich pellets in 5 ml of fresh PBS. Transfer the solutions to fresh ultracentrifuge tubes and centrifuge again for 70 min at 100,000 x g.
Discard supernatants. To store the exosomes long-term, resuspend the pellets in 50 µl of PBS and store at -80 °C. Alternatively, proceed directly to protein extraction as shown below.
3. Protein Extraction and Preparation
Dissolve isolated exosomal pellets in 100-200 µl RIPA lysis and extraction buffer with added protease inhibitor cocktails. The buffer is composed of 25 mM Tris-hydrochloride, 150 mM sodium chloride, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS (sodium dodecyl sulfate), at pH 7.6. The protease inhibitor cocktails should contain a variety of protease inhibitors, such as aprotinin, bestatin, leupeptin, pepstatin A and EDTA (ethylenediaminetetraacetic acid), that can inhibit a full range of proteases.
Centrifuge the dissolved solutions for 10 min at 13,000 x g (4 °C), and transfer the cleared supernatants to new 1.5 ml microcentrifuge tubes.
Quantify protein concentration of each sample of exosomes using bicinchoninic acid (BCA) or Bradford assay16.
Mix an equal amount of proteins (2 µg) from labeled and unlabeled samples and run the equal mixture on a 4-20% SDS-PAGE gel at 120 mA/200 V for 30 min.
Stain gel with Coomassie Blue followed by destaining17.
Using a razor blade, cut the sample lane from the gel. Then cut the gel lane into 10-15 equal pieces. Put each piece into a fresh 1.5 ml microcentrifuge tube for a total of 10-15 tubes.
Immerse cubes in 25 mM NH4HCO3 in 50% acetonitrile, vortex, and discard supernatants. Repeat twice. Dry cubes in a vacuum concentrator18,19.
Rehydrate cubes with 10 mM dithiothreitol (DTT), vortex, and centrifuge briefly. Incubate at 56 °C for 1 hr. Discard supernatant.
Immerse gel cubes in 55 mM iodoacetamide, vortex, and spin. Incubate at room temperature in the dark for 45 min. Discard supernatant.
Immerse cubes in 25 mM NH4HCO3, vortex, spin, and discard supernatant.
Immerse cubes in 25 mM NH4HCO3 in 50% acetonitrile, vortex, and spin. Repeat 3.9 and 3.10.
Dry the cubes in a vacuum concentrator. Add 25 µl sequencing grade modified trypsin in 25 mM NH4HCO3, incubate at 4 °C for 30 min, and discard the excess solution. Immerse cubes in 25 mM NH4HCO3 without trypsin, and incubate overnight at 37 °C.
Briefly spin cubes down and transfer the resulting peptide extract to a new tube. Add 30 µl of 5% formic acid in 50% acetonitrile to the cubes, vortex for 30 min, and spin. Combine the supernatant with the extract. Dry the peptide extracts with a vacuum concentrator to less than 5 µl.
Submit samples to mass spectrometry core facility for LC-MS/MS analysis. The MS analysis data, which can be generated from open source software, typically includes an accession number for each protein identified, labeled/unlabeled ratios and the number of unique peptides identified.
4. Western Blotting Verification
NOTE: Western blotting is recommended to verify mass spectrometry results.
Follow standard western blotting protocols and ensure that equal amounts of protein from each group are loaded. Use antibodies against proteins of interest (e.g. annexin A5, lactate dehydrogenase B chain) identified by MS. Western blotting detection, along with densitometry analysis, can reaffirm that MS protein identification and quantification are correct.
5. Proteomic Data Analysis
NOTE: The data quality assessment, data pretreatment, calculation and determination of significant protein candidates are done separately for each MS replicate. Once above analyses are completed, the analyzed data from replicates are compared and combined6,20,21.
- Assess the quality of MS data.
- First, log2 transform the SILAC-MS labeled/unlabeled ratios of quantified proteins in a spreadsheet program.
- In scientific graphing and statistical software, group the ratios into 40-100 ratio bins and plot the number of ratios per bin to generate a histogram. A normal distribution of histogram indicates good quality of MS data6. Do this step for all the MS replicates.
To increase the confidence and accuracy of the MS peptide ratios, consider removing proteins that have less than two quantified peptides.
- To determine significantly up- and down-regulated protein candidates, use the following steps to calculate significance thresholds22.
- In scientific graphing and statistical software, generate a non-linear regression or curve-fit to the frequency distribution data. This step yields values that can be used to calculate the cutoff values. Calculate the cut-off values as median ± 1.96 σ for 95% confidence limits or 2.56 σ for 99% confidence limits. NOTE: Specific details of calculating the cutoffs can be found at Emmott et al.22.
- Select the protein candidates whose ratios are either greater than 1.96 (or 2.56) standard deviations above the median (significantly over-expressed) or lower than 1.96 (or 2.56) standard deviations below the median (significantly under-expressed).
Compare the replicates of the above-identified significant candidates to achieve consistency. Ensure that they meet the following criteria to pass this final selection step: candidates must be consistently identified in all replicates; and replicates of a candidate must be consistent in their direction of regulation (preferably, at least 2 out of 3 replicates of a candidate should either be up-regulated or down-regulated).
Finalize data for candidates that meet the above-mentioned criteria by merging their replicates' data.
6. Bioinformatics Verification and Characterization
NOTE: Existing genomic and bioinformatic information offers a wealth of information for almost every protein. Data mining and bioinformatics analysis on that information can help in gaining a great deal of insight into the property and functions of the significant candidates. This process is usually necessary to design proper downstream wet-lab experiments.
- Using their GenBank Accession numbers or UniProt IDs, search the candidates against current exosome databases (Exocarta23, Evpedia24) to verify that the candidate proteins have been previously found in exosome. This step adds a layer of confidence that the candidates are indeed in exosomes.
- Access http://www.exocarta.org/, click “Query” and input either the gene or protein name/accession number. A summary page will appear and the evidence in exosome will be shown, provided if there is any.
- Search the candidates against the HIV-1 and the Human Protein Interaction Database25. Alternatively, search the specific databases that can provide information about the interaction of the candidate proteins and the test condition.
- Access the database at www.ncbi.nlm.nih.gov/RefSeq/HIVInteractions. On the ‘protein domain name’ entry, enter the candidates’ names or accession numbers, and click search. The search results can provide insight into the interactions between HIV-1 and the protein candidates, and suggest which of the candidates might be truly HIV-1 associated.
- Import GenBank Accession numbers or UniProt IDs of the candidates into GO analysis software (such as FunRich, Functional Enrichment analysis tool26) and run the software.
- Download the software at http://www.funrich.org/. After installation, open the software, under enrichment analysis, click “Add Data Set”, and upload the list of protein/genes. Next, select the chart type to visualize the GO analysis. NOTE: GO analysis results will be visualized in the form of pie charts. This step helps us to gain global insight associated with the candidates in the areas of Biological Process (BP), Cellular Component (CC), and Molecular Function (MF).
Access DAVID at http://david.ncifcrf.gov/27, click the “Functional Annotation Tool” on its website. Enter the candidate list, select the “Identifier” of the gene such as the “UniProt ID”, select “Gene List” and search. The enriched GO terms, p-values, and other parameters can be found by clicking the “Annotation Summary Results” page under the “Gene_Ontology” category.
- To investigate potential interactions of the candidates with other proteins, use openly accessible STRING database28 to elucidate potential protein-protein interactions and possible biochemical pathways. NOTE: GO Analysis and Functional Annotation (Section 6.3-6.4) can also be performed using the latest STRING software.
- Access the database at http://string-db.org/. Input the protein ID or sequence into the designated search box and select the correct species for analysis. Click “Search”. The results will give information on both direct (physical) and indirect (functional) associations. The top ten known matches for the exosomal candidate will be displayed and should be considered for significant candidate selection. NOTE: A great deal of information associated with the candidates can be analyzed using the steps above. The most significant candidates may be chosen for further downstream molecular and biochemical analysis.
Representative Results
Figure 1A is a flowchart outlining the SILAC labeling procedure21. In order to purify the exosomes, the samples must be spun down via centrifuge. Figure 1B shows the steps of exosome purification by serial ultracentrifugation21. Once purified, the exosomes are subject to experimental proteomic analysis as outlined in the procedure.
Figure 2A is a flowchart for determining significant protein candidates from proteomic data21. The selected candidates are then used for downstream proteomics analysis. Figure 2B is an example of SILAC histogram displaying representative ratios of typical protein quantification results (This figure has been modified from Li et al.6), and Table 1 is an example of determining cutoff values for significant candidates6 from these SILAC ratio histograms. By following the steps described in Section 6.3, the cutoff values for determining significantly up- or down-regulated proteins were calculated. In this representative dataset, proteins with a heavy/light (H/L) ratio above the upper cutoff value were considered as significantly up-regulated, while proteins with a H/L ratio below the lower cutoff value were considered as significantly down-regulated. Significantly regulated candidate proteins would be subjected to further investigation.
Figure 3 displays the overall bioinformatics analysis and data mining procedures for the selected significant candidates. Table 2 is a data mining example that utilizes exosome and HIV-1/host interaction databases6 to gather information on the candidates (This table has been modified from Li et al.6). By mining current exosome and HIV-1/host interaction databases, thirteen out of the fourteen candidates were found to associate with exosomes. Five out the fourteen candidates were also known to interact with HIV-1. Among them, four candidates (HSPA4, heat shock 70 kDa protein 4; NUTF2, nuclear transport factor 2; PTGES3, prostaglandin E synthase 3; LDHB, L-lactate dehydrogenase B chain) were found to associate with both exosome and HIV-1. Among HSPA4, NUTF2, PTGES3 and LDHB, only LDHB was consistently under-expressed in the infected fraction (heavy labeled). Through a series of analyses, we selected LDHB to be the most significant candidate, which could be subjected to further study. The downstream proteomics analyses are displayed in Figure 4. Figure 4A shows brief steps of gene ontology (GO) analysis; Figure 4B lists the steps of performing DAVID analysis; Figure 4C shows how to perform network analysis using STRING. Figures 5A, 5B, and 5C are DAVID analysis of a set of fourteen proteins in BP, CC and MF; Figure 5D shows the top ten interacting partners of LDHB; Figure 5E shows that the majority of interacting partners of LDHB are also associated with exosome and HIV-1. Initial DAVID analysis results suggested that many candidates are apoptosis-related and likely participate in protein binding. Further STRING analysis confirmed that LDHB binding partners are also functionally and locationally related to exosome and HIV-1. All these results give valuable information for potential downstream investigations.
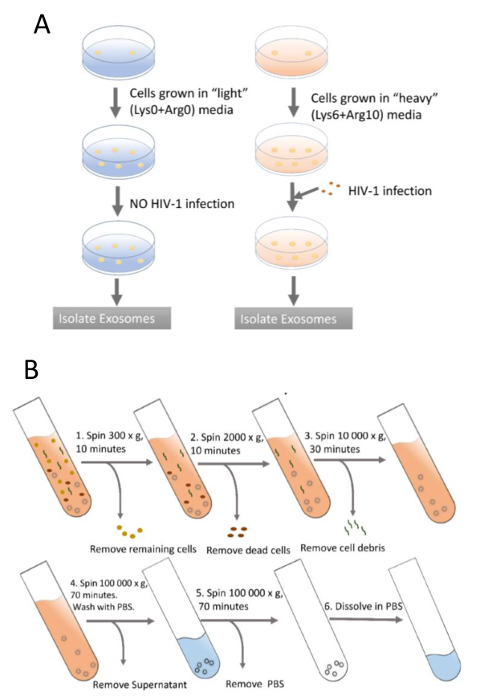 Figure 1: SILAC labeling and exosome purification using differential ultracentrifugation. (A) Two groups of cells are grown separately. One group is grown in labeled medium for six doublings for complete labeling. The other group is grown in the normal unlabeled medium for the same period. Next, the labeled cells are infected with HIV-1, while unlabeled cells are not. Finally, exosomes from both groups are isolated in parallel. (B) The samples (supernatants or pellets) used in the centrifugation at each step are indicated in the test tubes. Fractions to be discarded are noted on the right side of test tubes. The speed and length of each centrifugation are also shown. Please click here to view a larger version of this figure.
Figure 1: SILAC labeling and exosome purification using differential ultracentrifugation. (A) Two groups of cells are grown separately. One group is grown in labeled medium for six doublings for complete labeling. The other group is grown in the normal unlabeled medium for the same period. Next, the labeled cells are infected with HIV-1, while unlabeled cells are not. Finally, exosomes from both groups are isolated in parallel. (B) The samples (supernatants or pellets) used in the centrifugation at each step are indicated in the test tubes. Fractions to be discarded are noted on the right side of test tubes. The speed and length of each centrifugation are also shown. Please click here to view a larger version of this figure.
 Figure 2: Steps to validating proteomic dataset(s) and determining significant protein candidates. (A) Flowchart of determining significant protein candidates from proteomic dataset(s). These four steps ensure reliable selection of significant candidates. First, an SILAC ratio histogram is plotted to assess the quality of the data. Next, less ideal candidates that could reduce accuracy are removed. In the third step, statistical methods are used to set significance thresholds for potential protein candidates. Finally, the replicate data of the significant candidates are analyzed for consistency and are merged eventually. (B) Representative histogram of SILAC ratios. The histogram revealed symmetrical distribution along ratio = 1 (log2 = 0) trend line. The log2 transformed ratios are grouped into ratio bins, and the y-axis shows the relative number of detected ratios per bin. Please click here to view a larger version of this figure.
Figure 2: Steps to validating proteomic dataset(s) and determining significant protein candidates. (A) Flowchart of determining significant protein candidates from proteomic dataset(s). These four steps ensure reliable selection of significant candidates. First, an SILAC ratio histogram is plotted to assess the quality of the data. Next, less ideal candidates that could reduce accuracy are removed. In the third step, statistical methods are used to set significance thresholds for potential protein candidates. Finally, the replicate data of the significant candidates are analyzed for consistency and are merged eventually. (B) Representative histogram of SILAC ratios. The histogram revealed symmetrical distribution along ratio = 1 (log2 = 0) trend line. The log2 transformed ratios are grouped into ratio bins, and the y-axis shows the relative number of detected ratios per bin. Please click here to view a larger version of this figure.
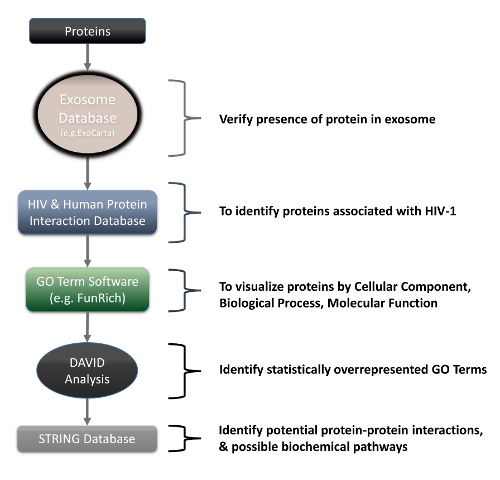 Figure 3: Overall bioinformatics analysis and data mining procedures for significant candidates. The procedures contain exosomal and HIV-1/host data mining, Gene Ontology characterization, DAVID analysis and network prediction. Please click here to view a larger version of this figure.
Figure 3: Overall bioinformatics analysis and data mining procedures for significant candidates. The procedures contain exosomal and HIV-1/host data mining, Gene Ontology characterization, DAVID analysis and network prediction. Please click here to view a larger version of this figure.
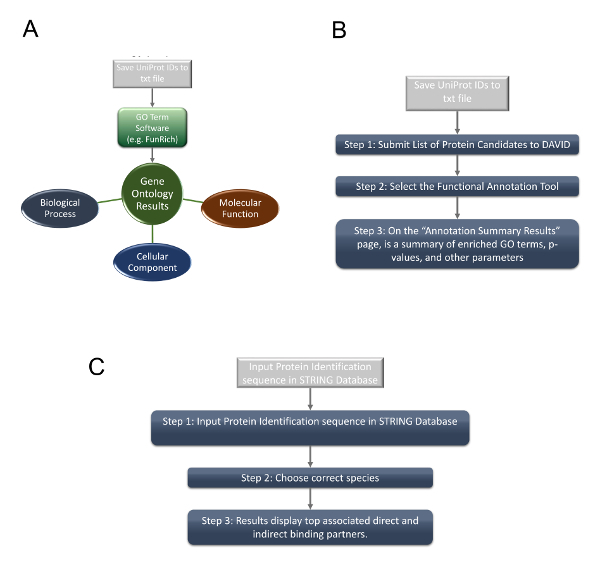 Figure 4: Steps to performing gene ontology characterization, enrichment, and pathway analyses. (A) Steps to performing gene ontology (GO) characterization. To gain insight of the functions and subcellular localizations of the significant candidates, steps to performing GO analysis using appropriate software26 are illustrated. (B) Steps to performing GO Term enrichment analysis. To gain enrichment information of the significant candidates, brief steps to performing DAVID analysis are shown. (C) Steps to performing interaction and pathway analysis. To elucidate possible pathways and find functional partners, steps to performing interaction or network analysis by STRING are shown. Please click here to view a larger version of this figure.
Figure 4: Steps to performing gene ontology characterization, enrichment, and pathway analyses. (A) Steps to performing gene ontology (GO) characterization. To gain insight of the functions and subcellular localizations of the significant candidates, steps to performing GO analysis using appropriate software26 are illustrated. (B) Steps to performing GO Term enrichment analysis. To gain enrichment information of the significant candidates, brief steps to performing DAVID analysis are shown. (C) Steps to performing interaction and pathway analysis. To elucidate possible pathways and find functional partners, steps to performing interaction or network analysis by STRING are shown. Please click here to view a larger version of this figure.
 Figure 5: Representative DAVID and STRING analysis results. (A) BP enrichment results of the fourteen candidates by DAVID analysis. Cell death related processes are significantly enriched. (B) CC enrichment results of the fourteen candidates by DAVID analysis. Many proteins have an intracellular origin. (C) BP enrichment results of the fourteen candidates by DAVID analysis. Most of the candidates participate in protein binding. (D) Top ten interacting partners of LDHB identified by STRING. (E) The majority of LDHB partners are also associated with exosome and HIV-1, which further supports the roles of LDHB in exosome/HIV-1. Please click here to view a larger version of this figure.
Figure 5: Representative DAVID and STRING analysis results. (A) BP enrichment results of the fourteen candidates by DAVID analysis. Cell death related processes are significantly enriched. (B) CC enrichment results of the fourteen candidates by DAVID analysis. Many proteins have an intracellular origin. (C) BP enrichment results of the fourteen candidates by DAVID analysis. Most of the candidates participate in protein binding. (D) Top ten interacting partners of LDHB identified by STRING. (E) The majority of LDHB partners are also associated with exosome and HIV-1, which further supports the roles of LDHB in exosome/HIV-1. Please click here to view a larger version of this figure.
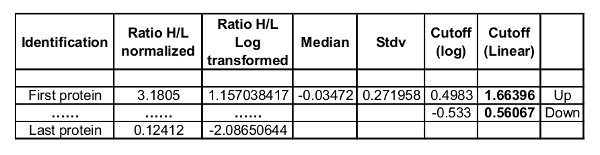 Table 1: Representative calculation of the cutoff values for significant protein perturbation.
Table 1: Representative calculation of the cutoff values for significant protein perturbation.
 Table 2: Associations between SILAC candidates and their exosomal localizations and interactions with HIV-1.
Table 2: Associations between SILAC candidates and their exosomal localizations and interactions with HIV-1.
Discussion
In the procedures described in this paper, we demonstrated the application of the SILAC technique to investigate the effect of HIV-1 infection on the host exosomal proteome. Initially, uninfected and HIV-1 infected cells are differentially isotope-labeled. The differentially labeled exosomes are then purified before performing protein extraction. Next, liquid chromatography-tandem mass spectrometry is employed to analyze the exosomal proteome. Finally, the resulting mass spectrometry data and potential candidate proteins are subjected to statistical and bioinformatics analyses before downstream biochemical dissections.
Critical steps throughout the protocol need to be followed to achieve optimized results. In the initial stages of the protocol, SILAC labeling could be affected by the cell type and the labeling medium. Healthy and highly proliferative cells should be used to obtain a high labeling efficiency. Critically, the labeling medium should be freshly prepared using dialyzed fetal bovine serum (FBS) rather than regular FBS. Dialyzed FBS is depleted of amino acids and peptides and is, therefore, less likely to interfere with the SILAC labeling. It should, however, be noted that dialyzed serum may not be suitable for some cell lines, especially primary cells. Normal growth in the medium with dialyzed FBS should be confirmed prior to employing the SILAC strategy. In addition, using double "heavy" isotope labeling (13C6 L-lysine and 13C615N4 L-arginine) instead of using 13C6 L-lysine singly, can increase the number of quantified peptides in the mass spectrometry analysis. The minimum number of cells needed for successful subsequent proteome analysis depends on many factors, such the sensitivity of the mass spectrometer, the mass of each cell, and proteome targeted (whole proteome or post-translational modified proteins). Based on our findings, we recommend ten million cells as an initial amount in estimating the appropriate number of cells required for the mass spectrometry.
Exosome isolation from cell culture supernatants can be improved by adding a filtration step as follows. For the suspension of cells, an initial centrifugation step at 200 x g for 10 min is done to remove cells suspended in the supernatant, followed by filtration through a 0.22 µm filter29,30. For adherent cells, the supernatant can be collected directly from the culture and filtered in the same way. Filtration is a critical step for separating exosomes from contaminating materials, such as small molecules and peptides. The exosomes can then be isolated from the supernatant using the classical ultracentrifugation method, or using commercially available exosome isolation reagents kits. The purity of the exosome can be verified by nanoparticle tracking analysis or electron microscopy31.
Since HIV-1 virions are typically similar in size to exosomes, they may contaminate exosomes purified from HIV-1 infected samples. In proteomic screens, potential viral contaminants can be filtered out by limiting the database search only to human proteins. Further isolation techniques using established methodologies such as iodixanol density gradients and immunoaffinity isolation can also be used to separate HIV-1 virions from exosomes32,33.
Next, we discuss some suggestions to ensure reliable data analysis to identify potentially significant candidates once MS results are obtained from isolated exosomes. In the initial step of the analysis, the SILAC ratio histogram should follow a normal distribution. If the data distribution is skewed in a particular direction, the reader would need to determine the cause before proceeding with analysis. For instance, the labeled and unlabeled samples may not have mixed in equal proportions. Also, some peptides may not have heavy/light SILAC ratio values, and should be eliminated from the potential protein candidates. After candidates are selected, software and databases can be employed to verify exosome enrichment, interactions with test conditions, and possible pathways. The interactions between the candidate proteins and test condition should be mined through specific databases before bioinformatics analysis. For bioinformatics characterizations, gene ontology annotations can be identified using various software and databases
The SILAC technique employed here offers a systematic and straightforward approach to analyzing host exosomal proteome. Most biomedical laboratories would be able to adopt the procedures with minimal additional equipment and extra cost. The SILAC technique, however, is primarily designed for studies using cell lines. As such, other methods may need to be employed to study different biological samples. For instance, the label-free MRM (multiple reaction monitoring) method can be used for targeted quantification of proteins and peptides in clinical samples9,34. While in the case of determining protein content from multiple studies, the SILAC technique can also be used to investigate two or more samples. The chemical labeling method iTRAQ (isobaric tags for relative and absolute quantification) or TMT-based (tandem mass tag) methods allow much higher multiplexing and would be better-suited for multiple samples.
The procedure described here is not limited to proteomic characterization of exosomes from HIV-1 infected cells. With modifications, it can be adapted to analyze exosomes under a wide range of test and stress conditions such as bacterial infection, viral infection, and radiation. It is also suitable for studying other subcellular compartments.
Disclosures
The authors have declared no conflict of interest.
Acknowledgments
This work was supported by an ARRA supplement to the Lifespan/Tufts/Brown CFAR, P30AI042853-13S1, NIH P20GM103421, P01AA019072, R01HD072693, and K24HD080539 to BR. This work was also supported by Lifespan Pilot Research Fund (#701- 5857), Rhode Island Foundation Medical Research Grant (#20133969), and NIH COBRE URI/RIH Pilot Research Grant (P20GM104317) to ML. We thank James Myall and Vy Dang for help with the manuscript and figure preparation.
References
- Kowal J, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):968–977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008;9(6):871–881. doi: 10.1111/j.1600-0854.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- Booth AM, et al. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol. 2006;172(6):923–935. doi: 10.1083/jcb.200508014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Engelenburg SB, et al. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science. 2014;343(6171):653–656. doi: 10.1126/science.1247786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, et al. Quantitative proteomic analysis of exosomes from HIV-1-infected lymphocytic cells. Proteomics. 2012;12(13):2203–2211. doi: 10.1002/pmic.201100376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Ross PL, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- Strober W. Current Protocols in Immunology. John Wiley & Sons, Inc; 2001. [Google Scholar]
- Verma A, et al. Evaluation of the MTT lymphocyte proliferation assay for the diagnosis of neurocysticercosis. J Microbiol Meth. 2010;81(2):175–178. doi: 10.1016/j.mimet.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Cepko C, Pear W. Retrovirus infection of cells in vitro and in vivo. Curr Protoc Mol Biol. 2001. Chapter 9 Unit9.14. [DOI] [PubMed]
- Lennette ET, Karpatkin S, Levy JA. Indirect immunofluorescence assay for antibodies to human immunodeficiency virus. J Clin Microbiol. 1987;25(2):199–202. doi: 10.1128/jcm.25.2.199-202.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. p. 22. Chapter 3 Unit 3. [DOI] [PubMed]
- Olson BJSC, Markwell J. Current Protocols in Protein Science. John Wiley & Sons, Inc; 2001. [Google Scholar]
- Gauci VJ, Padula MP, Coorssen JR. Coomassie blue staining for high sensitivity gel-based proteomics. J Proteom. 2013;90:96–106. doi: 10.1016/j.jprot.2013.01.027. [DOI] [PubMed] [Google Scholar]
- Soldi M, Bonaldi T. The ChroP approach combines ChIP and mass spectrometry to dissect locus-specific proteomic landscapes of chromatin. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed]
- Trompelt K, Steinbeck J, Terashima M, Hippler M. A new approach for the comparative analysis of multiprotein complexes based on 15N metabolic labeling and quantitative mass spectrometry. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed]
- Li M, et al. Stem-loop binding protein is a multifaceted cellular regulator of HIV-1 replication. J Clin Invest. 2016;126(8):3117–3129. doi: 10.1172/JCI82360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Ramratnam B. Proteomic Characterization of Exosomes from HIV-1-Infected Cells. Methods Mol Biol. 2016;1354:311–326. doi: 10.1007/978-1-4939-3046-3_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmott E, Goodfellow I. Identification of protein interaction partners in mammalian cells using SILAC-immunoprecipitation quantitative proteomics. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed]
- Keerthikumar S, et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J Mol Biol. 2016;428(4):688–692. doi: 10.1016/j.jmb.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK, et al. EVpedia: a community web portal for extracellular vesicles research. Bioinformatics. 2015;31(6):933–939. doi: 10.1093/bioinformatics/btu741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, et al. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009;37:417–422. doi: 10.1093/nar/gkn708. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathan M, et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15(15):2597–2601. doi: 10.1002/pmic.201400515. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:561–568. doi: 10.1093/nar/gkq973. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. Chapter 3 Unit 3.22. [DOI] [PubMed]
- Lobb RJ, et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles. 2015;4:27031. doi: 10.3402/jev.v4.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber J, Clayton A. How pure are your vesicles. J Extracell Vesicles. 2013;2 doi: 10.3402/jev.v2i0.19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin R, Diou J, Belanger D, Tremblay AM, Gilbert C. Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J Immunol Methods. 2008;338(1-2):21–30. doi: 10.1016/j.jim.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Chertova E, et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J Virol. 2006;80(18):9039–9052. doi: 10.1128/JVI.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov M, Schmidt C, Urlaub H. Quantitative mass spectrometry-based proteomics: an overview. Methods Mol Biol. 2012;893:85–100. doi: 10.1007/978-1-61779-885-6_7. [DOI] [PubMed] [Google Scholar]