

Abstract
Glucosinolates are a well-studied and highly diverse class of natural plant compounds. They play important roles in plant resistance, rapeseed oil quality, food flavoring, and human health. The biological activity of glucosinolates is released upon tissue damage, when they are mixed with the enzyme myrosinase. This results in the formation of pungent and toxic breakdown products, such as isothiocyanates and nitriles. Currently, more than 130 structurally different glucosinolates have been identified. The chemical structure of the glucosinolate is an important determinant of the product that is formed, which in turn determines its biological activity. The latter may range from detrimental (e.g., progoitrin) to beneficial (e.g., glucoraphanin). Each glucosinolate-containing plant species has its own specific glucosinolate profile. For this reason, it is important to correctly identify and reliably quantify the different glucosinolates present in brassicaceous leaf, seed, and root crops or, for ecological studies, in their wild relatives. Here, we present a well-validated, targeted, and robust method to analyze glucosinolate profiles in a wide range of plant species and plant organs. Intact glucosinolates are extracted from ground plant materials with a methanol-water mixture at high temperatures to disable myrosinase activity. Thereafter, the resulting extract is brought onto an ion-exchange column for purification. After sulfatase treatment, the desulfoglucosinolates are eluted with water and the eluate is freeze-dried. The residue is taken up in an exact volume of water, which is analyzed by high-pressure liquid chromatography (HPLC) with a photodiode array (PDA) or ultraviolet (UV) detector. Detection and quantification are achieved by conducting comparisons of the retention times and UV spectra of commercial reference standards. The concentrations are calculated based on a sinigrin reference curve and well-established response factors. The advantages and disadvantages of this straightforward method, when compared to faster and more technologically advanced methods, are discussed here.
Keywords: Chemistry, Issue 121, Arabidopsis, Brassica, cabbage, European community official method, mustard, high-pressure liquid chromatography-photodiode array (HPLC-PDA), indole glucosinolates, oilseed rape, response factors
Introduction
It is estimated that plants produce over 200,000 different types of chemical compounds1. Only the minority of these compound seems to play a role in the plants' primary metabolism, which fuels growth and reproduction; the majority are so-called secondary metabolites. Despite their name, secondary metabolites are often critical for plant survival and reproduction, as they serve to attract pollinators or to defend the plant against pathogens and herbivores1.
Glucosinolates are a very diverse class of secondary metabolites that have been studied for over 150 years2. To date, more than 130 structurally different glucosinolates have been identified3. Broadly, glucosinolates can be subdivided into different classes based on the amino acid from which they are synthesized. Indole glucosinolates, for example, are synthesized from the amino acid tryptophan, whereas phenylalanine provides the base skeleton for the synthesis of aromatic glucosinolates4. Within classes, there is a high level of structural diversity, which is brought about by sequential chain elongation steps in the biosynthetic pathways, such as in the class of aliphatic glucosinolates, or by side chain modifications (e.g., hydroxylation)4,5. One glucosinolate plant species may contain up to 37 different glucosinolates belonging to different structural classes6. Even though plant species have typical glucosinolate profiles, intraspecific variation for the types of glucosinolates is frequently found among individuals and populations6,7. Intact glucosinolates are stored in the vacuoles of plant cells and can be found in any aboveground or belowground organ7,8,9. Upon cell rupture (e.g., by herbivory), glucosinolates are released and are mixed with the enzyme myrosinase, setting off a mustard oil bomb10. Due to the activity of myrosinase, and depending on the structure of the glucosinolate, the reaction conditions, and the presence of modifying enzymes, different pungent, toxic, or noxious compounds are formed, such as nitriles and (iso)thiocyanates11. The reaction products have high biological activities; for example, they serve as repellents of generalist insect herbivores12. The heritability and biosynthetic pathways of glucosinolates are well studied, mainly because of their importance for herbivore and pathogen resistance, their value as flavor components in mustards and cabbages, and their negative (progoitrin) and positive (glucoraphanin) effects on human health5,13,14.
Because of the great interest in glucosinolates as biologically active compounds, extraction and detection methods based on reversed-phase high-pressure liquid chromatography (HPLC) equipped with ultraviolet (UV) or photodiode array (PDA) detectors have been commonly used since the 1980s15. Based on this method, the European Communities issued a standard protocol that was tested and validated in several labs for the analysis of glucosinolates in oilseeds (Brassica napus, colza, Canola16). Others added to this method (e.g., by determining additional response factors for glucosinolates that are not present in oilseed rape)17. Despite the increasing availability of liquid chromatography-mass spectrometry (LC-MS) platforms and high-throughput protocols for glucosinolate analysis18,19, the original HPLC-UV/PDA method is still widely used by scientists. The main reasons are that this method is straightforward, cost-effective, and relatively accessible to labs without an extensive chemical-analytical knowledge infrastructure. To serve this community, we here detail the protocol for the extraction of glucosinolates from plant materials and the analysis of their desulfo-forms with HPLC-PDA.
Protocol
1. Preparation of Solutions Needed for the Glucosinolate Extraction
Prepare 500 mL of 70% methanol (MeOH) in water (ultrapure) in a glass bottle. For 100 samples and 5 standards, 420 mL of 70% MeOH is needed.
Prepare 20 mM sodium acetate (NaOAc) (pH = 5.5) by adding 0.82 g of NaOAc or 1.36 g of NaOAc x 3 H2O in 500 mL of water; adjust the pH with hydrogen chloride (HCl). Store the NaOAc in the refrigerator. For 100 samples and 5 standards, 370 mL of 20 mM NaOAc (pH = 5.5) in water is needed.
To prepare the column material, mix 10 g of cross-linked dextran gel (type G-25) with 125 mL of ultrapure water. Store the resulting mixture in a refrigerator (4 °C).
- Preparation of the sulfatase solution
- Dissolve 10,000 units of aryl sulfatase (type H-1 from Helix pomatia) in 30 mL of ultrapure water and add 30 mL of absolute ethanol (EtOH). Mix the solution well and transfer it to one 250-mL centrifuge tube or multiple smaller tubes, depending on availability.
- Centrifuge the mixture at 2,650 x g for 20 min at room temperature (RT). Transfer the supernatant(s) to a beaker; add 90 mL of EtOH and mix it. Centrifuge the mixture in one or more centrifuge tubes at 1,030 x g for 15 min at RT.
- Discard the supernatants. Dissolve and combine the pellets in a total of 25 mL of ultrapure water. Vortex well, dispense in 1-mL tubes, and store in a freezer at -20 °C; they will keep for at least one year.
- Preparation of the sinigrin reference solution
- Weigh in around 9 mg of sinigrin monohydrate with 1-µg accuracy (e.g., 8.769 mg). Transfer it into a 10-mL volumetric flask and dissolve the sinigrin monohydrate in 10.0 mL of ultrapure water.
- Calculate the molarity of the stock solution and prepare five sinigrin references between 50-750 µM by diluting of the stock solution. For a detailed example, see Supplementary File S1.
- Dispense the different sinigrin references in 1.5-mL reaction tubes and freeze them at 20 °C. Include a series of five sinigrin references in each extraction batch. Always use the correct molarity values for the currently used sinigrin references when calculating the concentrations.
2. Preparation of the Extraction Setup
Prepare a column rack for the Pasteur pipette columns (for the preparation of columns, see step 3.1). Label the rack or the columns to keep track of the sample numbers (see Figure 1).
Prepare a block holding the same number of labelled 2-mL microcentrifuge tubes (see step 3.3 and Figure 1)
 Figure 1: Rack for glucosinolate extraction. Front view (left) and top view (right) of a self-made column rack and block for the glucosinolate extraction. Height: 100 mm, distance between shifted holes (to hold the Pasteur pipette columns): 30 mm (x-axis) x 15 mm (y-axis). Please click here to view a larger version of this figure.
Figure 1: Rack for glucosinolate extraction. Front view (left) and top view (right) of a self-made column rack and block for the glucosinolate extraction. Height: 100 mm, distance between shifted holes (to hold the Pasteur pipette columns): 30 mm (x-axis) x 15 mm (y-axis). Please click here to view a larger version of this figure.
3. Preparation of the Columns and Microcentrifuge Tubes
- Prepare one column per sample and five extra columns for the five sinigrin reference samples.
- In each glass pipette, place a piece of glass wool approximately 1 cm x 1 cm. Use a wooden or glass stick to push the glass wool down to the point where the pipette narrows. The glass wool should prevent the column material from running out, but do not use too much, as it will slow the elution of the columns. Wear gloves when working with the glass wool.
Place the columns on the column rack (see Figure 1). Place the column rack in a lab tray (waste tray) to catch the eluents used for the consecutive column washes.
- Cut the first 5 mm off the tip of a plastic 1-mL pipette tip and pipette 0.5 mL of the prepared column material (G-25 gel in water, see step 1.3) into each column. Shake the bottle containing the column material well before pipetting. Discard leaking columns (G-25 material running out through the glass wool layer) and replace with new ones.
- After pipetting the column material into the columns, add 1 mL of ultrapure water to flush down the material.
Prepare one 2-mL reaction tube per sample and five tubes for the five sinigrin reference samples; label them. Use a dissecting needle to make holes in the caps for later freeze-drying and place the prepared tubes into the tube block in the exact same pattern as the columns (step 2.2, see Figure 1).
4. Extraction of Glucosinolates
Weigh freeze-dried and finely-ground plant material (usually 50.0-100.0 mg of dry weight; the final glucosinolate concentrations in the extract should be in the range of the reference curve) to the nearest 0.1 mg in 2-mL, labeled, round-bottom reaction tubes. Add two small metal balls (3 mm in diameter) as boiling retardants to each tube. NOTE: The protocol can also be applied to fresh, flash-frozen materials, that have been ground under liquid nitrogen and kept frozen until extraction. Increase the amount of material weighed for extraction and the percentage of MeOH in the extraction liquid to 85% to compensate for dilution by the water in the materials19.
Pipette 1 mL of 70% MeOH into each tube and vortex briefly. Close the tubes and seal them with safety caps before placing them as quickly as possible into a hot water bath (90-92 °C) for a few minutes (~5 min), until the 70% MeOH just boils. Caution: Wear safety goggles during this step!
Place the sample tubes in an ultrasonic bath for 15 min. Meanwhile, take the sulfatase and the five sinigrin reference samples out of the freezer to thaw them at RT.
- After ultra-sonication, centrifuge the sample tubes at 2,700 x g in a benchtop centrifuge for 10 min at RT; a pellet should form in each tube. Add the supernatants to the labelled columns and pipette the five reference samples onto separate columns.
- While pipetting the supernatants, keep the tip well over the pellet to avoid pipetting plant materials. Note that when dried samples are used, the supernatant volume will be less than 1.0 mL.
Add 1 mL of 70% MeOH to the remaining pellets in the sample tubes and vortex the tubes before placing them in an ultrasonic bath for 15 min. Centrifuge the tubes again, as in step 4.4, and add the supernatants to the respective columns; due to the properties of the column material, the negatively charged sulfate group of the glucosinolates will be specifically retained on the column.
- Wash the columns with the extracts in three sequential steps.
- Pipette 2 x 1 mL of 70% MeOH onto each column. Wait for the column to run dry before adding the next 1 mL; this will remove more apolar compounds from the extracts (e.g., chlorophyll).
- Flush out the MeOH by adding 1 mL of ultrapure water to each column.
- Pipette 2 x 1 mL of 20 mM NaOAc buffer to each column to create the optimal conditions for the sulfatase reaction.
Take the rack with the columns out of the waste tray and dry the feet of the rack with a tissue. Place the rack over the block with vials and labeled tubes. Make sure that each column tip is in the corresponding, labeled, 2-mL tube (see Figure 1).
Add 20 µL of sulfatase solution to the columns. Ensure that the sulfatase reaches the surface of the column material. Pipette 50 µL of NaOAc buffer onto each column to flush down the sulfatase. Cover the columns with aluminum foil and let stand overnight. NOTE: Due to the activities of the sulfatase, the sulfate group will be removed, releasing the desulfoglucosinolates from the column so that they can elute with the water. For a detailed description, see Crocoll et al.19.
- The next day, elute the desulfoglucosinolates by pipetting 2 x 0.75 mL of ultrapure water onto each column. When all columns have run dry, lift the column rack and remove it from the reaction tubes.
- Cap the tubes (make sure that there are holes in the caps) and freeze them in liquid nitrogen or in a -80 °C freezer for 30 min. Freeze-dry the samples for 12-24 h (depending on the number of samples and the capacity of the freeze-drier) to remove all water.
After freeze-drying, re-dissolve the residue in an exact volume (usually 1.0 mL) of ultrapure water. Transfer the samples and the five sinigrin references to labeled HPLC vials. Keep the samples in a refrigerator (4 °C) for up to two weeks or a freezer (-20 °C) for up to one year before analyzing them with HPLC.
Let the glass columns dry under the hood overnight and dispose them when dry. Recover the metal balls from the sample tubes used in step 4.5 for reuse and put the tubes in the waste disposal bin.
5. HPLC Measurements of Extracted Samples
Separate the glucosinolates on a reversed-phase C18 column (4.6 x 150 mm, 3 µm, 300 Å) with an acetonitrile-water gradient (see Table 1) and a flow of 0.75 mL/min and a column temperature of 40 °C. Perform detection and quantification at 229 nm.
Measure the sinigrin references on the same column but with an adapted gradient method to reduce solvent use (see Table 2). Start with injecting the five sinigrin references, beginning with the reference with the lowest concentration. Then inject the samples, including a methanol injection (blank) after every tenth sample to clean the column and to prevent the carryover of peaks.
| Time [min] | Flow [mL/min] | % A Water | % B ACN |
| 1 | 0.750 | 98 | 2 |
| 35 | 0.750 | 65 | 35 |
| 40 | 0.750 | 98 | 2 |
| Column Temperature 40 °C. |
Table 1: Acetonitrile-water gradient for glucosinolate separation and analysis on reversed-phase HPLC.
| Time [min] | Flow [mL/min] | % A Water | % B ACN |
| 1 | 0.750 | 98 | 2 |
| 10 | 0.750 | 89.3 | 10.7 |
| 11 | 0.750 | 98 | 2 |
| Column Temperature 40 °C. |
Table 2: Shortened acetonitrile-water gradient for quantification of the five sinigrin references used for the quantification of glucosinolates.
6. Glucosinolate Identification and Quantification
- Identification
- Compare the UV spectra and retention times of the peaks in the samples with commercially available, pure glucosinolate references. See Figure 2 for examples of UV spectra from the various glucosinolate classes.
- Identify unknown glucosinolates with nuclear magnetic resonance (NMR), if possible.
- Compare the UV spectra and retention times of the peaks with those in Figure 2 and Table 3, databases, or literature references.
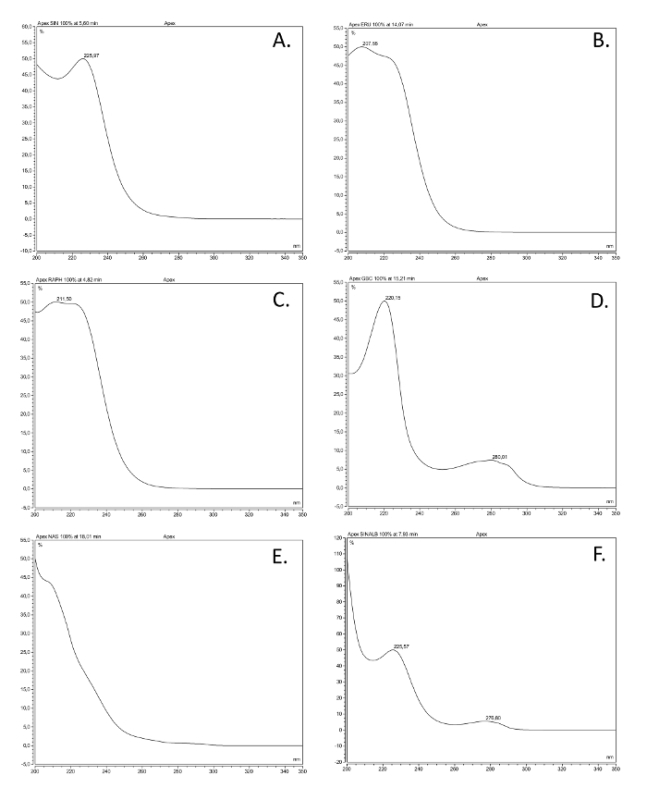 Figure 2. UV spectra of the most common glucosinolate classes. UV absorption spectra (200-350 nm) of six desulfoglucosinolates (GSL), based on solutions made of commercially available reference compounds extracted as described here, representing the most common structural classes. The common name, structural name (between brackets), and structural class are given. (A) Sinigrin (2-propenyl GSL), alkenyl; (B) glucoerucin (4-methylthiobutyl GSL), thioalkenyl; (C) glucoraphanin (4-methylsulfinlybutyl GSL), sulfinyl; (D) glucobrassicin (indol-3-ylmethyl GSL), indole; (E) gluconasturtiin (2-phenylethyl GSL), aromatic; (F) sinalbin (4-hydroxybenzyl GSL), aromatic. Please click here to view a larger version of this figure.
Figure 2. UV spectra of the most common glucosinolate classes. UV absorption spectra (200-350 nm) of six desulfoglucosinolates (GSL), based on solutions made of commercially available reference compounds extracted as described here, representing the most common structural classes. The common name, structural name (between brackets), and structural class are given. (A) Sinigrin (2-propenyl GSL), alkenyl; (B) glucoerucin (4-methylthiobutyl GSL), thioalkenyl; (C) glucoraphanin (4-methylsulfinlybutyl GSL), sulfinyl; (D) glucobrassicin (indol-3-ylmethyl GSL), indole; (E) gluconasturtiin (2-phenylethyl GSL), aromatic; (F) sinalbin (4-hydroxybenzyl GSL), aromatic. Please click here to view a larger version of this figure.
- Quantification
- Integrate the area under the peaks of the five sinigrin references and the samples.
- Calculate a calibration curve of the five measured sinigrin references based on the integrated area. Use the equation of the regression line (see Equation 1) to calculate the interpolated amount of glucosinolates (see Equation 2).
- To calculate the concentration of the different glucosinolates, multiply the interpolated amount (x) by the response factor (M) for detection at 229 nm from EC (1990)16, Buchner (1987)15, or Brown et al. (1993)17; the consensus response factors of the most commonly detected desulphoglucosinolates are given in Table 3. NOTE: Response factors may vary between systems-this may be the reason that there are different values given by different references (see References 15-17). Nevertheless, the values are mostly quite close and in a similar range with structural classes. Following the convention, set the response factor of unidentified glucosinolates to 1, although for unknown indole glucosinolates with a UV spectrum similar to that of glucobrassicin and other indole glucosinolates (Figure 2), it would be advisable to use 0.25 as a response factor to obtain a realistic estimate of the concentration (Table 3).
- To calculate the final amount of glucosinolates (xt) in the sample, take the dilution factor (D) and the mass of sample (w) used for the analysis into account (see Equation 3).
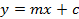 (1)
(1) 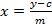 (2)
(2) 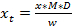 (3) y = Peak area m = slope of the reference 5-point regression curve c = the intercept of the regression line with the y-axis x = amount of glucosinolates in the extract xt = concentration of glucosinolates in the plant sample D = dilution factor M = response factor for the detection at 229 nm from Buchner (1987) or Brown et al. (1993) w = mass of sample used for extraction
(3) y = Peak area m = slope of the reference 5-point regression curve c = the intercept of the regression line with the y-axis x = amount of glucosinolates in the extract xt = concentration of glucosinolates in the plant sample D = dilution factor M = response factor for the detection at 229 nm from Buchner (1987) or Brown et al. (1993) w = mass of sample used for extraction
Representative Results
This method enables the detection and separation of commonly found desulfoglucosinolates in all structural classes (Figure 3). The detrimental 2-hydroxyglucosinolate progoitrin comes relatively early in the chromatogram and is clearly separated from the beneficial glucosinolate glucoraphanin, the only methylsulfinylglucosinolate in this sample. Sinigrin, gluconapin, and glucobrassicanapin form an eluotropic series of alkenyl glucosinolates with increasing side-chain lengths (C3, C4, and C5, respectively). A similar logical series is visible for the two methylthioalkenyl glucosinolates, glucoiberverin (C3) and glucoerucin (C4). The peaks of the three indole glucosinolates, glucobrassicin and its derivatives 4-hydroxy and 1-methoxyglucobrassicin (neoglucobrassicin), are also clearly separated. It should be noted that the peaks of neoglucobrassicin and glucoerucin, as well as gluconasturtiin, the only aromatic glucosinolate in this sample, are particularly high in this Brassica extract due to the addition of root extracts to the mix9.
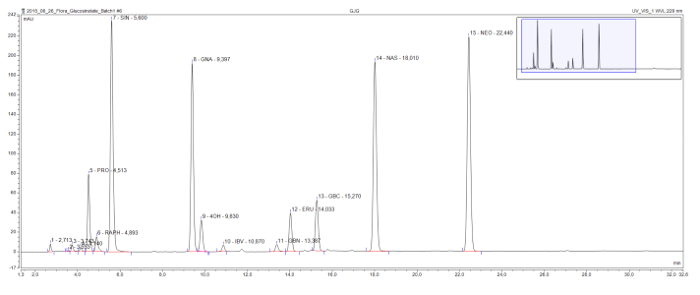 Figure 3: Chromatogram of a glucosinolate extract. Detail (1-32 min) of an HPLC chromatogram resulting from the analysis of combined root and shoot samples from Brassica nigra, B. rapa, and B. oleracea. The peak labels indicate the retention time and the abbreviations for identified desulfoglucosinolates (GSL). PRO = progoitrin (2-OH-3-butenyl GSL); RAPH = glucoraphanin (4-methylsulfinlybutyl GSL); SIN = sinigrin (2-propenyl GSL), GNA = gluconapin (3-butenyl GSL); 4OH = 4-hydroxyglucobrassicin; IBV = glucoiberverin (3-methylthiopropyl GSL); GBN = glucobrassicanapin (4-pentenyl GSL); ERU = glucoerucin (4-methylthiobutyl GSL); GBC = glucobrassicin (indol-3-ylmethyl GSL); NAS = gluconasturtiin (2-phenylethyl GSL); NEO = neoglucobrassicin (1-MeOH-glucobrassicin). Please click here to view a larger version of this figure.
Figure 3: Chromatogram of a glucosinolate extract. Detail (1-32 min) of an HPLC chromatogram resulting from the analysis of combined root and shoot samples from Brassica nigra, B. rapa, and B. oleracea. The peak labels indicate the retention time and the abbreviations for identified desulfoglucosinolates (GSL). PRO = progoitrin (2-OH-3-butenyl GSL); RAPH = glucoraphanin (4-methylsulfinlybutyl GSL); SIN = sinigrin (2-propenyl GSL), GNA = gluconapin (3-butenyl GSL); 4OH = 4-hydroxyglucobrassicin; IBV = glucoiberverin (3-methylthiopropyl GSL); GBN = glucobrassicanapin (4-pentenyl GSL); ERU = glucoerucin (4-methylthiobutyl GSL); GBC = glucobrassicin (indol-3-ylmethyl GSL); NAS = gluconasturtiin (2-phenylethyl GSL); NEO = neoglucobrassicin (1-MeOH-glucobrassicin). Please click here to view a larger version of this figure.
Longer chain methylsulfinyl glucosinolates, which are commonly found in the model plant Arabidopsis, also show an eluotropic series, as seen in a root extract of Rorippa austriaca. Glucohesperalin (C6), glucosiberin (C7), glucohirsutin (C8), and glucoarabin (C9) appear at regular intervals on the chromatogram (Figure 4). Together with the UV spectra of the peaks, such eluotropic logical series may be used to classify, and provisionally identify, unknown glucosinolates.
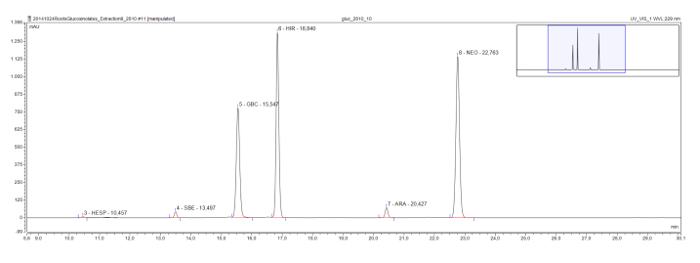 Figure 4: Chromatogram (frame 9-30 min) of the desulfoglucosinolates in a Rorippa austriaca root extract. The peak labels indicate the retention time and the abbreviations for identified desulfoglucosinolates (GSL). HES = glucohesperin (6-methylsulfinylhexyl GSL); SBE = glucosiberin (7-methylsulfinylheptyl GSL); GBC = glucobrassicin (indol-3-ylmethyl GSL); HIR = glucohirsutin (8-methylsulfinlyoctyl GSL); ARA = glucoarabin (9-methylsulfinylnonylglucosinolate); NEO = neoglucobrassicin (1-MeOH-glucobrassicin). Please click here to view a larger version of this figure.
Figure 4: Chromatogram (frame 9-30 min) of the desulfoglucosinolates in a Rorippa austriaca root extract. The peak labels indicate the retention time and the abbreviations for identified desulfoglucosinolates (GSL). HES = glucohesperin (6-methylsulfinylhexyl GSL); SBE = glucosiberin (7-methylsulfinylheptyl GSL); GBC = glucobrassicin (indol-3-ylmethyl GSL); HIR = glucohirsutin (8-methylsulfinlyoctyl GSL); ARA = glucoarabin (9-methylsulfinylnonylglucosinolate); NEO = neoglucobrassicin (1-MeOH-glucobrassicin). Please click here to view a larger version of this figure.
| Common name | Side chain structure | Rt (min)* | 229 nm | reference# |
| aliphatic glucosinolates | ||||
| Glucocapparin | methyl | 3.5 | 1 | Brown |
| Sinigrin | 2-propenyl | 5.5 | 1 | Brown, EC |
| Gluconapin | 3-butenyl | 9.5 | 1.11 | EC |
| Glucobrassicanapin | 4-pentenyl | 13.5 | 1.15 | EC |
| Glucoiberverin | 3-methylthiopropyl | 10.9 | 0.8 | Brown |
| Glucoerucin | 4-methylthiobutyl | 14.0 | 0.9 | Brown |
| Glucoiberin | 3-methylsulfinylpropyl | 3.7 | 1.2 | Brown |
| Glucoraphanin | 4-methylsulfinylbutyl | 4.9 | 0.9 | Brown |
| Glucoalyssin | 5-methylsulfinylpentyl | 7.6 | 0.9 | Brown |
| Glucohesperin | 6-methylsulfinylhexyl | 10.5 | 1 | Brown |
| Glucosiberin | 7-methylsulfinylheptyl | 13.5 | 1 | Brown |
| Glucohirsutin | 8-methylsulfinyloctyl | 16.8 | 1.1 | Brown |
| Glucoarabin | 9-methylsulfinylnonyl | 20.5 | 1 | |
| Glucocheirolin | 3-methylsulfonylpropyl | 4.2 | 0.9 | Brown |
| Progoitrin | 2(R)-OH-3-butenyl | 4.5 | 1.09 | Buchner, EC |
| Gluconapoleiferin | 2-OH-5-pentenyl | 8.3 | 1 | EC |
| indole glucosinolates | ||||
| 4-hydroxyglucobrassicin | 4-hydroxyindol-3-ylmethyl | 11.2 | 0.28 | Buchner, EC |
| Glucobrassicin | indol-3-ylmethyl | 15.3 | 0.29 | Buchner, EC |
| 4-Methoxyglucobrassicin | 4-methoxyindol-3-ylmethyl | 18.2 | 0.25 | Buchner, EC |
| Neoglucobrassicin | 1-methoxyindol-3-ylmethyl | 22.5 | 0.2 | Buchner, EC |
| aromatic glucosinolates | ||||
| Sinalbin | 4-hydroxybenzyl | 8.1 | 0.5 | Buchner |
| Glucosibarin | 2(R)-OH-2-phenylethyl | 12.1 | 0.95 | see next |
| Glucobarbarin | 2(S)-OH-2-phenylethyl | 12.7 | 0.95 | Buchner |
| Glucotropaeolin | benzyl | 13.8 | 0.95 | Buchner, EC |
| Gluconasturtiin | 2-phenylethyl | 18.0 | 0.95 | Buchner, EC |
| unknown - aliphatic/aromatic like UV spectrum | 1 | EC | ||
| unknown - indole like spectrum | 0.25 | EC | ||
| * approximate retention time (Rt) rounded to nearest 0.1 min (± 0.3 min depending on the column, eluent quality). Retention times are determined on ThermoFisher/Dionex Ultimate HPLC platforms equipped with an C18 column (150 x 4.6 mm, 3 micrometer particle size) plus C18 precolumn (10 x 4.6 mm, 5 micrometer particle size) with a gradient program as in Table 1. | ||||
| # References for response factors: Buchner, R. in Glucosinolates in rapeseed (ed J.P. Wathelet) 50-58 (Martinus Nijhoff Publishers, 1987); Brown, P. D., Tokuhisa, J. G., Reichelt, M. & Gershenzon, J. Variation of glucosinolate accumulation among different organs and developmental stages of Arabidopsis thaliana. Phytochemistry. 62 (3), 471-481, doi:10.1016/S0031-9422(02)00549-6, (2003); EC. Oil seeds - determination of glucosinolates High Performance Liquid Chromatography. Official Journal of the European Communities. L 170/28. Annex VIII 03.07.27-34 (1990). |
Table 3: Response factors of the most commonly found desulfo-glucosinolates in plant extracts and their approximate retention times on C18 columns. Eluents, gradient, column temperature, and flow rate as in Table 1.
Discussion
The greatest advantages of this established and widely used method are that it is robust, rather straightforward, and relatively low-cost per sample. Most of the equipment needed for the extraction and analysis should be available in a standard laboratory or can be self-built, with the exception of the HPLC-PDA. Another advantage is that desulfoglucosinolates dissolved in water are chemically quite stable when kept cool and in air-tight (HPLC) vials, so the extracts could easily be shipped for HPLC analysis elsewhere. In contrast to LC-MS platforms, which require specialized training and extensive hands-on experience for managing the software and analyzing the data, HPLC-UV/PDAs can be easily run after a short training period. This not only reduces the costs of the procedure, but also makes this method more accessible to a broad range of scientists, including students.
Generally, when the procedures described above are followed carefully, few problems should occur. In general, the glucosinolate peaks are very well separated in the chromatogram. If this is not the case, the gradient program could be adapted by decreasing the rate of increase of acetonitrile in the eluent. Alternatively, building in a new pre-column (200-500 injections) or column (1,500 -2,000 injections) may solve the issue. Occasionally, chromatograms of single samples in a batch may show very small or no peaks. This is usually due to pipetting errors when adding the sulfatase (e.g., a column has been skipped or the sulfatase was not properly washed down into the column). Alternatively, the glucosinolate concentration in the experimental materials may have been lower than expected and too little material was used for the extraction. If the latter is the case, the injection volume may be increased to 100 µL, or an exact aliquot (e.g., 800 µL) of the extract could be concentrated. The latter could be achieved by freeze-drying the extract, dissolving the residue in a smaller volume (e.g., 100 µL) of water, and reinjecting using the same reference curve. In the calculations for the original concentration of the extract, the numbers should be multiplied by the dilution factor. If this does not solve the issue, the materials should be extracted again using more starting material. If this is more than 100 mg, the volume of the extraction solvents and the size of the tubes should be adjusted proportionally to maintain the extraction efficiency.
An additional advantage is that this method has been well-validated. This is because it has been described as a standard method for the quantification of glucosinolates in rapeseed, for which the procedures and accuracy were confirmed in several laboratories16. In addition, the genetic background, biosynthesis, and biological functions of glucosinolates are subject to intense research efforts, in the model plant species Arabidopsis thaliana among others4,6,12. Therefore, many response factors for the exact quantification of desulfoglucosinolates in relation to sinigrin are well defined and publicly available15,17. Even though LS-MS-based protocols are more high-throughput, more sensitive, and are able to (tentatively) identify glucosinolates for which no standards are available18,19,20, the lack of universal response factors for LC-MS limits the exact quantification of glucosinolate concentrations18. Moreover, these methods usually do not include a freeze-drying step, and the amount of water in the fresh plant material is unaccounted for in the calculations, making exact quantification difficult. Lastly, because our extraction method involves a column-based purification and concentration step, it can also be applied to "dirty" samples with low concentrations of glucosinolates, such as soils21.
Compared to LC-MS-based methods that usually extract freshly frozen materials, use 96-well plates for extraction, and do not include a sulfatase step18,19, our method is relatively time-consuming and labor intense. With the column racks described in this paper, a single person can extract about 100-150 samples in one day. Elution (next day), freeze drying (overnight), and re-dissolving can take place within the following two days. With an automated HPLC injector, a run and equilibration time of 40-45 min per injection, and no unforeseen events, it would take 3-4 days to acquire the data for this sample set. When the HPLC software allows automatic quantification based on the sinigrin curve, a manual check of the chromatograms and peak assignments for 100 samples may only take another 1 or 2 h before the data can be used for statistical analyses.
Despite the increasing availability of glucosinolate standards, only a small fraction of the more than 130 candidates can currently be commercially bought. However, with a few references for each of the biosynthetic classes; access to literature databases specifying the compounds previously found in the plant species (e.g., Fahey et al.22); basic knowledge of chromatographic principles, such as the logic of eluotropic series (e.g., for increasing numbers of Cs on the side chain in aliphatic compounds, Figures 3 and 4); and the validation of single samples on LC-MS19 or isolated glucosinolates on NMR23, one may easily overcome this limitation. Most protocols for glucosinolate analyses use internal reference curves (i.e., a certain concentration for the extraction of sinigrin or sinalbin to the extraction solvent16,17,19). Principally, internal reference curves are more appropriate to correct for individual sample processing errors and thus theoretically yield a higher precision. Despite this advantage, we prefer to use a five-point external reference curve, as we often analyze different wild species, some of which contain high levels of sinigrin (e.g., Brassica nigra24) or sinalbin (e.g., Sinapis alba25), the two glucosinolate references for which response factors are available. Moreover, adding internal standards to each sample increases the cost of the analyses, as high-grade glucosinolate reference standards are usually quite expensive.
In conclusion, despite the time-consuming steps, this protocol provides a straightforward and accessible method to extract and quantify glucosinolates in plant samples. However, it is important to consider that the glucosinolate levels themselves are only an indication of the potential biological activity, seen as the necessity to react with myrosinase, and variation in the reaction products may arise from a single glucosinolate11. Validation assays must be performed to confirm the biological relevance.
Disclosures
The authors have nothing to disclose.
Acknowledgments
NMvD thanks Dr. Michael Reichelt (Max-Planck-Institute for chemical Ecology, Jena, Germany) for providing the first reference samples when she started using this method 16 years ago. Ciska Raaijmakers (NIOO-KNAW, Wageningen, the Netherlands), Sebastian Krosse (B-Ware, Nijmegen, the Netherlands), and Christian Ristok (iDiv, Leipzig) are acknowledged for improving the protocol over the course of the years. Mirka Macel and Martine Huberty (University Tübingen, Germany) are acknowledged for their permission to use the Rorippa chromatogram. The authors gratefully acknowledge the support of the German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig, funded by the German Research Foundation (FZT 118).
References
- Hartmann T. From waste products to ecochemicals: Fifty years research of plant secondary metabolism. Phytochemistry. 2007;68(22-24):2831–2846. doi: 10.1016/j.phytochem.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Will H, Laubenheimer A. Ueber das Glucosid des weissen Senfsamens. Justus Liebigs Annalen der Chemie. 1879;199(1):150–164. [Google Scholar]
- Agerbirk N, Olsen CE. Glucosinolate structures in evolution. Phytochemistry. 2012;77:16–45. doi: 10.1016/j.phytochem.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Halkier BA, Gershenzon J. Biology and biochemistry of glucosinolates. Ann Rev Plant Biol. 2006;57:303–333. doi: 10.1146/annurev.arplant.57.032905.105228. [DOI] [PubMed] [Google Scholar]
- Sønderby IE, Geu-Flores F, Halkier BA. Biosynthesis of glucosinolates - gene discovery and beyond. TIPS. 2010;15(5):283–290. doi: 10.1016/j.tplants.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Kliebenstein DJ, et al. Genetic control of natural variation in Arabidopsis glucosinolate accumulation. Plant Physiol. 2001;126(2):811–825. doi: 10.1104/pp.126.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leur H, Raaijmakers CE, van Dam NM. A heritable glucosinolate polymorphism within natural populations of Barbarea vulgaris. Phytochemistry. 2006;67(12):1214–1223. doi: 10.1016/j.phytochem.2006.04.021. [DOI] [PubMed] [Google Scholar]
- Kelly PJ, Bones A, Rossiter JT. Sub-cellular immunolocalization of the glucosinolate sinigrin in seedlings of Brassica juncea. Planta. 1998;206(3):370–377. doi: 10.1007/s004250050412. [DOI] [PubMed] [Google Scholar]
- van Dam NM, Tytgat TOG, Kirkegaard JA. Root and shoot glucosinolates: a comparison of their diversity, function and interactions in natural and managed ecosystems. Phytochem Rev. 2009;8(1):171–186. [Google Scholar]
- Ratzka A, Vogel H, Kliebenstein DJ, Mitchell-Olds T, Kroymann J. Disarming the mustard oil bomb. Proc Natl Acad Sci U S A. 2002;99(17):11223–11228. doi: 10.1073/pnas.172112899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittstock U, Kliebenstein DJ, Lambrix V, Reichelt M, Gershenson J. Romeo JT, editor. Integrative Phytochemistry: From ethnobotany to molecular ecology: Recent Advances in Phytochemistry. 2003. Ch. 5 Pergamon.
- Hopkins RJ, van Dam NM, van Loon JJA. Role of glucosinolates in insect-plant relationships and multitrophic interactions. Ann Rev Entomol. 2009;54(1):57. doi: 10.1146/annurev.ento.54.110807.090623. [DOI] [PubMed] [Google Scholar]
- Kliebenstein DJ, Gershenzon J, Mitchell-Olds T. Comparative quantitative trait loci mapping of aliphatic, indolic and benzylic glucosinolate production in Arabidopsis thaliana leaves and seeds. Genetics. 2001;159(1):359–370. doi: 10.1093/genetics/159.1.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithen R, Bennett R, Marquez J. Glucosinolate biochemical diversity and innovation in the Brassicales. Phytochemistry. 2010;71(17-18):2074–2086. doi: 10.1016/j.phytochem.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Buchner R. In: Glucosinolates in rapeseed. Wathelet JP, editor. Martinus Nijhoff Publishers; 1987. pp. 50–58. [Google Scholar]
- Oil seeds - determination of glucosinolates High Perfomance Liquid Chromatography. Official Journal of the European Communities. 1990. pp. 27–34. L 170/28. Annex VIII 03.07.
- Brown PD, Tokuhisa JG, Reichelt M, Gershenzon J. Variation of glucosinolate accumulation among different organs and developmental stages of Arabidopsis thaliana. Phytochemistry. 2003;62(3):471–481. doi: 10.1016/s0031-9422(02)00549-6. [DOI] [PubMed] [Google Scholar]
- Glauser G, Schweizer F, Turlings TCJ, Reymond P. Rapid Profiling of Intact Glucosinolates in Arabidopsis Leaves by UHPLC-QTOFMS Using a Charged Surface Hybrid Column. Phytochem Analysis. 2012;23(5):520–528. doi: 10.1002/pca.2350. [DOI] [PubMed] [Google Scholar]
- Crocoll C, Halkier BA, Burow M. Current Protocols in Plant Biology. John Wiley & Sons, Inc; 2016. [DOI] [PubMed] [Google Scholar]
- Griffiths DW, Bain H, Deighton N, Botting NP, Robertson AAB. Evaluation of liquid chromatography-atmospheric pressure chemical ionisation-mass spectrometry for the identification and quantification of desulphoglucosinolates. Phytochem Analysis. 2000;11(4):216–225. [Google Scholar]
- Gimsing AL, Kirkegaard JA. Glucosinolates and biofumigation: fate of glucosinolates and their hydrolysis products in soil. Phytochem Rev. 2009;8(1):299–310. [Google Scholar]
- Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. 2001;56:5–51. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- de Graaf RM, et al. Isolation and identification of 4-α-rhamnosyloxy benzyl glucosinolate in Noccaea caerulescens showing intraspecific variation. Phytochemistry. 2015;110(0):166–171. doi: 10.1016/j.phytochem.2014.11.016. [DOI] [PubMed] [Google Scholar]
- van Dam NM, Raaijmakers CE. Local and systemic induced responses to cabbage root fly larvae (Delia radicum) in Brassica nigra and B. oleracea. Chemoecology. 2006;16(1):17–24. [Google Scholar]
- Gols R, et al. Temporal changes affect plant chemistry and tritrophic interactions. Bas Appl Ecol. 2007;8(5):421–433. [Google Scholar]