

Abstract
Cardiomyocytes are prone to variations of the cell cycle, such as endoreduplication (continuing rounds of DNA synthesis without karyokinesis and cytokinesis) and acytokinetic mitosis (karyokinesis but no cytokinesis). Such atypical cell cycle variations result in polyploid and multinucleated cells rather than in cell division. Therefore, to determine cardiac turnover and regeneration, it is of crucial importance to correctly identify cardiomyocyte nuclei, the number of nuclei per cell, and their cell cycle status. This is especially true for the use of nuclear markers for identifying cell cycle activity, such as thymidine analogues Ki-67, PCNA, or pHH3. Here, we present methods for recognizing cardiomyocytes and their nuclearity and for determining their cell cycle activity. We use two published transgenic systems: the Myh6-H2B-mCh transgenic mouse line, for the unequivocal identification of cardiomyocyte nuclei, and the CAG-eGFP-anillin mouse line, for distinguishing cell division from cell cycle variations. Combined together, these two systems ease the study of cardiac regeneration and plasticity.
Keywords: Developmental Biology, Issue 120, Myocardial biology, Cardiomyocyte, Proliferation, Genetically altered mice, Cardiac development, Cell biology, Cell culture
Introduction
The correct identification of cardiomyocyte nuclei and the cell cycle status is of crucial importance for the determination of cardiac muscle turnover and regeneration. This is especially true for the use of nuclear markers, such as pHH3, Ki-67, or Thymidine analogs, for identifying cell cycle activity. As the proliferative capacity of adult mammalian cardiomyocytes is very small1, a false identification of a nucleus positive for a proliferation marker of a cardiomyocyte nucleus could make a crucial difference in the outcome of a proliferation assay. Moreover, cardiomyocytes are prone to variations in the cell cycle, such as endoreduplication and acytokinetic mitosis, which result in polyploid and multinucleated cells rather than in cell division. To this end, the interpretation of antibody staining against common cell cycle markers is not conclusive in all cases.
Here, we present methods for the straight-forward recognition of mouse cardiomyocytes and their nuclearity in native isolated cells and thick tissue sections at postnatal and adult stages by the unequivocal identification of their nuclei. For that purpose, a transgenic mouse line with cardiomyocyte-specific expression of a fusion protein consisting of human histone H2B and mCherry under the control of the Myh6 promoter (Myh6-H2B-mCh) was used2. Cross-breeding this mouse line with a transgenic proliferation indicator mouse line, in which the expression of an eGFP-anillin fusion protein is under the control of the ubiquitous chicken actin promoter with a CMV enhancer (CAG-eGFP-anillin), allows for the determination of the cell cycle status. The scaffolding protein anillin is specifically expressed in cell-cycle active cells3, and its differential subcellular localization during the cell cycle allows for live-tracking cell-cycle progression with a high resolution of M-Phase4. Therefore, the double transgenic mice can be used to discriminate between proliferating cardiomyocytes and those that undergo cell-cycle variations. This proves especially useful in screening for proliferation-inducing substances in vitro.
Protocol
All procedures of this protocol involving animals were in accordance with the ethical standards of the University of Bonn and complied with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
1. In Vitro Visualization of Cell Cycle Activity in Postnatal Cardiomyocytes
- Postnatal cardiomyocyte dissociation
- Pre-experimental preparations
- Prepare culture media (IMDM, 1% Penicillin/Streptomycin, 1% non-essential amino acids, 0.1% β-Mercaptoethanol); medium 1: add FCS to 2%; medium 2: add FCS to 20%.
- Coat a 96-well culture dish (half-growth area: 15 mm2) with 0.1% gelatin in PBS solution.
- Heart dissection
- Prepare a Petri dish with 15 mL of PBS and store it on ice. Sterilize two tweezers and small scissors by putting them in a dry glass bead sterilizer.
- Sacrifice neonatal transgenic mice (CAG-eGFP-anillin/Myh6-H2B-mCh) by decapitation. Open the thorax, dissect the heart out, as described in Ehler et al., 2013 (J Vis Exp.; (79):50154. doi:10.3791/50154), and transfer it to the ice-cold PBS. Move on to the next mouse.
- If using non-homozygous breeding pairs, identify the transgenic hearts with a fluorescent microscope. Put the hearts in PBS under a fluorescent microscope. Check for eGFP-anillin expression (Ex.: 488 nm; Em.: 509 nm) and H2B-mCh expression (Ex.: 587 nm; Em.: 610 nm) with the appropriate filter sets.
- Cardiomyocyte isolation
- Dissociate the selected hearts by using the neonatal heart dissociation kit. Incubate the enzyme mix 1 at 37 °C for 5 min. To obtain 1 mL of the final enzyme mix, add 945 µL of enzyme mix 1 to 55 µL of enzyme mix 2.
- Transfer up to 4 hearts from P1 - P3 mice or up to 2 hearts from P4 - P6 mice to a 1.5-mL reaction tube containing 1 mL of enzyme mix and cut the heart into small pieces with scissors. Transfer the solution into 15 mL reaction tubes.
- Incubate at 37 °C for 15 min. To maximize the contact between the tissue and the enzymes, put the 15-mL tube almost horizontally in the incubator. Mix by pipetting 5 - 10 times up and down with a 5-mL pipette. Repeat this step three times.
- Add 7.5 mL of medium 2 (20% FCS) to stop the enzyme reaction. Filter the cell suspension through a 70-µm cell strainer. Rinse the cell strainer with 3 mL of medium 2.
- Centrifuge the cell suspension at 300 × g for 15 min and discard the supernatant. Resuspend the pellet in 500 µL of medium 1; red blood cell lysis is not required for further experiments. Pipet 10 µL of the cell suspension into a cell counting chamber and determine the number of cells.
- For a 96-well plate: seed 10,000 cells per well in 120 µL of medium 1 (2% FCS). Place the cells into an incubator (37 °C and 5% CO2) and proceed immediately to the transfection.
- Transfection of neonatal cardiomyocytes with siRNAs or miRNAs
- Wipe a bench with RNase decontamination solution to remove the RNases. Clean the following material with RNase decontamination solution: 10-µL, 100-µL, and 200-µL pipettes and an ice box. Thaw siRNA stock aliquots (100 µM) on ice.
- Prepare 2 µM working stocks of si/miRNAs in reduced serum medium (98 µL of reduced serum medium + 2 µL of siRNA 100 µM stock). The working stock should be used the same day. Freezing is not recommended.
- Add 4 µL of the 2 µM stock to 6 µL of reduced serum medium to a 0.5-mL reaction tube (amount for one 96-well; final si/miRNA concentration in 140 µL: ~ 57 nM). Choose an appropriate volume for the number of wells to be transfected with a certain si/miRNA.
- Prepare a master mix of the transfection reagent. Use 10 µL (0.6 µL of transfection reagent + 9.4 µL of reduced serum medium) for each well (96 wells total).
- Add the si/miRNA mix to the transfection reagent mix. Incubate for 5 min on ice. Pipet 20 µL into each well. Incubate the cells for 48 h at 37 °C and 5% CO2 and then replace the medium in each well with 120 µL of medium 1. After 24 h or more, proceed to fixation and immunofluorescence staining.
- Fixation and immunofluorescence staining
- Remove the medium and wash the cells once with PBS. Cover the cells with 4% formaldehyde solution for 20 min. Take off the formaldehyde solution and wash the cells once with PBS, and then cover them with PBS for storage or proceed to immunostaining.
- Incubate with primary antibody (concentration according to the manufacturer's instructions) in 0.2% Triton-X and 5% donkey serum for at least 2 h. If staining for α-actinin (dilution 1:400; monoclonal anti-α-actinin), stain overnight at 4 °C.
- Remove the primary antibody and wash 3 times with PBS. Dilute the secondary antibody in Hoechst solution 1:400 and incubate for 1 h at RT protected from light. Remove the antibody, wash with PBS 3 times, and cover the cells with PBS for storage at 4 °C.
- Confocal video microscopy
- Use a confocal microscope for the image acquisition. Open the imaging software. Open "A1plus Settings" and check the boxes for Ch1, Ch2, and Ch3. Set Ch1 to DAPI, Ch2 to eGFP, Ch3 to Cy3, and Ch4 to Cy4 by clicking on the pulldown menu.
- For each channel, click on HV and set it to 80 by using the slider bar. Set the Offset to 0 by using the slider bars, and click on the "Home" button to set the pinhole to home position. Set the scan size to 1,024 x 1,024 by clicking on the pulldown menu.
- Click optimize to open "XYZ Size Setup" window. Check "Perfect Voxel" box under "Suggested Step (z)". Increase the laser intensities until the picture is neither underexposed nor overexposed. NOTE: For example; Ch1 (DAPI): 16%, Ch2 (eGFP): 12%, Ch3 (Cy3): 100%, Ch4 (Cy5): 1%. Set the Offset for all channels to 0.
- Take pictures using a 20X objective (200X magnification). Scan a large image consisting of tile images (e.g., 3 x 3).
- In the imaging software, go to "Acquire" and choose "Scan large image" from the pulldown menu. Under "Area," choose "current position is at top left corner" from the pulldown menu and set "Number of fields in X and Y" to 3 x 3. Click "Scan."
- Image analysis
- Define the number of cardiomyocyte nuclei by counting H2B-Ch signals and the number of cardiomyocytes by counting α-actinin signals.
- Click on "Measure" and choose "Manual Measurement" from the pulldown menu. Choose "Counts" from the next pulldown menu. Click on nuclei with H2B-mCh signals, thereby marking and counting them. Do the same for α-actinin-positive cells to get the number of cardiomyocytes.
- Count S/G2 phase cardiomyocytes with nuclear eGFP-anillin signals (eGFP-anillin+/ H2BmCh+ nuclei). Click on "Measure" and choose "Manual Measurement" from the pulldown menu. Choose "Counts" from the next pulldown menu. Mark the nuclei with eGFP-anillin signals and H2B-mCh signals by clicking on them.
- Count the cardiomyocytes with mitosis-specific eGFP-anillin signals (e.g., Figure 1F and G), such as cytoplasmatic signals, contractile rings, and midbodies. Click on "Measure" and choose "Manual Measurement" from the pulldown menu. Choose "Counts" from the next pulldown menu. Mark cardiomyocytes with mitosis-specific eGFP-anillin signals (e.g., Figure 1F and G), cytoplasmatic signals, contractile rings, and midbodies by clicking on them.
2. Determination of Nucleation in Adult Cardiomyocytes by Langendorff Dissociation and Thick Tissue Sections
- Isolation of adult cardiomyocytes by Langendorff dissociation
- Preparations before heart dissection
- Fill an insulin syringe (30-gauge needle) with heparin (20 units/g body weight). Grasp the mouse by the scruff of the neck. Lift and turn the mouse backwards to expose the abdomen for injection.
- Perform an intraperitoneal injection. Put the heparinized mouse back into the cage and wait at least 15 min before starting the heart dissection. Rinse and ventilate the Langendorff apparatus with 5 - 10 mL of perfusion buffer (perfusion buffer: 135 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2.5 mM HEPES, 5 mM Glucose, and 25 mM BDM in ddH2O, adjust to pH 7.4 with NaOH).
- Heart dissection
- Prepare a 10-cm Petri dish with chilled (4 °C) PBS and place it on ice. Fill and ventilate a 1-mL syringe connected to a cannula (20 gauge) with PBS. Fix the cannula with modeling clay on the border of the Petri dish. Place the cannula tip slightly below the PBS surface.
- Sacrifice the mouse by cervical dislocation. Wipe the abdomen with 70% ethanol solution. Make an incision from the mid abdomen to the sternum with surgical scissors. Remove the diaphragm and cut the thorax bilaterally with surgical scissors.
- Lift and fix the sternum with a 20-gauge needle, open the thoracic cavity, and gently grasp the heart with anatomical forceps. Lift the heart and remove it from the lungs and vasculature by cutting the vessels of the outflow tract. NOTE: To identify the aorta, position the heart with its ventral side up. This decreases the distance between both atria, so the aorta with its branches can be found between the atria. Depending on the individual heart architecture, the branches can be removed if the principal branch of the aorta is still long enough.
- Transfer the heart to the 10-cm Petri dish containing chilled PBS (see step 2.1.2.1). Cannulate the heart by pulling the ascending aorta on an G20x1 ½ injection cannula, which was blunted by an oscillating multi-tool to avoid tissue damage. Fix the aorta with a thread on the cannula. Gently rinse the heart with PBS to remove excess blood by pushing the syringe plunger. A slight enlargement of the heart should be visible.
- Langendorff heart dissociation
- Connect the cannulated heart rapidly to the Luer lock adapter at the Langendorff perfusion apparatus. Perfuse the heart with 30 mL of oxygenated perfusion buffer at 37 °C for 5 min with a flow rate of 1 drop/s. The flow rate can be adapted by regulating the O2 flow within the Langendorff apparatus using the pressure-reducing valve (pressure between 0 and 200 mbar).
- Prepare digestion buffer immediately before use: perfusion buffer with 6 U Collagenase B, 10,000 units Trypsin, and 50 µM CaCl2. Remove the perfusion buffer and start the enzymatic digestion by adding 30 mL of warm (37 °C) and oxygenated digestion buffer. NOTE: The amount of Collagenase B needs to be titrated depending on the activity of different batches.
- Adjust the flow to a rate of 1 drop/s and digest the heart for 10 - 13 min. To avoid contamination, do not use the efflux again. Transfer the heart to a 10-cm Petri dish with 5 mL of digestion buffer. Dissect the tissue manually with forceps by holding the heart with one pair of forceps and using the other pair to rip the heart into small pieces. NOTE: At this step, the atria can be separated, cut into small pieces (with iris scissors), and digested for 30 min in 750 µL of digestion buffer in the incubator at 37 °C in order to obtain isolated atrial cells. To get pure atrial cardiomyocytes, pipet up and down several times using a 1-mL pipette. Stop the enzyme reaction by adding 750 µL of stop solution.
- Stop the digestion by adding 5 mL of stop solution (perfusion buffer with 5% FBS and 50 µM CaCl2). Using a serological pipette, filter the cell suspension through a 100-µm cell strainer and collect the cells in a 50-mL centrifuge tube. Centrifuge the filtered cell suspension at 80 x g for 1 min at room temperature.
- Discard the supernatant and gently resuspend the cell pellet in 10 mL of stop solution using a 10 mL serological pipette. Centrifuge the cell suspension at 80 x g for 1 min at room temperature.
- Following centrifugation, discard the supernatant and resuspend the cells in 1 mL of culture medium (IMDM with 20% FCS, 0.1 mM non-essential amino acids, 50 µg/mL each of penicillin and streptomycin, and 0.1 mM β-mercaptoethanol) for culturing. Alternately, proceed to step 2.2, "immunofluorescence staining," and fix the cells.
- For fixation on 24-well plates, plate 10,000 of the isolated cells per well on 8 µg/mL laminin-coated coverslips in 24-well plates with 500 µL of culture medium at 37 °C and 5% CO2 for at least 3 h.
- Immunofluorescence staining on Langendorff-isolated cardiomyocytes in suspension
- Fix Langendorff-isolated cardiomyocytes in 1 mL of 4% PFA in PBS, pH 7.4, for 15 min at room temperature. Centrifuge the cell suspension at 800 x g for 2 min at room temperature, remove the fixative, and wash the cells twice with PBS.
- Resuspend the fixed cardiomyocytes in 1 mL of PBS and either proceed with the immunofluorescence staining or store them in 500 µL of PBS per well on a 24-well plate at 4 °C. Transfer a part of the cell suspension (~ 100 - 200 µL) to a microcentrifuge tube. Centrifuge the cell suspension at 800 x g for 2 min at room temperature and discard the supernatant.
- Permeabilize, block, and stain the cells with primary antibody against α-actinin by adding 200 µL of primary antibody diluted 1:400 in PBS with 0.2% Triton X-100 and 5% donkey serum for 1 h at room temperature.
- Centrifuge at 800 x g for 2 min at room temperature and discard the supernatant. Wash the cell pellet with PBS and centrifuge at 800 x g for 2 min at room temperature. Discard the supernatant and incubate in 200 µL of Alexa-Fluor-conjugated secondary antibody (anti-mouse IgG1) diluted 1:400 in Hoechst dye (working solution: 1 µg/mL) for 1 h at room temperature, protected from light.
- Centrifuge at 800 x g for 2 min at room temperature and discard the supernatant. Repeat this step. Protect the sample from light. Resuspend the cells in 200 - 500 µL of PBS and transfer 100 - 300 µL of the cell suspension to a well of a microscopy chamber. Close the lid of the chamber and store the cells at 4 °C until imaging. Protect from light.
- Analysis of the nuclearity of Langendorff-isolated cardiomyocytes
- Take images from α-actinin-stained cardiomyocytes with a 25X objective, corresponding to a 250X magnification. Count the number of H2B-mCh+ nuclei only in those cardiomyocytes that are regularly stained by α-actinin and show typical rod shaped morphology and are therefore assumed to be intact.
- Count at least 100 cardiomyocytes from three different hearts. Calculate the percentages of mononuclear, binuclear, and trinuclear cardiomyocytes. NOTE: Typically, binuclear cells should make up around 80 - 90%, while trinuclear cells and those with greater numbers of nuclei are rare (< 1% in Langendorff-isolated cardiomyocytes, < 3% in thick slices). Atrial cardiomyocytes are significantly smaller than ventricular cardiomyocytes. Typically, mononuclear cells make up 90% of the total population.
- Isolation, fixation, and cryopreservation of adult hearts as preparation for thick slices
- Assemble a perfusion apparatus for fixation by connecting a 50-mL syringe to a 3-way stopcock with a Heidelberger extension tube and a second 3-way stopcock. Fix the syringe in a stand in such a way that flow-through is enabled by gravity. Fill the system with 15 mL of PBS.
- Follow steps 2.1.2.1-2.1.2.4, "heart preparation." Connect the G20x1 ½ injection cannula with the heart to the Luer lock adapter of the bubble-free-PBS-filled perfusion apparatus and perfuse it with PBS. Fill the system with 10 - 15 mL of 4% formaldehyde in PBS, which is then perfused through the heart. Immersion fix the heart in 4% formaldehyde solution overnight.
- Replace the 4% formaldehyde solution with PBS and wash the heart in PBS on a horizontal shaker at 150 rpm for 8 h at room temperature. Exchange the PBS with 20% sucrose solution to dehydrate the heart overnight at 4 °C.
- Freeze the heart in cryo-embedding medium in a small mold.
- Set up a beaker filled with 2-methylbutane and place it in a box with dry ice. Put the heart into a freezing mold previously half-filled with cryo embedding medium and cover it completely with cryo embedding medium. Carefully place the mold into the cold beaker, preventing contact of the cryo-embedding medium with the 2-methylbutane. Store the frozen tissue at -80 °C until use.
- Preparation of thick heart slices
- Use the following setup in a cryotome: an object temperature of -18 to -20 °C, a chamber temperature of -21 to -23 °C, a clearing angle on the knife holder of 4°, and Feather R35 microtome blades.
- Take the cryopreserved organ from the freezing container and fix it on the specimen disc with cryo-embedding medium using the quick freezing shelf. Position the heart in such a way that sectioning begins at the apex.
- Trim away 50-µm slices until an even cutting plane is achieved and the organ becomes visible. Cut 50-µM tissue slices by using the cryotome in manual mode with a slow velocity and with the anti-roll plate placed on the knife. Mount slices on saline-treated microscope slides. Let the slices dry for 30 min at room temperature and store the slides at -80 °C or stain directly.
- Staining of thick heart slices (nuclei and cell membranes)
- Treat heart slices with RNAse A (20 µg/mL) in washing buffer (0.5 M NaCl, 0.1 M Tris pH 7.5, and 50 mM EDTA) in a cuvette for 1 h at 37 °C. Incubate the slices in 0.2% Triton-X in washing buffer for 30 min at room temperature. Wash the slices twice for 5 min each in washing buffer in a cuvette on a horizontal shaker at room temperature.
- Stain overnight with 1 µM TO-PRO3 iodide (642/661) and fluorescein-coupled wheat germ agglutinin (WGA, 1:100) in washing buffer at 4 °C. Wash the slices three times for 5 min each with washing buffer in cuvettes on a horizontal shaker and cover them with polyvinyl alcohol mounting medium with an anti-fading reagent and a glass coverslip.
- Image acquisition
- Ideally, use an inverted confocal laser scanning microscope equipped with a 40X/1.15 NA water-dipping objective. Search by eye for an area within the slice that consists of longitudinally lined cardiomyocytes; for that purpose, the fluorescein-WGA channel (Ex.: 488 nm) is best suited. NOTE: This step is essential, as the upper and lower limits of the cardiomyocytes should be visible within the z-stack for later analyses. As the imaging depth is limited to ~ 30 nm and the thickness of the slice is 50 µm, it is not possible to image cross sections of these cells (cell length: ~ 120 µm).
- Adjust the setup within the imaging software. Open the imaging software. Open A1plus Settings and check the boxes for Ch2, Ch3, and Ch4. Set Ch2 to EGFP, Ch3 to Alx546, and Ch4 to Alx647 by clicking on the pulldown menu.
- For each channel, click on HV and set it to 80 by using the slider bar. Set the Offset to 0 by using the slider bars. Click on the "Home" button to set the pinhole to the home position. Set the scan size to 1,024 x 1,024 by clicking on the pulldown menu and click optimize to open the "XYZ Size Setup" window. Check the "Perfect Voxel" box under "Suggested Step (z)".
- Click on "live scan" and adjust the laser intensities by clicking on the slider bars on the individual channels. Check the "Z Series" box on the "ND Acquisition" window and click the "defined by top button" box. Define the top and button of the z-stack by clicking on the appropriate buttons after adjusting the focus on the microscope. Set the z-step width to 0.5 µm. NOTE: The depth of the z-stack is limited by the optical penetration within the tissue and the signal-to-noise ratio.
- Go to "A1plus Settings" and set the Line Average/Integral to 2-4 by clicking on the pulldown menu. Fine-tune the laser intensities; the pinhole should be as small as possible in order to image in a focal plane.
- Analysis of nucleation
- Open the image analysis software and load the z-stack of interest.
- Manually determine nucleation in the z-stacks by scrolling through the different layers of the stack in order to identify cells that lie completely within the stack; this is the case when the WGA staining is visible in all dimensions. Count the number of nuclei (most of them will be binuclear) and mark the analyzed cell by an annotation in the software.
- Determine the number of CM nuclei (H2B-mCh+) and total nuclei (TO-PRO3+) in 3D reconstructions for the single channels (Figure 2D). In the imaging software, click on "Binary" and choose "Define Threshold 3D" from the pulldown menu. Choose from the channel pulldown menu either Alx546 for the number of CM nuclei or Alx647 for the number of total nuclei. By clicking the appropriate arrows, set the program to "Smooth 4x", Clean 1x, and Fill Holes ON. Check the "Size" box and set it to 5 µm by using the slider bar. Set the threshold by using the slider bar until separate objects appear. NOTE: After applying the fragmentation, the results window shows a table of quantified events and a colored fragmentation of the image. As some nuclei directly contact each other, manually correct the automatic measurement result for doublets, which might not be correctly separated by the software.
Representative Results
In order to analyze the effects of siRNAs/miRNAs on the cell cycle activity of postnatal cardiomyocytes in vitro, cardiomyocytes of double-transgenic Myh6-H2B-mCh/CAG-eGFP-anillin mice were isolated on postnatal day 3 (P3) and transfected with cell cycle activity-inducing miR1995, siRNA p27, and siRNA Fzr1. Compared to the negative control (Figure 1A), the pictures of miR199- (Figure 1B) and siRNA p27- (Figure 1C) transfected cardiomyocytes show an induction of cell cycle activity. In the eGFP-anillin mouse model, siRNAs against Fzr1 can be used as transfection controls, as the inhibition of Fzr1 leads to the accumulation of eGFP-anillin fusion protein in the nuclei of transfected cells and a loss of Fzr1 leads to the inhibition of APCFzr1. Fzr1 is a cofactor of the anaphase-promoting complex E3 ligase, which targets anillin for proteasomal degradation. Figure 1D shows a confocal overview picture of siRNA Fzr1-transfected cardiomyocytes 3 days after transfection, indicating a transfection efficiency of ~ 45%. Cardiomyocytes that perform endoreduplication (e.g., after knockdown of p276) show exclusively nuclear eGFP-anillin expression (Figure 1E) or are eGFP-anillin negative (see discussion). They do not express eGFP-anillin in M-phase-typical localizations (Figure 1F and G), as endoreduplication only consists of an endo-S phase (eGFP-anillin-positive) and an endo-G phase (eGFP-anillin-negative). In the endo-G phase, the APC is active, resulting in the ubiquitination and degradation of eGFP-anillin in the proteasome.
Quantification of mono- and binucleated cardiomyocyte portions at the adult stage can be performed either at the single-cell level after the Langendorff dissociation of Myh6-H2B-mCh transgenic hearts or in thick cryoslices of transgenic hearts. After the enzymatic digestion of the heart tissue at the Langendorff apparatus, atria and ventricles can be mechanically separated and analyzed independently of each other. Figure 2A shows a representative picture of non-fixed H2B-mCh transgenic ventricular cardiomyocytes after Langendorff isolation with a high degree of binucleated cardiomyocytes, as indicated by the nuclear H2B-mCh expression. By contrast, the majority of atrial cardiomyocytes are mononucleated (Figure 2B). As the enzymatic digestion does not result in 100% single cells, the pattern of cross-striation revealed by α-actinin staining facilitates discrimination between binucleated cardiomyocytes (continuous pattern of cross-striation, Figure 2C) and cell doublets. Figure 2D shows an example of the identification of a binucleated cardiomyocyte in a thick slice.
3D reconstructions of thick slices of adult Myh6-H2B-mCh transgenic hearts can be used to determine the proportion of cardiomyocyte nuclei under physiological conditions within the tissue. Using the 3D module of imaging software, Hoechst-stained nuclei and H2B-mCh-positive nuclei can be detected and counted automatically, as illustrated in Figure 2E. The final result should be corrected manually for doublets, meaning nuclei that directly touch each other, in this case. To analyze the nucleation index of cardiomyocytes in thick slices, it is necessary to manually scroll through the stack, as the nuclei do not necessary lie within one z-plane. WGA staining allows for the detection of cell borders.
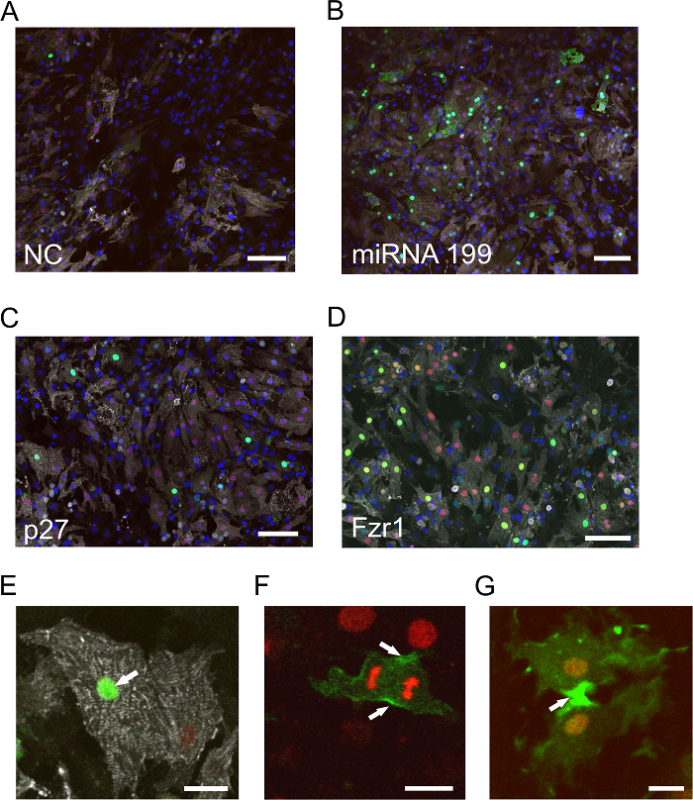 Figure 1:Examples ofIn Vitro Visualization of Cell Cycle Activity in Postnatal Cardiomyocytes after Transfection with siRNAs. (A-D) P3 cardiomyocytes from eGFP-anillin/Myh6-H2B-mCh hearts stained for α-actinin (white). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red), cell cycle activity by eGFP-anillin signals (green), and nuclei by Hoechst nuclear dye (blue). (A) P3 cardiomyocytes transfected with scramble siRNA serve as the negative control. The bar is 100 µm. (B) P3 cardiomyocytes transfected with miRNA-199 display significantly more eGFP-anillin signals than the control (A). The bar is 100 µm. (C) Example of exclusively nuclear eGFP-anillin signals after transfection with p27 siRNAs, indicating endoreduplication. The bar is 80 µm. (D) Transfection with siRNA against Fzr1 for the determination of transfection efficiency. As eGFP-anillin accumulates, the number of eGFP-anillin+ cardiomyocytes indicates the transfection efficiency. The bar is 80 µm. (E-G) Different localizations of eGFP-anillin (green) in cardiomyocytes during the cell cycle: nuclear localization (arrow in E), contractile ring (arrows in F), and midbody localization (arrow in G). The bar is 20 µm in (E) and 10 µm in (F and G). Please click here to view a larger version of this figure.
Figure 1:Examples ofIn Vitro Visualization of Cell Cycle Activity in Postnatal Cardiomyocytes after Transfection with siRNAs. (A-D) P3 cardiomyocytes from eGFP-anillin/Myh6-H2B-mCh hearts stained for α-actinin (white). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red), cell cycle activity by eGFP-anillin signals (green), and nuclei by Hoechst nuclear dye (blue). (A) P3 cardiomyocytes transfected with scramble siRNA serve as the negative control. The bar is 100 µm. (B) P3 cardiomyocytes transfected with miRNA-199 display significantly more eGFP-anillin signals than the control (A). The bar is 100 µm. (C) Example of exclusively nuclear eGFP-anillin signals after transfection with p27 siRNAs, indicating endoreduplication. The bar is 80 µm. (D) Transfection with siRNA against Fzr1 for the determination of transfection efficiency. As eGFP-anillin accumulates, the number of eGFP-anillin+ cardiomyocytes indicates the transfection efficiency. The bar is 80 µm. (E-G) Different localizations of eGFP-anillin (green) in cardiomyocytes during the cell cycle: nuclear localization (arrow in E), contractile ring (arrows in F), and midbody localization (arrow in G). The bar is 20 µm in (E) and 10 µm in (F and G). Please click here to view a larger version of this figure.
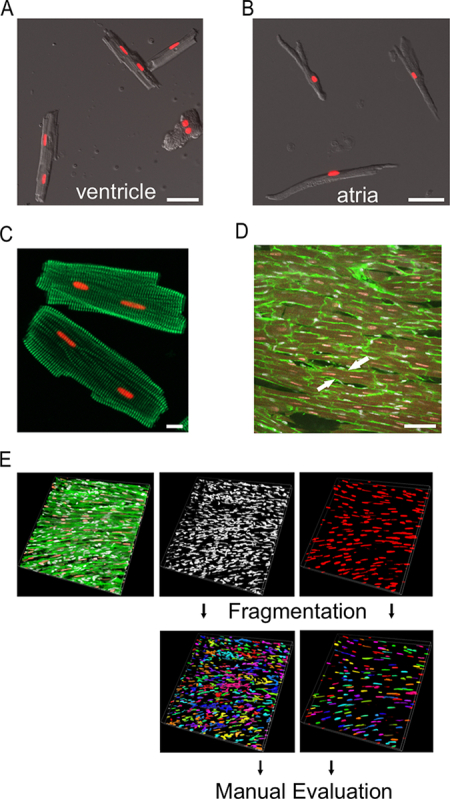 Figure 2: Examples for the Assessment of Multinucleation by the Langendorff Isolation of Cardiomyocytes from H2B-mCh Mice and by the 3D Analysis of Thick Slices. (A,B) Ventricular (A) and atrial (B) cardiomyocytes from hearts from adult H2B-mCh transgenic mice after isolation by Langendorff dissociation. The bars are 50 µm. (C) Cardiomyocytes from Myh6-H2B-mCh hearts stained for α-actinin (green). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red). The bar is 10 µm. (D) Binucleated cardiomyocyte in a thick slice (arrows). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red), cell borders by WGA staining (green), and nuclei by ToPro3 (white). The bar is 50 µm. (E) Workflow for the 3D analysis of binucleation in thick slices. Please click here to view a larger version of this figure.
Figure 2: Examples for the Assessment of Multinucleation by the Langendorff Isolation of Cardiomyocytes from H2B-mCh Mice and by the 3D Analysis of Thick Slices. (A,B) Ventricular (A) and atrial (B) cardiomyocytes from hearts from adult H2B-mCh transgenic mice after isolation by Langendorff dissociation. The bars are 50 µm. (C) Cardiomyocytes from Myh6-H2B-mCh hearts stained for α-actinin (green). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red). The bar is 10 µm. (D) Binucleated cardiomyocyte in a thick slice (arrows). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red), cell borders by WGA staining (green), and nuclei by ToPro3 (white). The bar is 50 µm. (E) Workflow for the 3D analysis of binucleation in thick slices. Please click here to view a larger version of this figure.
Discussion
There is a controversy over whether cardiomyocytes are able to reenter the cell cycle and divide after injury and during tissue homeostasis. Values for the basic turnover of cardiomyocytes have been given in the range between 1%1 and 80%7. Also after a cardiac lesion, the induction of cell cycle activity and the generation of new cardiomyocytes has been reported in the border zone, with values between 0.0083%8 and 25 - 40%7. These discrepancies can partly be explained by different experimental approaches to identify cardiac nuclei, a process that is very challenging on histological sections9 and during cell division. As cardiomyocytes are prone to variations of the cell cycle, it is of crucial importance to distinguish authentic cell division from endoreduplication and acytokinetic mitosis, which lead to polyploid and multinucleated cells. For the identification of cell division, it is necessary to visualize hallmarks of cell division, such as contractile rings and midbodies, as well as to determine the percentage of binucleated cardiomyocytes. We have developed techniques to analyze cell cycle activity in cardiomyocytes in detail and to determine their degree of nuclearity in isolated cells or in thick sections.
It is important to note that the eGFP-anillin system provides direct proof of cell division, through the visualization of a symmetrical contractile ring (Figure 1F) and the midbody (Figure 1G), and indirect proof for endoreduplication and acytokinetic mitosis. As previously described, the occurrence of a unilateral ingression of the contractile ring is an indicator of binucleation10, and this can be observed with the eGFP-anillin system.
Endoreduplication is suggested as long as no contractile ring or midbody are observed, which makes calculations of the probabilities of detecting these localizations mandatory. Assuming a cell cycle duration of 25 h, an average duration of contractile ring visibility of 20 min, and a midbody persistence of 1 h, there should be 1 contractile ring in 100 dividing eGFP-anillin-positive cells and 4 midbodies. Statistically speaking, a minimum of 25 eGFP-anillin-positive cells would need to be analyzed to distinguish cell division from variations of the cell cycle. This number changes accordingly as the cell cycle duration (which is often unknown) increases or decreases. This also implies that the majority of eGFP-anillin signals will be nuclear (Figure 1E), as M-phase, the only phase with non-nuclear localizations, lasts only approximately 1 h.
During postnatal development, binucleation takes place in mouse hearts and increases to 90% in ventricular cardiomyocytes (~ 25% in humans)11. For the analysis of regeneration in the heart, it is important to determine the degree of binucleation. We describe two methods for addressing this important morphological feature: namely, Langendorff dissociation and the creation of thick sections of cardiac tissue. While Langendorff isolation is easier and faster, the morphology of thick sections is more time consuming but also more accurate. Interestingly, we have found that Langendorff isolation overestimates the percentage of binuclear cardiomyocytes. This could be due to different survival rates of mononuclear and binuclear cells during this rather rigid procedure.
As the induction of proliferation in postnatal cardiomyocytes is an emerging approach in cardiac regeneration, this protocol describes a screening system by combining two transgenic mouse lines, Myh6-H2BmCh and CAG-eGFP-anillin. Cardiomyocytes isolated from hearts on postnatal days P1 - P6 can be easily cultured and transfected with miRNAs or siRNAs or treated with libraries of small molecules. Cardiomyocyte nuclei can be manually or automatically detected by software algorithms, and potential "hits" can be determined by an increase in the mCh+ nuclei number. Discrimination of cell division from endoreduplication and acytokinetic mitosis can be done by quantification of the different localizations of the eGFP-anillin signals. This system should shed some new light onto the regulatory mechanisms underlying postnatal cardiomyocyte proliferation and may lead to the discovery of new therapeutic agents for the treatment of cardiac diseases.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank S. Grünberg (Bonn, Germany) and P. Freitag (Bonn, Germany) for their technical assistance.
References
- Bergmann O, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raulf A, et al. Transgenic systems for unequivocal identification of cardiac myocyte nuclei and analysis of cardiomyocyte cell cycle status. Basic Res.Cardiol. 2015;110(3):33. doi: 10.1007/s00395-015-0489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CM, Alberts BM. Anillin, a contractile ring protein that cycles from the nucleus to the cell cortex. J.Cell Biol. 1995;131(1):165–178. doi: 10.1083/jcb.131.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse M, et al. Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle. Nat.Commun. 2012;3:1076. doi: 10.1038/ncomms2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492(7429):376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- Di SV, Giacca M, Capogrossi MC, Crescenzi M, Martelli F. Knockdown of cyclin-dependent kinase inhibitors induces cardiomyocyte re-entry in the cell cycle. J.Biol.Chem. 2011;286(10):8644–8654. doi: 10.1074/jbc.M110.184549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda T, et al. Clonality of mouse and human cardiomyogenesis in vivo. Proc.Natl.Acad.Sci.U.S.A. 2009;106(40):17169–17174. doi: 10.1073/pnas.0903089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am.J.Physiol. 1997;272(1 Pt 2):H220–H226. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- Ang KL, et al. Limitations of conventional approaches to identify myocyte nuclei in histologic sections of the heart. Am.J.Physiol Cell Physiol. 2010;298(6):C1603–C1609. doi: 10.1152/ajpcell.00435.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel FB, Schebesta M, Keating MT. Anillin localization defect in cardiomyocyte binucleation. J Mol Cell Cardiol. 2006;41(4):601–612. doi: 10.1016/j.yjmcc.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Bergmann O, et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell. 2015;161(7):1566–1575. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]