

Abstract
Higher-order nucleic acid structures called G-quadruplexes (G4s, G4 structures) can form in guanine-rich regions of both DNA and RNA and are highly thermally stable. There are >375,000 putative G4-forming sequences in the human genome, and they are enriched in promoter regions, untranslated regions (UTRs), and within the telomeric repeat. Due to the potential for these structures to affect cellular processes, such as replication and transcription, the cell has evolved enzymes to manage them. One such enzyme is G4 Resolvase 1 (G4R1), which was biochemically co-characterized by our laboratory and Nagamine et al. and found to bind extremely tightly to both G4-DNA and G4-RNA (Kd in the low-pM range). G4R1 is the source of the majority of G4-resolving activity in HeLa cell lysates and has since been implicated to play a role in telomere metabolism, lymph development, gene transcription, hematopoiesis, and immune surveillance. The ability to efficiently express and purify catalytically active G4R1 is of importance for laboratories interested in gaining further insight into the kinetic interaction of G4 structures and G4-resolving enzymes. Here, we describe a detailed method for the purification of recombinant G4R1 (rG4R1). The described procedure incorporates the traditional affinity-based purification of a C-terminal histidine-tagged enzyme expressed in human codon-optimized bacteria with the utilization of the ability of rG4R1 to bind and unwind G4-DNA to purify highly active enzyme in an ATP-dependent elution step. The protocol also includes a quality-control step where the enzymatic activity of rG4R1 is measured by examining the ability of the purified enzyme to unwind G4-DNA. A method is also described that allows for the quantification of purified rG4R1. Alternative adaptations of this protocol are discussed.
Keywords: Biochemistry, Issue 121, G4 Resolvase1, G4R1, RHAU, DHX36, G-quadruplex, ATP-dependent elution, G-quadruplex affinity protein purification, DNA helicase, RNA helicase
Introduction
G4 structures are highly stable nucleic acid secondary structures that form within guanine-rich regions of DNA and RNA. G4 structures are stabilized via Hoogsteen-bonding interactions and coordinate bonding within the central cavity with monovalent cations (i.e. K+ and Na+) that significantly contribute to the remarkable thermal stability of G4 structures1,2. Early bioinformatics studies suggested that the human genome contains >375,000 “potential G4-forming motifs”3,4. More recent study estimates suggest that the number of G4 motifs is higher by a factor of 2-55, while another study predicts 716,310 distinct potential G4-forming sequences in the human genome6. G4-forming sequences are evolutionarily conserved and not randomly dispersed in the genome. G4 motifs are enriched in gene coding regions, and upwards of 40% of all gene promoters contain G4 motifs7. Interestingly, the degree of enrichment of G4 motifs in a gene has been demonstrated to suggest the function of the gene. For example, proto-oncogenes and genes involved in development have significantly greater enrichment of G4 structures than tumor suppressor genes8,9.
With high thermal stabilities, a nearly ubiquitous presence throughout the genome, and the potential to significantly affect major cellular processes, it is unsurprising to find that the cell has evolved enzymes to manage these structures. One such enzyme is G4 Resolvase1 (G4R1; also called RHAU and DHX36), which we characterized as the source of the majority of tetramolecular G4-DNA resolving activity in human (HeLa) cells10. Since then, it has been shown that G4R1 tightly binds and catalytically unwinds tetramolecular and unimolecular G4-DNA and G4-RNA with the tightest reported KDs for a G4-binding protein11,12,13. Additionally, the G4-resolving activity of G4R1 has been implicated in a wide range of biochemical and cellular processes, including telomere/telomerase biology11,14,15,16, transcription and splicing17,18,19,20, development21, hematopoiesis21, and immune regulation22,23. With a preponderance of G4 sequences specifically situated throughout the genome and the diverse cellular processes that G4R1 has recently been implicated to be involved with, the ability to express and efficiently purify highly active rG4R1 will be of the utmost importance for elucidating the biochemical mechanisms and behaviors of this protein.
Here, we demonstrate a novel expression and purification scheme (Figure 1) that takes advantage of the ATP-dependent, G4-resolving activity of rG4R1 to efficiently isolate active enzyme. This scheme could be adapted to purify other ATP-dependent nucleic acid enzymes for which the product of the enzymatic reaction is no longer a substrate for binding, as is the case for G4R1.
Protocol
1. Preparation of G4-DNA Structures to be Used for the Purification of rG4R1 (Formation of Biotinylated G4-DNA G-Quadruplex)
Order the following DNA oligomer, called Z33-Bio, at a 1 µmole-scale: 5’ AAA GTG ATG GTG GTG GGG GAA GGA TTC GGA CCT-biotin 3’. Ensure that the biotin moiety is at the 3’ end of the oligomer.
Prepare 10x G4 buffer: 450 mM Tris-HCl pH 8, 25 mM EDTA, and 2,500 mM NaCl.
Resuspend the Z33 oligomer in 250 µL of water (so that the oligomer concentration is ~2.5 mM). Add 25 µL of 10x G4 buffer and mix. Incubate the oligomer at 50 °C for ~48 h.
- Briefly spin down the tube to collect the condensation (~1,000 x g, 10 s). Add ~50 µL of a high-mass, hydrophilic polysaccharide (30% Ficoll in H2O) and mix. Pour a 10% acrylamide/1x TBE/10% glycerol gel (gel dimensions: 16 cm x 16 cm x 1 mm). Once the gel is polymerized, wash the wells with 1x TBE and load the formed Z33-Bio oligomer evenly across most of the gel (sample lane loading can be visualized with Schlieren lines).
- In one lane at the end of the gel, load a small fraction of the oligomer that has been heated at 98 °C for 10 min (this will serve as an unstructured control), as well as a small amount of gel loading dye in another lane to monitor how far the gel has run.
Use 1x TBE as the gel running buffer and run the gel at 120 V until the dye front has traversed at least 1/3 of the gel.
Place a thin layer chromatography plate with its rough side face up in a sheet protector (or cover with plastic wrap). Remove one of the glass plates and transfer the gel to the surface of the sheet protector. Turn off the overhead lights and UV shadow the gel (long wavelength: 365 nm). Use a fresh razor blade and cut out the G-quadruplex-Z33-Bio band, which will be seen as running higher up in the gel (having slower mobility) relative to the melted control.
Place the gel slice containing the formed G4-DNA in a 50 mL tube and add just enough soaking buffer to cover the gel slice (soaking buffer consists of 1/3 volume 10x TBE, 1/3 volume saturated sodium acetate, and 1/3 volume H2O). Place the tube in a 37 °C incubator (nonhumidified) and incubate O/N.
- Transfer the solution to a 15 mL tube and add 1/10 volume glycogen (glycogen stock at 20 mg/mL in 10 mM Tris-HCl pH 8, 2.5 mM EDTA pH 8, and 0.05% sodium azide) and ~1.2x volume isopropanol. NOTE: Sodium azide is acutely toxic, so use with caution!
- Mix and place the tube at -20 °C for at least 2 h. Spin the tube at 2,700 x g in a tabletop centrifuge cooled to 4 °C for 12 min. Gently decant the solution from the pelleted G4-DNA. Wash the pellet 3 times with 70% EtOH + 50 mM NaCl (the salt in the wash is critical for G4 stability). Wick away excess liquid with lint-free tissue paper.
Place the washed pellet in the refrigerator for at least 2 h to hydrate the pellet and allow for easier resuspension. Resuspend the pellet in 50 µL of TNE buffer (10 mM Tris-HCl pH 8, 50 mM NaCl, and 0.05 mM EDTA pH 8).
Place 5 µL of this formed DNA G-quadruplex into 495 µL of H2O and quantify by using a spectrophotometer that allows the extinction coefficient to be determined by entering the nucleotide composition of the oligomer (the extinction coefficient of the Z33 DNA oligomer with a base composition of A = 8, C = 3, G = 15, and T = 7 is 341,946 L/mol/cm). Enter the dilution factor as 25 (not 100), as the formed G-quadruplex consists of 4 strands of DNA.
Aliquot the volume equivalent of 3 ODs (OD260 units) per tube and store at -20 °C; for example, if the 5 µL that was added to the 495 µL of H2O gives an OD260 reading of 3, then prepare 5 µL aliquots.
2. Preparation of G4-DNA Structures to be Used in An Enzymatic Activity Assay of rG4R1 (Formation of TAMRA-labeled G4-DNA)
Order the following DNA oligomer, called Z33-TAM, at a 1 µmole-scale: 5’ TAMRA-AAA GTG ATG GTG GTG GGG GAA GGA TTC GGA CCT 3’. Ensure that the TAMRA moiety is at the 5’ end of the oligomer.
Follow the same procedure as outlined in Section 1 for the formation of the G-quadruplex, except that ultraviolet (UV) shadowing is not necessary because the tetramethylrhodamine (TAMRA)-labeled G-quadruplex will be readily visible with the naked eye. Again, make sure to include a melted control on the gel.
After taking a spectrophotometer reading of the TAMRA-labeled DNA G-quadruplex, as in step 1, dilute the formed G-quadruplex to 0.2 pmol/µL with TNE buffer. Finally, add glycerol to a 10% final concentration and store at -20 °C.
3. Transform (DE3) PlysS Competent Cells with pTriEx4-DHX36 (Plasmid Encoding Human G4R1) and Grow/Induce Large Bacterial Cultures
Obtain the TriEx4-DHX36 plasmid.
Aliquot the bacteria into 20 µL aliquots and store them at -80 °C. Aliquot the SOC medium (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose) into 80 µL aliquots and store at -80 °C.
Prepare carbenicillin (CARB)/chloramphenicol (CAM) LB-agar plates. Stock concentrations of CARB and CAM are 50 mg/mL in H2O and 35 mg/mL in 70% EtOH, respectively, and these are 1,000x concentrated; thus, the final concentrations in the LB agar selection plates are 50 µg/mL and 35 µg/mL, respectively.
Place 1 aliquot of bacteria on ice and place 1 aliquot of SOC medium at RT. Add 1 µg of pTriEx4-DHX36 plasmid to the bacteria and swirl gently with the pipette tip to mix. Incubate on ice for a further 5 min and then heat shock at 42 °C for 30 s. Place on ice for 2 min and add the SOC medium.
Plate the transformed bacteria on prewarmed selection plates (typically, plate a small volume, such as 5 - 10 µL, to ensure low colony density so that single colonies can be easily inoculated into large cultures) and incubate at 37 °C O/N.
Prepare Terrific Broth according to the manufacturer’s instructions24. Make 500 mL per large flask (make sure to add glycerol) and autoclave on a short liquid cycle.
Add CARB/CAM (50 µg/mL/ 35 µg/mL) to the broth and then inoculate the cultures by taking agar plugs (using a p1000 pipette) containing a single bacterial colony and pipetting into the broth. Swirl the cultures vigorously to aerate them and then incubate the cultures at 37 °C O/N without shaking.
The next day, place the cultures in a 37 °C shaker and shake at ~225 rpm. Grow the cultures until the OD600 is about 0.4 - 0.6, which will typically take about 4 - 6 h, depending on the initial cell density. NOTE: This will require periodic density monitoring via spectrophotometric readings, and it is critical to not grow the cultures to much above 0.5 ODs. As a blank for spectrophotometric readings, spin down 1 mL of bacteria and use the clarified broth as the blanking solution.
Once the proper OD600 is obtained, immediately place the cultures on ice and quickly cool them to 10 °C. Monitor the temperature using a thermometer wiped clean with 70% EtOH. Expedite the cooling by quickly rotating the flasks while on ice, as this will limit further bacterial growth prior to recombinant protein induction.
Add IPTG to a 1 mM final concentration (~120 mg/500 mL culture) and then shake at 80 rpm at 14 °C for ~17 - 18 h. The ideal temperature for protein induction has been found to be 14 °C; to best achieve this, use a cooled water-bath shaker located in a conventional cold room and manually check the temperature of the water bath with a thermometer. NOTE: Alternatively, use an air-cooled shaker/incubator, although it is necessary to test what digitally-set temperature setting will yield the desired liquid temperature (test this by incubating a flask of water with a thermometer in it and shaking at 80 rpm for a few hours, adjusting the digital temperature setting accordingly until 14 °C is reached).
Place the cultures on ice and cool them quickly, as in step 3.9. Take a small aliquot of bacteria and take an OD600 reading; ideally, the cultures will have doubled during induction to about 0.8 - 1.2 OD600.
Transfer the bacteria to 500 mL centrifuge tubes and manually balance them using bacterial culture to even the weight between tubes. Once balanced, centrifuge them at 3,840 x g for 20 min at 4 °C. Pour off the clarified broth and freeze the bacterial pellets at -80 °C until the time of protein purification.
4. Purification of Human rG4R1
- Thaw and lyse bacterial pellets.
- Thaw the bacterial pellet in 3 mL of TN buffer at RT (TN buffer consists of 100 mM Tris pH 7.5 and 50 mM NaCl). Hold the bottles in hand and swirl to thaw/resuspend the bacterial pellet. Once thawed and relatively evenly suspended, bring the volume up to 5 mL with TN buffer (further suspension can be accomplished via pipetting up and down with a 5 mL pipette). NOTE: It is typical to thaw/prep either 2 or 4 bottles on the day of preparation, depending on the user’s level of comfort with the purification procedure.
- Dissolve 20 mg of lysozyme in 250 µL of H2O (per culture) and add to the resuspended bacteria. Allow the lysozyme to digest the bacteria for ~5 - 10 min while swirling the bottles in hand. NOTE: The color of the suspension will begin to lighten slightly as the digestion proceeds, and the suspension will be relatively non-viscous. If the cultures are too overgrown (i.e. much greater than 1.2 OD600), then chromosomal bacterial DNA can cause unwanted viscosity at this stage.
- Add 250 µL of protease inhibitor cocktail (PIC) and one 10 µL aliquot of leupeptin. Mix thoroughly.
- Place on ice and add 10 mL of cold TN buffer and 22 µL of BME. Transfer to a 50 mL tube.
- Sonicate the bacteria on ice with a digital sonicator set to 30% amplitude. Pulse at 2 s ON and 2 s OFF for 1 min. Repeat the sonication 3 times, allowing the samples to cool on ice for at least 2 min between sonication steps. NOTE: With multiple cultures, sonicate each one in turn before repeating the sonication iterations. During sonication, be careful to keep the sonication tip sufficiently submerged in the bacterial lysate, but not touching the sides of the tube, as this will generate excess heat.
- Add an equal volume (15 mL) of cold 4x SSC + 20 µL of BME + 50 µL of PIC + 1 aliquot of leupeptin and mix.
- In a tabletop centrifuge pre-cooled to 4 °C, centrifuge the lysates at 2,300 x g for 20 min. Transfer the supernatant to fresh, pre-cooled 50 mL tubes (1 tube per large culture being prepped).
- Prepare G4-magnetic beads.
- Take 1 mL of streptavidin paramagnetic bead (SPB) suspension (per 1 L culture) and transfer it to a 1.5 mL microcentrifuge tube. Pellet the SPB with a magnet and wash 2x with 2x SSC + 5 mM EDTA pH 8. Resuspend the washed SPB in 200 µL of the same solution used for washing the beads.
- Add one 3 OD aliquot of G4-DNA (Z33-Bio G4 prepared in Section 1) to the SPB suspension and mix quickly by pipetting up and down several times. Place the tubes on a rotator and rotate at RT for at least 30 min (and up to 60 min).
- After this period of rotation, block the G4-bound SPB by adding 1 mL of 0.4% lactalbumin, and keep on ice until needed. Dilute the 0.4% lactalbumin from the 4% lactalbumin stock using 1x Tris-glycine. NOTE: The stock 4% lactalbumin (w/v) is initially prepared by dissolution in 2 M glycine pH 7.5, with the further addition of 1x PIC and 0.05% sodium azide.
- Bind histidine-tagged rG4R1 to cobalt beads (CB) (continued from step 4.1).
- Add 1 mL of CB slurry (equivalent to a 0.5 mL bead volume) to each 50 mL tube containing clarified bacterial lysate. Incubate for 20 min at RT on a rotator. NOTE: 0.5 mL bead volume is used per 1 L of clarified lysate; e.g., if two 500 mL bacterial cultures are prepared, only add CB to the 50 mL tube containing clarified lysate from one of the cultures at this point, as lysate from the second culture will be serially bound with the same 0.5 mL of CB in step 4.3.3).
- Spin down CB at 110 x g for 5 min in a 4 °C tabletop centrifuge. Aspirate the liquid carefully, and leave a couple mL of liquid so as to not disturb the pelleted CB. Wash with 10 - 15 mL of cold 4x SSC + BME (0.5 µL of BME/mL of 4x SSC). NOTE: For washes, pour the 10-15 mL directly onto the pelleted cobalt beads instead of pipetting, as pipetting will cause the beads to get stuck to the inside of the pipette and protein will be lost.
- Pellet the CB again via centrifugation and then pour the next clarified lysate (30 mL, representing the 2nd large induced bacterial culture) onto the pelleted CB. Incubate for a further 20 min at RT on the rotator and then wash twice with 10-15 mL of 4x SSC + BME. Pellet at 110 x g for 5 min at 4 °C.
- After the second wash, aspirate the liquid and leave about 2 mL of beads/liquid at the bottom of the tube. Transfer the protein-bound CB to a pre-cooled 2-mL tube by using a 1 mL “wide-bore” pipette tip. NOTE: Use a fresh razor blade to cut the end of the tip prior to transfer, as the use of this “wide-bore” pipette tip will reduce the loss of protein-bound cobalt beads.
- Briefly centrifuge the beads in the 2 mL tube at high speed (~18,000 x g) in a microcentrifuge at 4 °C (~5 - 10 s is all that is necessary for quick pelleting). Gently remove and discard the supernatant by pipetting, being careful to not lose the protein-bound CB.
- Elute rG4R1 from CB.
- Incubate the CB in 0.5 mL of histidine elution buffer (HEB) on a rotator for 5 min in a cold room. Again, when adding HEB, just pipette the 0.5 mL onto the beads (to eliminate CB loss) and resuspend the beads by inverting the tube. HEB is 0.7 M L-histidine pH 6 (the pH is adjusted using acetic acid).
- Centrifuge at 18,000 x g in a 4 °C microcentrifuge for 1 min. Transfer the supernatant to a pre-cooled 15 mL tube. Gently remove and transfer the protein-containing supernatant by careful pipetting, leaving a small amount of liquid on top of the CB.
- Repeat steps 4.4.1 and 4.4.2 for a total of 3 HEB elutions.
- Elute once with 0.2 M EDTA pH 6.0; the color of the CB will change from pink to white as the EDTA chelates the cobalt.
- After this fourth and final elution has been transferred to the 15 mL tube, pipette the residual elution buffer from the CB by using a gel-loading tip on the end of a 1 mL pipette tip; by plunging the tip to the bottom of the tube, residual protein-containing elution buffer can be collected.
- Bind rG4R1 to G4-bound SPB and elute recombinant enzyme in an ATP-dependent manner.
- Prepare 5x Res Buffer: 250 mM Tris-Acetate pH 7.8, 250 mM NaCl, 2.5 mM MgCl2, and 50% glycerol. Prepare 3x Res Buffer: 0.915 mL of H2O, 2 mL of 5x Res Buffer, 0.33 mL of 0.4% lactalbumin (diluted from 4% lactalbumin with 1x Tris-glycine), 0.010 mL of BME, 0.015 mL of 1 M MgCl2, 0.050 mL of PIC, and 0.010 mL of leupeptin. Keep on ice. NOTE: The stock 4% lactalbumin (w/v) is initially prepared by dissolution in 2 M glycine pH 7.5, with the further addition of 1x PIC and 0.05% sodium azide.
- Pellet G4-SPB to the side of the tube with a magnet and discard the supernatant (G4-SPB, as prepared in step 4.2). Add 1 mL of 3x Res Buffer to the G4-SPB and pipette to mix.
- Pipette this 1 mL of G4-SPB suspended in 3x Res buffer into the 15 mL tube containing ~2 mL of protein-containing HEB/EDTA (eluate from step 4.4). Incubate for 15 min in a 37 °C water bath with occasional agitation so the beads do not settle.
- On ice, pellet the protein-bound G4-SPB to the side of the tube with a magnet. To wash, add 1 mL of 4x SSC + 0.4% lactalbumin (+ 0.5 µL/mL BME) and pipette to resuspend the beads. Pellet beads with the magnet on ice. Repeat this wash for a total of 2 washes. NOTE: Patience is required during the washing procedure to ensure that all of the magnetic beads have been pelleted between washes. This is especially true given the increased viscosity of the solution due to the glycerol contained in the Res buffers.
- Wash the G4-SPB 1x with 1 mL of 3x Res Buffer.
- Prepare elution buffer (EB): 0.5 mL of 3x Res Buffer, 0.1 mL of 0.1 M ATP, and 0.4 mL of H2O.
- Pre-warm 1 mL of EB to 37 °C in PCR tubes distributed across the heating block of a thermal cycler set to 37 °C.
- Pellet G4-SPB with the magnet on ice and resuspend the beads in 100 µL of pre-warmed EB. Immediately transfer the beads to a pre-warmed PCR tube and incubate for 30 s at 37 °C.
- Promptly add 12 µL of 5 M NaCl and pipette up and down vigorously 20 - 30 times. The combination of the high salt and ATP contained in the EB serves to elute rG4R1 from the G4-beads. NOTE: It is highly important to minimize the number of bubbles formed during the pipetting process, as bubbles can denature the protein.
- Immediately pellet the G4-SPB with a magnet and transfer the protein-containing eluate to a fresh PCR tube on ice (labeled with the date).
- Repeat the elution and transfer described in steps 4.5.8-4.5.10; the end product of this process is thus ~200 µL of EB containing purified rG4R1. Set aside two 7 µL aliquots (in PCR tubes labeled with the appropriate date) for use in the quality-control enzymatic activity assay that will test whether highly active rG4R1 has indeed been purified.
- Store the purified rG4R1 at -80 °C until the time of the activity assay.
5. Quality Control Enzymatic Activity Assay of Purified rG4R1
For each preparation of rG4R1 to be assayed, prepare 300 µL of resolvase assay buffer (RAB), where each 30 µL contains 0.2 pmol of TAMRA-labeled Z33 G4-DNA (prepared in step 2); the makeup of the RAB is as follows: 0.1 mL of 3x Res Buffer, 0.01 mL of 0.2 pmol/µL G4-Z33-TAM, 0.03 mL of 0.1 M ATP, and 0.16 mL of H2O. In a strip of 6 PCR tubes, pipette 40 µL of RAB into the first tube and 30 µL of RAB into the remaining tubes (keep the tubes on ice at this point).
Retrieve one of the 7 µL rG4R1 aliquots (from step 4.5.11) and place it on ice. With a p20 pipette set to 5 µL, gently pipette up and down a few times to mix the rG4R1 in the aliquot and transfer 5 µL to the first tube of the strip of 6 PCR tubes (the one that contains 40 µL of RAB). Next, set the pipette to 10 µL and serially transfer 10 µL to the remaining PCR tubes containing RAB.
Immediately incubate this strip of PCR tubes at 37 °C for 30 min in a thermal cycler, followed by a 4 °C hold. Open the tubes while they are being held at 4 °C and add 2 µL of 0.5 M EDTA pH 8 to each tube (to stop the enzymatic reaction).
- Pour a 10% acrylamide/1x TBE/10% glycerol gel. After removing the comb, wash the wells with 1x TBE. Add 5 µL of a high-mass, hydrophilic polysaccharide (30% Ficoll in H2O) to each tube and load 15 µL from each tube (the loading of wells can again be observed using Schlieren lines).
- As a control for G4-DNA mobility, add 4 µL of a high mass, hydrophilic polysaccharide to 20 µL RAB, and load 15 µL; as a control for unwound monomeric Z33, heat 20 µL of RAB at 98 °C for 10 min, add 4 µL of a high mass, hydrophilic polysaccharide, and load 15 µL.
Run the gel at 120 V for 2 h with 1x TBE as the running buffer. Image the gel on a multimodal imager with fluorescence-imaging capability using TAMRA-specific filters (excite with a 532 nm green laser and use a 580 nm band pass 30 filter (580 BP 30)). NOTE: A highly active preparation of rG4R1 will yield complete resolution of TAMRA-labeled Z33 in the first 3 lanes that were loaded on the gel, as shown in Figure 2.
6. Pooling of Highly Active rG4R1 Preps, Aliquoting, and Storage
NOTE: This step requires 2 people. The number and enzymatic requirements of the downstream assays that the purified rG4R1 will be used in will determine how many preparations are needed prior to aliquoting. A typical large preparation consists of the pooling of 8 highly active preparations (thus using a total of 16 induced 500 mL bacterial cultures), but this is an arbitrary number and is laboratory-specific.
Calculate the total volume of highly active, purified rG4R1 that is to be aliquoted. Typically, 7 µL aliquots are used in downstream assays. Fill the block of a thermal cycler set to 4 °C with strip PCR tubes (these will be aliquoted into, as the solid nature of the block will expedite the rapid closing of the PCR strips).
Retrieve the PCR tubes containing highly active rG4R1 from -80 °C and quickly thaw the tubes in hand; when a small amount of ice is left in the tube, place the tubes on ice. Combine all of the quickly-thawed preparations into a prechilled, 15 mL tube and mix well, making sure to minimize bubbles.
Using an automatic repeating pipette, dispense the desired aliquot volume into the pre-chilled PCR strip tubes in the thermal cycler. As soon as 1 - 2 strips are completed, have the second person close the lids and transfer them to 96-well wafers that are floating in liquid nitrogen (flash freezing is necessary to preserve enzymatic activity). NOTE: Do not take more than about 200 µL of rG4R1 into the tip of the automatic pipette at a time to reduce the time that the enzyme is not at 4 °C.
Once the prechilled PCR strip tubes in the thermal cycler have been filled with aliquots and properly frozen, quickly transfer more pre-chilled PCR strip tubes from ice to the thermal cycler and continue aliquoting. Continue in this manner until the remainder of the enzyme has been aliquoted, flash frozen in liquid nitrogen, and transferred to dry ice. Finally, transfer the aliquots to -80 °C for long-term storage.
7. Quantification of Purified rG4R1 Concentration
Pour a standard 5% stacking/12% resolving acrylamide/SDS gel for protein separation.
Thaw about 50 µL of aliquoted rG4R1, combine into one tube, and mix well. Measure the exact volume with a pipette and add an equal amount of 2x Laemmli sample buffer (65.8 mM Tris-HCl pH 6.8, 26.3% (w/v) glycerol, 2.1% SDS, and 0.02% bromophenol blue) + BME (50 µL/950 µL of 2x Laemmli buffer). Denature the protein at 98 °C for 10 min.
Prepare dilutions of broad-range MW markers in 1x sample buffer + BME such that the following protein amounts can be loaded onto the gel as standards: 31.3, 62.5, 125, 250, and 500 ng. NOTE: Each protein standard contained in the broad-range MW markers is present at 0.1 µg/µL, except for the 50 kDa protein, which is present at 0.3 µg/µL. These markers do not need to be heat-denatured.
Remove the comb from the gel and wash the wells with standard 1x SDS/glycine running buffer. Load the equivalent of 25 and 12.5 µL of rG4R1 (which is 50 and 25 µL of denatured protein), although the optimal volume of enzyme to load should be determined by the user, as the concentration of the enzymatic preparations will vary (e.g., 35 µL was loaded on the gel in Figure 3). Load the protein standards prepared in step 7.3. Ensure that the pipettes are properly calibrated, and make sure to be as precise as possible with pipetting technique to ensure accurate quantification.
Run the gel at 120 V until the dye front has traversed about 2/3 of the gel (to ensure proper separation of the proteins that make up the broad-range MW markers).
Remove the gel and dispose of the portion of the gel that will not contain protein by cutting it away with a razor blade. Place the gel in a glass casserole dish or an equivalent receptacle. Stain the gel with Coomassie dye (50 mg of Coomassie R-250 per 100 mL of 50:10:40 v/v methanol:acetic acid:H2O that has been filtered through a filter-paper funnel) O/N at RT on an orbital shaker set to ensure adequate agitation of the gel.
De-stain the gel with 30:10:60 v/v methanol:acetic acid:H2O using balled-up, lint-free tissue paper to expedite destaining. Change the destaining solution as needed. Continue destaining until the background is sufficiently low to visualize rG4R1 and protein standards; a typical gel at this stage is shown in Figure 3.
Place the destained gel in between a sheet protector and scan the gel at ≥300 dpi resolution. Open the scanned image in an image analysis software program (such as Fuji Multiguage or equivalent) and prepare standard curves from the protein standards that correspond to 75, 100, and 150 kDa (the curves represent gram amounts versus densitometric readings). Make sure to subtract the background from each lane being quantified during the process of obtaining densitometric values.
Assuming that the volume amount of rG4R1 that was loaded on the gel contains an amount of protein that is within the linear range of these standard curves, the gram amount of protein per microliter of rG4R1 loaded can be extrapolated.
Convert the extrapolated gram amount of rG4R1 into a molar amount using the molecular weight of G4R1, which is 120,000 g/mol. NOTE: For each gel, three extrapolated values will be generated (one for each of the three protein markers used to generate the standard curves: 75, 100, and 150 kDa). Run two further gels and stain and quantify them by repeating steps 7.1 - 7.10. With the results of the quantification for the first gel, the user will be able to gauge how much rG4R1 needs to be loaded on further gels to yield an amount that is within the linear range of the protein standards. Altogether, extrapolate nine values representing the concentration of highly active, purified rG4R1. Average these values to give the final batch-specific rG4R1 concentration, which is typically in the range of 20-100 nM. Calculate the standard deviation from these values (n = 9).
Representative Results
This protocol (Figure 1) routinely yields nearly pure, catalytically active rG4R1. As a measure of enzymatic activity, it is typically observed that 50% of 0.2 pmol of TAMRA-labeled tetramolecular G4-DNA is converted into monomers within the 0.2 - 0.013 µL range of rG4R1, as assayed by the G4 activity assay outlined above (Figure 2). Coomassie staining of purified rG4R1 indicates a single band at the expected 120 kDa size, with a minor contaminating band at ~75 kDa, which may represent truncated rG4R1 that has maintained its ability to bind G4-DNA beads and to elute in an ATP-dependent manner (Figure 3). The band of interest is quantified against a protein standard curve, and this protocol typically obtains 200 µL of 20-100 nM purified enzyme per 1 L of bacteria culture.
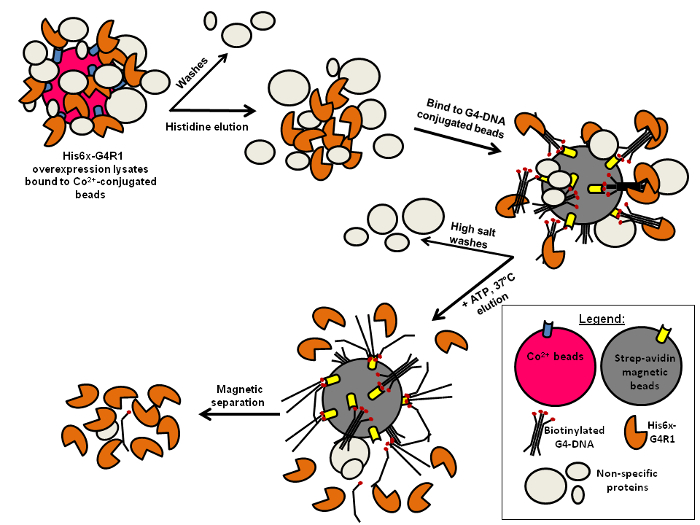 Figure 1: Schematic of a Two-step Purification of G4R1. A 6xHis-tagged rG4R1 is bulk-purified from E. coli lysates by first binding to and eluting from Co2+-conjugated beads. A binding step to G4-DNA-conjugated magnetic beads follows. Finally, an ATP-dependent elution step is necessary to obtain relatively pure and enzymatically active rG4R1. Please click here to view a larger version of this figure.
Figure 1: Schematic of a Two-step Purification of G4R1. A 6xHis-tagged rG4R1 is bulk-purified from E. coli lysates by first binding to and eluting from Co2+-conjugated beads. A binding step to G4-DNA-conjugated magnetic beads follows. Finally, an ATP-dependent elution step is necessary to obtain relatively pure and enzymatically active rG4R1. Please click here to view a larger version of this figure.
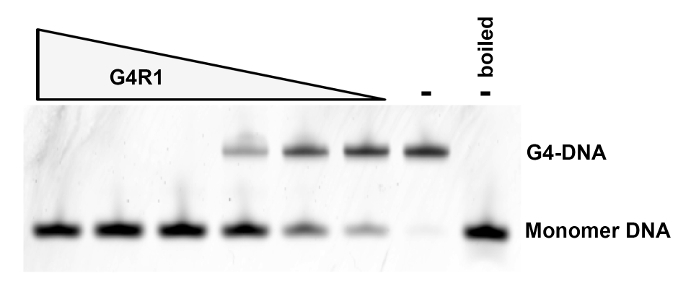 Figure 2: Quality-control G4-DNA Unwinding Assay. Lanes 1 - 6: A constant concentration of TAMRA-labeled tetramolecular G4-DNA was incubated at 37 °C for 30 min in the presence of 4x serial dilutions of purified rG4R1 representing 3.9 µL, 0.83 µL, 0.2 µL, 0.05 µL, 0.013 µL, and 0.003 µL, respectively. Lane 7: tetramolecular G4-DNA in the absence of rG4R1. Lane 8: tetramolecular G4-DNA boiled to reduce the G4 structure into monomers in the absence of rG4R1. Please click here to view a larger version of this figure.
Figure 2: Quality-control G4-DNA Unwinding Assay. Lanes 1 - 6: A constant concentration of TAMRA-labeled tetramolecular G4-DNA was incubated at 37 °C for 30 min in the presence of 4x serial dilutions of purified rG4R1 representing 3.9 µL, 0.83 µL, 0.2 µL, 0.05 µL, 0.013 µL, and 0.003 µL, respectively. Lane 7: tetramolecular G4-DNA in the absence of rG4R1. Lane 8: tetramolecular G4-DNA boiled to reduce the G4 structure into monomers in the absence of rG4R1. Please click here to view a larger version of this figure.
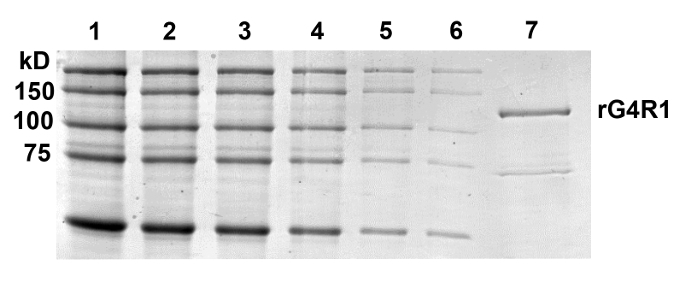 Figure 3: Quantification of Purified rG4R1 Concentration. Broad-range MW protein markers were loaded in the following quantities: 500, 250, 125, 62.5, 31.3, and 15.7 ng in Lanes 1 - 6, respectively, and were used to generate a standard curve of protein concentrations. Lane 7 represents 35 µL of rG4R1. This particular gel was quantified, as part of a triplicate set of gels, resulting in an average protein concentration of 62 ± 22 nM standard deviation (SD; N = 9). Please click here to view a larger version of this figure.
Figure 3: Quantification of Purified rG4R1 Concentration. Broad-range MW protein markers were loaded in the following quantities: 500, 250, 125, 62.5, 31.3, and 15.7 ng in Lanes 1 - 6, respectively, and were used to generate a standard curve of protein concentrations. Lane 7 represents 35 µL of rG4R1. This particular gel was quantified, as part of a triplicate set of gels, resulting in an average protein concentration of 62 ± 22 nM standard deviation (SD; N = 9). Please click here to view a larger version of this figure.
Discussion
This protocol represents a highly efficient expression, purification, and quantification scheme for the isolation of the DHX36 gene product, G4-Resolvase1 (G4R1, also called RHAU and DHX36) (Figure 1). This protocol utilizes two purification steps: His-tag affinity purification on cobalt affinity beads and enzymatic purification on G4-DNA-conjugated beads. The latter step is unique in that it takes advantage of the tight affinity, high specificity, and catalytic unwinding activity that rG4R1 has for G4 structures. The tight affinity for G4 structures (Kd in the low-pM range) allows for a high salt wash (near the solubility limit of NaCl) prior to elution from the G4 beads, greatly reducing the presence of non-specific proteins. The catalytic activity of G4R1 on G4 structures allows for the specific elution of active rG4R1 upon the addition of ATP and MgCl2. This two-step purification scheme consistently produces highly pure, catalytically active rG4R113 in quantities suitable for most biochemical analyses.
Several key steps will ensure a good yield of highly pure, catalytically active rG4R1. The first is to ensure the proper induction conditions of His-rG4R1 in the bacterial expression strain. We have found that the best yield of recombinant protein occurs when cells are grown to an OD600 of no more than 0.4-0.6 just prior to induction. Growing the cells beyond this point may result in an overall loss of protein recovery, possibly due to the incorporation of rG4R1 into inclusion bodies. Second, we obtained a higher concentration of purified protein by “serially-binding” the lysates to the cobalt affinity beads. For example, we bound the lysate from 0.5 L of induced cultures to the cobalt affinity beads and then performed a second binding of another lysate from an additional 0.5 L of culture to the same cobalt affinity beads. This step ensures a more concentrated preparation of protein by increasing the number of rG4R1 molecules bound to a given volume of beads, thus utilizing the full capacity of the cobalt affinity beads. Thirdly, the high salt wash after binding the cobalt affinity elutions to the G4 beads ensures that nearly all non-specific protein binding to the beads is removed. Fourthly, the ATP/MgCl2 elution step allows for rG4R1 bound to the G4 beads to catalytically unwind the tetramolecular structure into single strands, causing rG4R1 to be released from the beads. We cannot completely rule out the possibility that ATP elutes rG4R1 in a competitive rather than a catalytic manner; however, this is less likely to be the case, since we have previously shown that a non-hydrolyzable ATP analogue is not sufficient for competitive binding13,18. The affinity of rG4R1 for the unwound, single-stranded DNA is an order of magnitude less than the starting tetramolecular G4-DNA, and thus rG4R1 should not re-bind to the beads. In order to reduce this possibility, however, this step should be done at 37 °C, and the elution volume should be separated from the beads as quickly as possible. The elution step is repeated twice to ensure maximum recovery. If downstream applications require the protein to be free of DNA contaminants, we recommend an additional cleanup step in which the purified preparation is re-bound to streptavidin beads in order to remove any biotinylated DNAs, if present.
We have found that rG4R1 is susceptible to degradation if the proper conditions are not maintained throughout the protocol. In order to maintain the integrity and activity of the enzyme, we employ the following critical safeguards. Protease inhibitors are kept present throughout the purification procedure. The protocol is performed at 4 °C, unless otherwise noted. The protein is purified in the presence of lactalbumin and β-mercaptoethanol. The protocol is performed in a timely fashion (in 1 day for the purification). Additionally, we have found that multiple freeze-thaw cycles negatively impact the activity of the protein, so we aliquot the protein into “one-time use,” 7 µL aliquots following purification and store them at -80 °C.
Although the presence of lactalbumin in the preparation is required to maintain the integrity and activity of the protein, as mentioned above, we have found that this may also impede downstream applications. Other potential interfering molecules that are present in the purified rG4R1 preparation include ATP, β-mercaptoethanol, and singled-stranded DNA. For example, we have found this protein preparation to be incompatible with BIACORE analysis due to the high background signal from the buffer components. Also, the presence of lactalbumin in the protein preparation precludes the use of standard Bradford and BCA protein quantitation assays. However, we have developed an alternative gel-based quantitation method to circumvent this limitation.
This purification procedure, which harnesses the enzymatic activity of G4R1 as a means to specifically purify it, makes this method distinct from other methods. For example, other groups have expressed FLAG-tagged rG4R1 in human cells15,25 or GST-tagged rG4R1 in insect cells26 and purified it by FLAG- or GST-affinity chromatography, respectively. These methods have the advantage of being done in a eukaryotic expression system compared to a bacterial expression system. Estimated Kd values of the resulting purified GST-G4R1 for G4 structures were found to be an order of magnitude higher14 than our reported Kd values12,13. We attribute this discrepancy in Kd values to differences associated with a bulkier GST-tag versus a 6xHis-tag, differences in the purities obtained from these two purification schemes, and differences in the extent and type of post-translational modifications acquired in a bacterial versus an insect expression system. Our approach has a distinct advantage over the aforementioned alternatives because the purification of this protein directly hinges on its enzymatic activity. Therefore, we primarily obtain an enzyme that has been folded and modified in such a way that maintains its enzymatic properties. Other affinity-tag and/or size-exclusion techniques are unable to separate active enzyme from enzymatically dead enzyme. It would be useful for future protocol development to combine the strengths of other groups’ purification protocols15,25,26 (i.e. a human or insect cell expression system) with the strength of our protocol (i.e., catalytic-based purification) to further improve upon this method.
Although this protocol is currently specific for G4R1, it could easily be adapted to any ATP-dependent, G4-resolving proteins, including, but not limited to, BLM, WRN, FANCJ, hnRNP proteins, hPif1, and/or ChlR1/DDX1. By altering the sequence of the nucleic acid bound to the streptavidin beads, this protocol could be adapted to purify other ATP-dependent nucleic acid enzymes for which the product of the enzymatic reaction is no longer a substrate for the enzyme, including nucleic acid helicases and nucleases.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank our funding sources, including a generous gift from the Ware Foundation (to J.P.V.), The National Institutes of HealthGrant T32-CA079448 (to P.J.S.), and Ball State University startup funds (to P. J. S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Qin Y, Hurley LH. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie. 2008;90(8):1149–1171. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegle O, Payet L, Mergny JL, MacKay DJ, Leon JH. Predicting and understanding the stability of G-quadruplexes. Bioinformatics. 2009;25(12):i374–i382. doi: 10.1093/bioinformatics/btp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AK, Johnston M, Neidle S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005;33(9):2901–2907. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33(9):2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedrat A, Lacroix L, Mergny JL. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016;44(4):1746–1759. doi: 10.1093/nar/gkw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza O, Bourdoncle A, Boule JB, Brosh RM, Jr, Mergny JL. G-quadruplexes and helicases. Nucleic Acids Res. 2016;44(5):1989–2006. doi: 10.1093/nar/gkw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35(2):406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J, Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006;34(14):3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J, et al. G4 motifs correlate with promoter-proximal transcriptional pausing in human genes. Nucleic Acids Res. 2011;39(12):4975–4983. doi: 10.1093/nar/gkr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn JP, et al. The DEXH protein product of the DHX36 gene is the major source of tetramolecular quadruplex G4-DNA resolving activity in HeLa cell lysates. J Biol Chem. 2005;280(46):38117–38120. doi: 10.1074/jbc.C500348200. [DOI] [PubMed] [Google Scholar]
- Booy EP, et al. The RNA helicase RHAU (DHX36) unwinds a G4-quadruplex in human telomerase RNA and promotes the formation of the P1 helix template boundary. Nucleic Acids Res. 2012;40(9):4110–4124. doi: 10.1093/nar/gkr1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creacy SD, et al. G4 resolvase 1 binds both DNA and RNA tetramolecular quadruplex with high affinity and is the major source of tetramolecular quadruplex G4-DNA and G4-RNA resolving activity in HeLa cell lysates. J Biol Chem. 2008;283(50):34626–34634. doi: 10.1074/jbc.M806277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri B, et al. G4 resolvase 1 tightly binds and unwinds unimolecular G4-DNA. Nucleic Acids Res. 2011;39(16):7161–7178. doi: 10.1093/nar/gkr234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattmann S, Stadler MB, Vaughn JP, Akman SA, Nagamine Y. The DEAH-box RNA helicase RHAU binds an intramolecular RNA G-quadruplex in TERC and associates with telomerase holoenzyme. Nucleic Acids Res. 2011;39(21):9390–9404. doi: 10.1093/nar/gkr630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton AN, Collins K. The 5' guanosine tracts of human telomerase RNA are recognized by the G-quadruplex binding domain of the RNA helicase DHX36 and function to increase RNA accumulation. Mol Cell Biol. 2011;31(4):736–743. doi: 10.1128/MCB.01033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaldino PJ, et al. Mutational Dissection of Telomeric DNA Binding Requirements of G4 Resolvase 1 Shows that G4-Structure and Certain 3'-Tail Sequences Are Sufficient for Tight and Complete Binding. PLoS One. 2015;10(7):e0132668. doi: 10.1371/journal.pone.0132668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booy EP, et al. The RNA helicase RHAU (DHX36) suppresses expression of the transcription factor PITX1. Nucleic Acids Res. 2014;42(5):3346–3361. doi: 10.1093/nar/gkt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, et al. Yin Yang 1 contains G-quadruplex structures in its promoter and 5'-UTR and its expression is modulated by G4 resolvase 1. Nucleic Acids Res. 2012;40(3):1033–1049. doi: 10.1093/nar/gkr849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Schilling M, Wirbelauer C, Hess D, Nagamine Y. Facilitation of mRNA deadenylation and decay by the exosome-bound, DExH protein RHAU. Mol Cell. 2004;13(1):101–111. doi: 10.1016/s1097-2765(03)00481-7. [DOI] [PubMed] [Google Scholar]
- Yoo JS, et al. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog. 2014;10(3):e1004012. doi: 10.1371/journal.ppat.1004012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JC, et al. The DEAH-box helicase RHAU is an essential gene and critical for mouse hematopoiesis. Blood. 2012;119(18):4291–4300. doi: 10.1182/blood-2011-08-362954. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011;34(6):866–878. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2010;107(34):15181–15186. doi: 10.1073/pnas.1006539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BD Biosciences. 2016. Available from: http://www.bd.com/ds/technicalCenter/misc/difcobblmanual_2nded_lowres.pdf.
- Booy EP, McRae EK, McKenna SA. Biochemical characterization of G4 quadruplex telomerase RNA unwinding by the RNA helicase RHAU. Methods Mol Biol. 2015;1259:125–135. doi: 10.1007/978-1-4939-2214-7_9. [DOI] [PubMed] [Google Scholar]
- Lattmann S, Giri B, Vaughn JP, Akman SA, Nagamine Y. Role of the amino terminal RHAU-specific motif in the recognition and resolution of guanine quadruplex-RNA by the DEAH-box RNA helicase RHAU. Nucleic Acids Res. 2010;38(18):6219–6233. doi: 10.1093/nar/gkq372. [DOI] [PMC free article] [PubMed] [Google Scholar]