

Abstract
Gene expression measurements from bulk populations of cells can obscure the considerable transcriptomic variation of individual cells within those populations. Single-cell gene expression measurements can help assess the role of noise in gene expression, identify correlations in the expression of pairs of genes, and reveal subpopulations of cells that respond differently to a stimulus. Here, we describe a procedure to measure the expression of up to 96 genes in single mammalian cells isolated from a population growing in tissue culture. Cells are sorted into lysis buffer by fluorescence-activated cell sorting (FACS), and the mRNA species of interest are reverse-transcribed and amplified. Gene expression is then measured using a microfluidic real-time PCR machine, which performs up to 96 qPCR assays on up to 96 samples at a time. We also describe the generation and use of PCR amplicon standards to enable the estimation of the absolute number of each transcript. Compared with other methods of measuring gene expression in single cells, this approach allows for the quantification of more distinct transcripts than RNA FISH at a lower cost than RNA-Seq.
Keywords: Genetics, Issue 120, single cell, qPCR, fluorescence-activated cell sorting, stochasticity, internal standards, cell fate decisions
Introduction
Individual cells in a population can show widely differing responses to a uniform physiological stimulus1,2,3,4. The genetic variation of cells in a population is one mechanism for this variety of responses, but there are also several non-genetic factors that can increase the variability of responses, even in a clonal population of cells. For example, the levels of individual proteins and other important signaling molecules can vary on a cell-by-cell basis, giving rise to variation in downstream gene expression profiles. Additionally, gene activation can occur in short-duration bursts of transcripts5,6 that may be limited to a relatively small number of transcripts per burst7,8,9. Such stochasticity in gene activation can greatly contribute to variability in biological responses and can provide a selective advantage in microorganisms10 and in mammalian cells1,2 responding to a physiological stimulus. Due to both genetic and non-genetic sources of variation, the gene expression profile of any given cell in response to a stimulus may differ greatly from the average gene expression profile obtained from the measurement of the bulk response. Determining the extent to which individual cells show variability in response to a stimulus requires techniques for the isolation of individual cells, the measurement of the expression levels for transcripts of interest, and the computational analysis of the resulting expression data.
There are several approaches for assaying gene expression in single cells, covering a wide range of costs, number of transcripts probed, and accuracy of quantification. For example, single-cell RNA-Seq offers a wide depth of transcript coverage and the ability to quantify thousands of distinct transcripts for the most highly expressed genes in individual cells; however, the cost associated with such sequencing depth can be prohibitive, although costs continue to decrease. Conversely, single-molecule RNA fluorescence in situ hybridization (smRNA FISH) offers precise quantification of transcripts for even low-expressing genes at a reasonable cost per gene of interest; however, only a small number of target genes can be assayed in a given cell by this approach. Quantitative PCR-based assays, described in this protocol, provide a middle ground between these techniques. These assays employ a microfluidic real-time PCR machine to quantify up to 96 transcripts of interest at a time in up to 96 cells. While each of the aforementioned methods has requisite hardware costs, the cost of any individual qPCR assay is relatively low. This protocol is adapted from one suggested by a manufacturer of a microfluidic real-time PCR machine (Protocol ADP 41, Fluidigm). To enable the estimation of the absolute number of each transcript in a PCR-based approach, we have expanded the protocol to make use of internal controls of prepared target gene amplicons that can be used across multiple experiments.
As an example of this technique, the quantification of the expression of genes regulated by the tumor suppressor p53 in MCF-7 human breast carcinoma cells is described11. The cells are challenged with a chemical agent that induces DNA double-strand breaks. Previous studies have shown that the p53 response to DNA double-strand breaks exhibits a great deal of heterogeneity in individual cells, both in terms of p53 levels12 and in the activation of distinct target genes11. Furthermore, p53 regulates the expression of over 100 well-characterized target genes involved in numerous downstream pathways, including cell cycle arrest, apoptosis, and senescence13,14. Since the p53-mediated response in each cell is both complex and variable, the analysis of the system benefits from an approach in which nearly 100 target genes can be probed simultaneously in individual cells, such as that described below. With slight modifications (such as alternative methods for single-cell isolation and lysis), the protocol can be readily adapted to study a wide range of mammalian cell types, transcripts, and cellular responses.
With proper advance preparation, a round of cell sorting and gene expression measurement can be conducted according to this protocol over a period of three days. The following timing is suggested: in advance, select the transcripts of interest, identify and validate the primer pairs that amplify the cDNA from those transcripts, and prepare the standards and primer mixes using those primers. On Day 1, following cell treatment, harvest and sort the cells, perform reverse transcription and specific target amplification, and treat the samples with an exonuclease to remove unincorporated primers. On Day 2, perform quality control on sorted cells using qPCR. Finally, on Day 3, measure the gene expression in the sorted cells using microfluidic qPCR. Figure 1 summarizes the steps involved.
Protocol
1. Advance Preparation
Select up to 96 genes of interest whose expression will be measured. NOTE: At least one of these genes should be a "housekeeping gene," such as ACTB or GAPDH, that is known to be expressed at a relatively high and constant level under the conditions used in the experiment. This gene will be used to identify positively sorted wells (step 8.1) and amplified samples (step 10.1). NOTE: For the example experiment, well-characterized, direct targets of p53 with a variety of known functions11,14 and GAPDH as a housekeeping control were selected. Please see Reference 11 for the complete list of target genes and primer sequences used in this study.
Identify potential primer pairs specific to the genes of interest (e.g., using the scientific literature or a primer design tool such as Primer-BLAST)15. Have the primers synthesized and maintain them at a stock concentration of 100 μM in nuclease-free water. NOTE: These primers should be 15-25 bases long; produce an amplicon 90-130 base pairs (bp) long; span an exon junction, if one exists, to reduce the chance of amplifying genomic DNA; have melting temperatures of 60 ± 1 °C; and have minimal secondary structure16. These primers will be used to prime reverse transcription, amplify cDNA from the transcripts of interest, and measure gene expression in DNA binding dye-based qPCR.
- Validate the primers.
- First, obtain cDNA from the cells of interest. Harvest RNA from the cells using an RNA harvesting kit according to the manufacturer's instructions or standard molecular biology methods17. Reverse-transcribe the RNA into cDNA using a reverse transcription kit with random hexamers according to the manufacturer's instructions or standard methods17.
- For each primer pair, set up qPCR with DNA binding dye master mix, forward and reverse primers, and the cDNA from the previous step, following the master mix manufacturer's recommendations for suggested reaction conditions. Run these reactions on a real-time PCR machine using a thermal cycling protocol recommended by the master mix manufacturer that includes melt curve acquisition.
- After the qPCR, run the amplified DNA samples on a 2% w/v agarose gel and visualize the DNA via a DNA intercalating dye following a standard protocol17.
- Determine the efficiency of the primer pair by constructing a standard curve from serial dilutions of cDNA (made in step 1.3.1) according to a standard protocol16. NOTE: A good primer pair will yield a melt curve with a single, well-defined peak and a single band in the expected location on the gel16,17 (Figure 2). If the melt curve has multiple peaks or a "shoulder," or if the gel has multiple bands or a smear, the primers are amplifying an off-target sequence. Redesign any primers that have been observed to amplify off-target sequences and repeat the preceding validation steps as necessary. A good primer pair will also have an efficiency of 90-110%, with R2 ≥0.985 for the standard curve16; redesign any primers for which the efficiency or R2 fall outside these ranges.
- Prepare standards from purified DNA amplicons.
- For each validated primer pair:
- Amplify the region of interest from cDNA using a high-fidelity DNA polymerase according to the polymerase manufacturer's recommendations for primer and template DNA concentrations, reaction conditions, and PCR cycling conditions, or using a standard PCR protocol17.
- Run the amplified cDNA on a 2% w/v agarose gel and visualize the band with a DNA intercalating dye17. Excise the band representing the amplicon using a clean razor blade and place the gel piece in a microcentrifuge tube.
- Purify the amplicon from the gel using a standard gel extraction procedure, such as an agarase-based extraction17 or a spin-column gel extraction kit, according to the manufacturer's instructions.
- Measure the concentration of amplicon using a fluorescence-based dsDNA quantification kit according to the manufacturer's instructions. Divide the measured dsDNA concentration by the known molecular weight of the amplicon based on the amplicon sequence to obtain the number of amplicon molecules per µl.
- In a 2.0 ml low-binding microfuge tube, add 2 µl of purified amplicon to a volume of DNA suspension buffer such that the final concentration of amplicon is 5 x 108 molecules/µl.
- When all amplicons have been purified, combine 18 µl of each purified amplicon in a 2.0 ml low-binding microcentrifuge tube. Add DNA suspension buffer to make a total volume of 1,800 µl; thus, the concentration of each amplicon is 5 x 106 molecules/µl. NOTE: This mixture, which contains cDNA amplicons from all genes of interest in known equimolar amounts, will serve as a standard in single-cell qPCR.
- Vortex this "5e6" standard thoroughly, spin for 10 sec at 2,000 x g, and divide into 20 µl aliquots in low-binding PCR tubes. Store the aliquots at -80 °C.
- Prepare the specific target amplification (STA) primer mix (10x).
- In a 15 ml conical tube, combine 25 µl of each 100 µM stock primer (up to 192). Add DNA suspension buffer to make a total volume of 5,000 µl. NOTE: The concentration of each primer in this mixture is 500 nM. Each plate of sorted cells will use 105.6 µl of this primer mix to prime the reverse transcription and specific target amplification. The volume of primer mix given here is enough to make 45 plates to sort. It is advisable to make a large enough volume of primer mix so that it only needs to be made once; scale the volumes as necessary.
- Mix thoroughly by vortexing, divide into 110 µl aliquots, and store the aliquots at -20 °C.
- Prepare assay mixes, one for each primer pair, for use in microfluidic chip-based qPCR.
- In each well of a 96-well PCR plate, combine 3.0 µl of the 100 µM forward primer for a given gene, 3.0 µl of the corresponding 100 µM reverse primer, 24.0 µl of DNA suspension buffer, and 30.0 µl of 2x assay loading reagent.
- Vortex this plate, spin for 10 sec at 500 x g, and store at -20 °C.
Create two thermal cycling programs: RTSTA (reverse transcription and specific target amplification; Table 1) and EXOI (exonuclease I treatment; Table 2).
2. Treatment
Plate the mammalian cells on a tissue culture dish such that they will grow to the desired confluence at the time of treatment, depending on the conditions of the experimental study. For the example presented in this study, plate 4 x 105 MCF-7 p53-Venus cells18 per 6 cm dish in RPMI with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycin, and 250 ng/ml amphotericin B. Incubate the cells for two days at 37 °C with 5% CO2 until they reach 50% confluency.
Treat the cells to establish the condition of interest based on the experimental study. For the example presented in this study, incubate MCF-7 p53-Venus cells with 400 ng/ml neocarzinostatin for 3 hr, 8.5 hr, 14 hr, or 24 hr.
3. Lysis Buffer Preparation for Cell Sorting
NOTE: Making a single plate of lysis buffer takes about 1 hr. It is advisable to make and sort multiple plates, as cell sorting can be inefficient and yield many wells with no detectable cell.
Clean the bench, pipettes, tube rack, cold PCR plate holders, and gloves with DNA decontaminating solution in preparation for the PCR.
Dilute the RNAse inhibitor to 2.64 U/µl by adding stock RNAse inhibitor to nuclease-free water in a 1.5 ml tube. NOTE: This allows RNAse inhibitor to be added to the lysis buffer without pipetting sub-µl volumes.
Make lysis buffer for the cells by combining the components listed in Table 3.
Transfer 92.4 µl of the lysis buffer for the cells into a separate tube; this will be the lysis buffer for the amplicon standards.
Using low-binding pipette tips and microcentrifuge tubes, prepare 200 µl and 500 µl of E. coli DNA diluted to 3.1 pg/µl and 6.2 pg/µl, respectively, with DNA suspension buffer.
Add 184.8 µl of 3.1 pg/µl E. coli DNA to the lysis buffer for the cells. NOTE: The E. coli carrier DNA serves to broaden the scope of nonspecific primer binding possibilities and thereby reduce the likelihood that primer dimerization will lead to a PCR product.
- Prepare standards.
- Label six low-binding microcentrifuge tubes as 5e5, 5e4, … 5e0.
- Add 90 µl of DNA suspension buffer to the "5e5" tube.
- Add 90 µl of 6.2 pg/µl E. coli DNA to each of the other five tubes. Keep all tubes on ice. NOTE: Incorporating carrier DNA into the standards makes the total mass of DNA in the standard wells comparable to that in the 1-cell wells; this reduces the probability that the concentration of standards will be affected by DNA binding to tubes or pipette tips.
- Take an aliquot of the "5e6" standard (5 x 106 molecules/µl of each amplicon, prepared in step 1.4) from storage. Vortex briefly and spin briefly (5 sec at 500 x g) to ensure that the liquid is at the bottom of the tube.
- Using a low-binding pipette tip, add 10 µl of 5e6 standard to the "5e5" tube. Vortex briefly, spin (5 sec at 500 x g), and put the tube back on ice.
- Pipette 10 µl from the "5e5" tube into the "5e4" tube. Vortex briefly, spin (5 sec at 500 x g), and put back on ice.
- Repeat these serial dilutions, with each tube receiving 10 µl from the previous tube, until all tubes have a dilution of standard.
Prepare a row of 12 PCR tubes with caps. Distribute 67 µl of "cell" lysis buffer to each tube. Cap the tubes and spin down briefly (5 sec at 500 x g) if necessary.
Using a 12-channel pipette, distribute 9 µl of "cell" lysis buffer from the row of PCR tubes into each well of the first 7 rows of a PCR plate (Figure 3).
Distribute 7 µl of "standard" lysis buffer to each well of the last row of the plate (Figure 3).
Using low-binding pipette tips, add 2 µl of standard to each well of the bottom row according to the plate map (Figure 3). NOTE: 2 µl of 5 x 100 molecules/µl standard amounts to 10 molecules, etc.
Add 2 µl of 3.1 pg/µl E. coli DNA into the 10-cell and 100-cell wells; add 2 µl of 6.2 pg/µl E. coli DNA into the no-cell wells. NOTE: Each 1-cell, 10-cell, and 100-cell well has 6.2 pg of E. coli DNA, approximating the mass of genomic DNA in a single human cell. Each standard and no-cell well has 12.4 pg of E. coli DNA, approximately two human cells' worth.
Seal the plate using a sterile thermal seal, spin for 5 sec at 500 x g, and store at 4 °C until the cells are ready for sorting.
4. Cell Sorter Setup
Program the fluorescence-activated cell sorter to sort into a 96-well plate according to the plate map (Figure 3); no cells should be sorted into wells H1-H8 or H11-H12.
- Verify that the cell sorter is well aimed for sorting into a PCR plate.
- Set the angle of the sort stream to the minimum possible; this setting increases the chance that the cells will be sorted into the bottoms of wells rather than hit the sides.
- To aim the machine, seal an empty PCR plate and use the test stream to "sort" droplets of PBS onto the seal of the empty plate, according to the instructions of the machine19; the droplets should land on the surface of the seal at a position above the center of the well. For the best results, verify the aim across the length and width of the plate.
- If the droplets do not land in the correct position, wipe them off the surface of the seal, recalibrate the cell sorter according to the machine manufacturer's instructions, and repeat until the droplets in the sort stream are deposited correctly.
5. Cell Harvest and Sorting
Trypsinize the cells as appropriate for the cell line. For example, for MCF-7 cells, aspirate the medium from the cells, rinse the cells with 1 ml of 0.05% trypsin, and incubate the cells with 2 ml of 0.05% trypsin for 5 min at 37 °C and 5% CO2.
Add 8 ml of cell culture medium to the trypsinized cells and transfer all 10 ml of cell suspension to a 15 ml tube. Centrifuge for 4 min at 400 x g.
Aspirate the supernatant and resuspend the cell pellet in PBS + 2% FBS. NOTE: A smaller resuspension volume concentrates the cells and facilitates cell sorting; a 500 µl resuspension volume works well for a 6 cm dish of cells at 50% confluency.
Transfer the cell suspension to a 5 ml flow cytometry tube, pipetting through a 40 µm filter to remove clumps of cells.
Centrifuge the plate of lysis buffer at 400 x g for 30 sec at 4 °C before sorting the cells to ensure that the liquid is at the bottom of the wells.
Insert the tube with the cell suspension into the cell sorter. Open the seal on the plate of lysis buffer and carefully sort the cells of interest into the plate according to the plate map and following the cell sorter manufacturer's instructions19. To ensure that single cells are sorted at the maximum purity, run the machine in "single cell" mode and use standard flow cytometry gating based on forward and side scatter19. NOTE: In the example experiment, gate the cells based on p53-Venus fluorescence to isolate the brightest 15% of individual cells11.
Seal the plate with a new, sterile thermal seal and centrifuge at 400 x g for 1 min at 4 °C. Place the plate in the thermal cycler and run the RTSTA program (see step 1.7). NOTE: This performs reverse transcription on the mRNA from the sorted cells and then amplifies the cDNA of the transcripts of interest.
6. Exonuclease Treatment
Mix dilute exonuclease I by combining the components listed in Table 4. Prepare a row of 12 PCR tubes with caps. Distribute 30.5 µl of dilute exonuclease I into each tube. Cap the tubes and spin briefly (5 sec at 500 x g) to ensure that the liquid is at the bottom of the tube.
When the RTSTA is finished, spin the sample plate for 5 sec at 500 x g. Using a 12-channel pipette, distribute 3.6 µl of dilute exonuclease I into each well of the plate.
Seal the plate with a sterile thermal seal and spin briefly (5 sec at 500 x g). Place the plate in the thermal cycler and run the EXOI program (see step 1.7). NOTE: This step degrades any primers remaining after specific target amplification so that they will not interfere with future PCR.
7. Sample Dilution
When the exonuclease treatment is finished, add 32.4 µl of TE buffer to each well of the sample plate; this is an optional stopping point, and samples can be stored at -20 °C for several days.
8. Sort Quality Control Using qPCR
NOTE: Because cell sorting is not perfectly efficient, this step is necessary to identify which wells of the sorted plate actually received a cell. These samples can then be used for further analysis.
- Measure the housekeeping gene expression in each sample by qPCR.
- Prepare 900 µl of DNA binding dye qPCR master mix for 96 reactions according to the polymerase manufacturer's protocol using primers for one of the housekeeping genes selected in step 1.1; the exact concentrations of reaction buffer, polymerase, primers, etc. for this reaction will depend on the specific polymerase being used.
- Distribute 9 µl of master mix to every well of a 96-well PCR plate. Add 1 µl of sample from the sample plate to the corresponding well of the PCR plate.
- Run the plate on a real-time PCR machine using a thermal cycling protocol suggested by the master mix manufacturer or following a standard protocol20.
- Analyze the qPCR data20; an example of qPCR data from the sort quality-control steps listed below is shown in Figure 4.
- Verify that the measurements from the no-cell wells show a low (background) level of gene expression or no expression at all.
- Verify that measurements from 1-cell wells divide clearly into two groups: positive samples with high gene expression and negative samples with background levels of expression or no expression at all, similar to the no-cell wells. NOTE: The samples with high gene expression represent actual sorted cells; those with background expression should be excluded from further analysis.
- Verify that measurements from the 10-cell and 100-cell wells reflect higher gene expression than in the positive 1-cell wells, allowing for the possibility that inefficient sorting could cause those wells to have fewer cells than desired.
- Verify that measurements from the standard wells are evenly distributed in logarithmic space (i.e., that the Ct values are evenly distributed).
After identifying all positive 1-cell samples, make a "Sample Mixes" plate map to map positive 1-cell samples onto a new 96-well plate. Include eight wells for standards on this plate map. See Figure 5 for an example.
9. Gene Expression Measurement Using Microfluidic qPCR
NOTE: For every step in this section, pipette only to the first stop to minimize the formation of bubbles in the reagents.
Open a row of 8 PCR tubes. If there are multiple plates of samples, open a second row of 8 PCR tubes and label the tubes 1-8. This row will be for pooling standards from multiple plates.
In a 1.5 ml tube, mix 360 µl of DNA binding dye qPCR master mix with 36 µl of DNA binding dye sample loading reagent. Vortex briefly and spin for 10 sec at 2,000 x g. Divide this mixture (396 µl) into the unlabeled row of 8 PCR tubes, placing 48 µl in each tube.
Using an 8-channel pipette, distribute 3.3 µl of the master mix/sample loading reagent mixture into each well of a new 96-well PCR plate. Label this plate, "Sample Mixes."
- For each PCR plate of samples:
- Vortex the plate for 10 sec and spin for 10 sec at 500 x g. Remove the cover and transfer 2.7 µl of each positive 1-cell sample to its corresponding well on the Sample Mixes plate for the 1-cell wells indicated on the plate map.
- If there is only one plate of samples, transfer 2.7 µl of each amplified standard to its corresponding well on the Sample Mixes plate according to the plate map.
- If there are multiple plates of samples, transfer 2.7 µl of each amplified standard to its corresponding position in the labeled row of PCR tubes. Add standards from the current plate of samples to any standards already there. Seal the plate of samples when done using a sealing film.
- If there are multiple plates of samples:
- Vortex the row of PCR tubes containing standards for 10 sec and spin for 10 sec at 2,000 x g. Transfer 2.7 µl from each tube of mixed standards into the corresponding well on the Sample Mixes plate according to the plate map.
Load the NTC wells with 2.7 µl of nuclease-free water where indicated on the plate map. Seal the Sample Mixes plate and store at 4 °C until needed.
Peel the sticker off the bottom of a new microfluidic qPCR chip. Using a syringe of control line fluid with the cap still on, push down each accumulator spring to loosen it, and inject each accumulator with 150-200 µl of control line fluid.
Insert the chip in the loading machine and run the "Prime" script. This takes ~20 min.
When priming is done, vortex and spin down the plates of assay mixes (prepared in step 1.6) and sample mixes.
Using an 8-channel pipette, transfer 4.5 µl of each assay mix into the left side of the microfluidic qPCR chip according to the diagram in Figure 6. Pipette very carefully to avoid creating air bubbles at the bottom of the wells. When finished, seal the plate of assay mixes and store at -20 °C for future use.
Transfer 4.5 µl of each sample mix to the right side of the microfluidic qPCR chip according to Figure 6. Insert the chip into the loading machine and run the "Load Mix" script. This takes ~90 min.
Turn on the microfluidic real-time PCR machine, start the data collection software, and turn on the lamp to warm it up. This takes ~20 min.
When the Load Mix script is done, verify that there are no lines across the chip-this would indicate a loading problem. Carefully remove any dust particles from the chip using Scotch tape or lab tape. Insert the chip into the microfluidic real-time PCR machine and run the data collection script. Use the analysis software to inspect the results and export the data for further analysis.
10. Data Analysis
Remove the measurements for which the qPCR amplification curves show poor quality (i.e., for which the curves do not resemble the standard sigmoidal amplification curve)20.
Remove samples with more than 30 poor-quality or zero measurements or for which housekeeping gene expression (see step 1.1) is much lower than the majority of other samples (Figure 7).
For each gene, estimate the correct melt peak location as the median of the melt peak locations from each sample. If a measurement has a melt peak located more than 1 °C from the median, remove that measurement from consideration20.
- For each gene, create a standard curve to estimate the mRNA count in each sample using a spreadsheet or scripting language20. NOTE: The standards in this experiment consist of 10, 100, 1,000, or 10,000 molecules of dsDNA corresponding to each transcript of interest. If reverse transcription were perfectly efficient, the standards would correspond to 20, 200, 2,000, and 20,000 molecules of mRNA, since mRNA and cDNA are single-stranded. In practice, this is not the case, so we express measurements as an estimate in units of molecules × ERT, where ERT is the efficiency of reverse transcription.
- For each standard well, identify m, the number of molecules of ssDNA (twice the number of molecules of dsDNA) before amplification (e.g., 20 molecules, 200 molecules, etc.).
- For each standard well, identify the Ct value from qPCR20.
- With the values for m and Ct, perform a linear regression using the following equation to generate a standard curve with parameters a and b for each gene, and find the R2 value as an indicator of the goodness of fit:
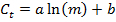
- From the standard curve, compute the PCR efficiency E for each gene:
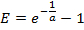 NOTE: A value of E much greater than 1.0 (e.g., E > 1.1) is not physically realistic; an R2 value for the linear regression much less than 1.0 (e.g., R2 < 0.985) means that the standards are not working reliably. These problems generally indicate that the lowest amplicon standard falls below a threshold of reliable quantification, and thus the lowest standard should be excluded from the linear regression. If an adequate linear regression cannot be made using at least three unique standards, discard the expression measurements for the gene.
NOTE: A value of E much greater than 1.0 (e.g., E > 1.1) is not physically realistic; an R2 value for the linear regression much less than 1.0 (e.g., R2 < 0.985) means that the standards are not working reliably. These problems generally indicate that the lowest amplicon standard falls below a threshold of reliable quantification, and thus the lowest standard should be excluded from the linear regression. If an adequate linear regression cannot be made using at least three unique standards, discard the expression measurements for the gene. - If the linear regression is adequate, use the resulting fit parameters a and b for each gene to estimate the number of mRNA molecules mest (in units of molecules × ERT) for the corresponding gene in each single-cell sample from the measured Ct value:
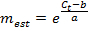
- Designate any measurement mest < 1 as mest = 0. Also, designate any measurement with no amplification in qPCR and thus no Ct value as mest = 0. NOTE: Measurements with an mest less than the lowest or greater than the highest standard used in the regression are found by extrapolation from the standard curve. These estimates are not necessarily invalid but should be regarded with caution.
- Visualize the single-cell gene expression data.
- Make a beeswarm plot (Dorn, J. and Holy, T., 2012, http://www.mathworks.com/matlabcentral/fileexchange/37105) in which each point represents the gene expression in a particular cell; an example is shown in Figure 8.
- Make a violin plot (Dorn, J., 2012, http://www.mathworks.com/matlabcentral/fileexchange/23661) in which the width of the “violin” represents the frequency of gene expression measurements around a particular level in the population of cells analyzed; an example is shown in Figure 8. NOTE: More sophisticated analyses can probe the relationships in expression between pairs of genes21,22 and can infer gene expression networks.
Representative Results
A general overview of the protocol is shown in Figure 1, including steps for cell treatment, the isolation of single cells by FACS, the generation and pre-amplification of cDNA libraries from single-cell lysates, the confirmation of single-cell cDNA libraries in sorted wells, and the measurement of gene expression by qPCR.
In preparation for single-cell isolation and gene expression analysis, it is necessary to first identify valid oligonucleotide primer pairs for each target gene of interest. Figure 2 shows two examples of primer quality-control methods. Melt curves are generated following qPCR with the primer pair being tested. Valid primers result in a melt curve with a single peak (Figure 2A, green curve). If multiple peaks (Figure 2A, blue curve) or a curve with a pronounced shoulder (Figure 2A, red curve) are present, this indicates the presence of multiple PCR products, and the primers should be redesigned. As a second quality-control method, agarose gel electrophoresis can be used to visually verify that a single band of the correct size is present for each primer pair tested (Figure 2B). Multiple bands or bands of the wrong size indicate that the primers are not valid (Figure 2B).
Following primer validation and the generation of libraries of qPCR standards from the correct amplicons, cell sorting can be performed. An example sorting scheme is indicated in Figure 3. Each sorted plate should include wells containing a dilution series of amplicon standards, with 10, 100, 1,000, or 10,000 copies of each target amplicon. It is also recommended to include wells into which no cells, 10 cells, or 100 cells are sorted, in addition to the single-cell wells. Due to sorting inefficiency, it is often necessary to identify the wells of the plate in which single cells have successfully been deposited. For this validation step, following the procedure for reverse transcription and specific target amplification, qPCR should be performed to measure the expression of a housekeeping gene (Figure 4). The wells for the amplicon standards (Figure 4, black curves) should show evenly spaced amplification curves. There should also be clear separation in the curves corresponding to the "no-cell" wells (Figure 4; red curves), "10-cell" wells (Figure 4; blue curves), and "100-cell" wells (Figure 4; purple curves). A clear separation should also occur for the "1-cell" wells corresponding to successful cell deposition (Figure 4; green amplification curves with Ct values less than those for the no-cell controls but greater than those for the 10-cell controls) versus unsuccessful cell deposition (Figure 4; green curves with Ct values near those for the no-cell controls). Once wells with a single cell lysate have been identified, cDNA from the wells (potentially from multiple plates) can be transferred to a microfluidic qPCR chip, with one single-cell lysate per well. For example, Figure 5 shows a hypothetical array that combines cDNA from single cells from three different sorted 96-well plates into a single new plate for qPCR analysis.
To analyze the microfluidic qPCR results, the samples that likely did not receive a cell should first be removed, as indicated by many missing or zero gene expression measurements (Figure 7, rows indicated by arrows) or unusually low housekeeping gene expression. For each assay, any measurements with melt peaks that do not match those of the other samples in the same assay should also be removed. After removing bad samples and measurements, linear regression can be performed on the measurements corresponding to the amplicon standards to estimate the absolute count of each transcript of interest in each single-cell sample. Figure 8 shows a visualization of these gene expression estimates, measured in units of molecules × reverse transcription efficiency, as beeswarm and violin plots. In this example, untreated cells show a broad range of CDKN1A expression levels, with many cells not expressing CDKN1A at all and with others expressing the gene at levels spanning two orders of magnitude. After treatment with neocarzinostatin (NCS) for 3 hr, most cells express CDKN1A at much higher levels, and the variability decreases to roughly a single order of magnitude. As the duration of treatment increases, the average level of CDKN1A expression decreases and the variability in expression expands.
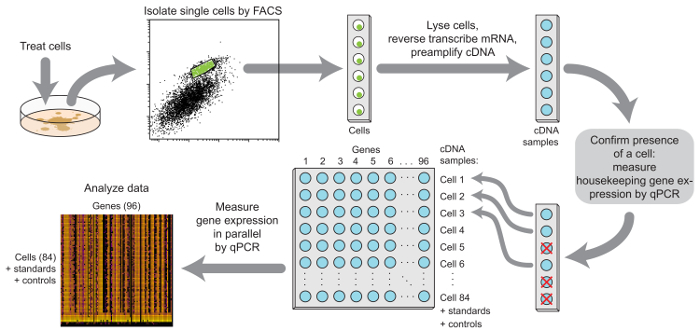 Figure 1:Overview of the steps in single-cell gene expression profiling. Treated cells are harvested and sorted via FACS directly into lysis buffer. mRNA species of interest are reverse-transcribed, and the resulting cDNA is amplified by PCR. Housekeeping gene expression in each sample is measured by qPCR to determine which samples actually received a cell. In samples passing this quality-control test, the expression of up to 96 genes is measured using microfluidic qPCR. This figure has been modified from a previous publication11. Please click here to view a larger version of this figure.
Figure 1:Overview of the steps in single-cell gene expression profiling. Treated cells are harvested and sorted via FACS directly into lysis buffer. mRNA species of interest are reverse-transcribed, and the resulting cDNA is amplified by PCR. Housekeeping gene expression in each sample is measured by qPCR to determine which samples actually received a cell. In samples passing this quality-control test, the expression of up to 96 genes is measured using microfluidic qPCR. This figure has been modified from a previous publication11. Please click here to view a larger version of this figure.
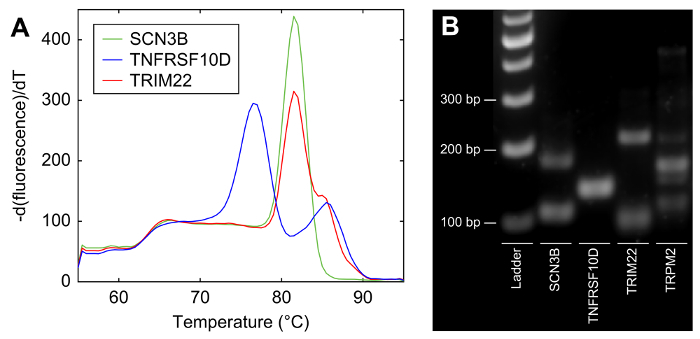 Figure 2:Example results of primer testing. (A) Melt curves from qPCR. The blue melt curve (TNFRSF10D) has multiple peaks and the red melt curve (TRIM22) has a "shoulder," which amounts to a second peak close to the first peak. These indicate that the primers used in those reactions amplify multiple targets and need to be redesigned. The green melt curve (SCN3B) has a single peak, consistent with primers that amplify a single target. (B) Amplified DNA run on an agarose gel. The lanes for SCN3B, TRIM22, and TRPM2 contain multiple bands; the primers used to make those DNA amplicons amplify multiple targets and need to be redesigned. The lane for TNFRSF10D has a single band, consistent with primers that amplify a single target. It is worth noting that the primers for SCN3B pass the melt curve test but fail the gel test, while those for TNFRSF10D pass the gel test but fail the melt curve test; these two methods of primer validation are complementary. In this example, one can conclude that all four primer sets need to be redesigned. Please click here to view a larger version of this figure.
Figure 2:Example results of primer testing. (A) Melt curves from qPCR. The blue melt curve (TNFRSF10D) has multiple peaks and the red melt curve (TRIM22) has a "shoulder," which amounts to a second peak close to the first peak. These indicate that the primers used in those reactions amplify multiple targets and need to be redesigned. The green melt curve (SCN3B) has a single peak, consistent with primers that amplify a single target. (B) Amplified DNA run on an agarose gel. The lanes for SCN3B, TRIM22, and TRPM2 contain multiple bands; the primers used to make those DNA amplicons amplify multiple targets and need to be redesigned. The lane for TNFRSF10D has a single band, consistent with primers that amplify a single target. It is worth noting that the primers for SCN3B pass the melt curve test but fail the gel test, while those for TNFRSF10D pass the gel test but fail the melt curve test; these two methods of primer validation are complementary. In this example, one can conclude that all four primer sets need to be redesigned. Please click here to view a larger version of this figure.
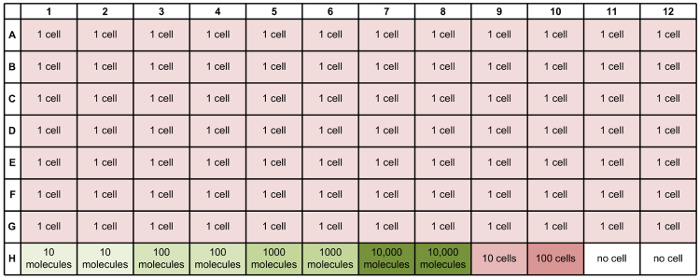 Figure 3: Plate map for cell sorting. This plate map includes 1-cell wells (red), 10- and 100-cell wells (darker red), standards (green), and no-cell wells (white). Including two replicates of each standard on each plate helps to compensate for variability in standards. Including 10-cell and 100-cell wells is helpful as a sanity check for gene expression. The no-cell wells serve as a negative control. Please click here to view a larger version of this figure.
Figure 3: Plate map for cell sorting. This plate map includes 1-cell wells (red), 10- and 100-cell wells (darker red), standards (green), and no-cell wells (white). Including two replicates of each standard on each plate helps to compensate for variability in standards. Including 10-cell and 100-cell wells is helpful as a sanity check for gene expression. The no-cell wells serve as a negative control. Please click here to view a larger version of this figure.
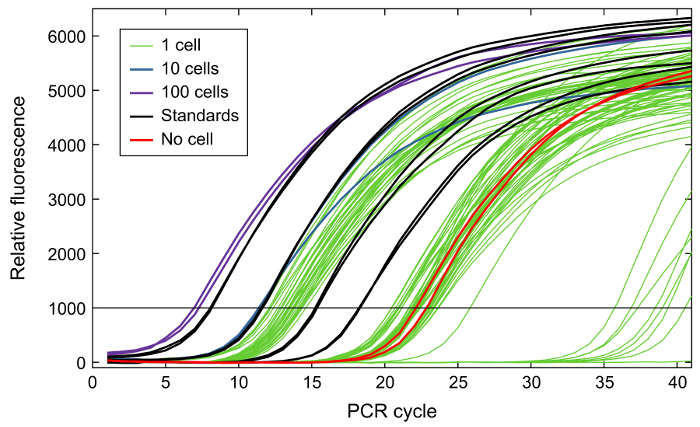 Figure 4: Example of qPCR amplification curves from sort quality control measuring GAPDH expression. No-cell samples (red) show a background level of expression. 1-cell samples (green) divide into two distinct groups, one with high expression and one with low expression similar to the no-cell samples. The high-expression samples most likely received a real cell during sorting; the low-expression samples most likely did not. 10-cell (blue) and 100-cell (purple) samples show higher expression than the 1-cell samples; the 10-cell samples are close to the highest-expressing 1-cell samples because of sort inefficiency. Standards (black) are evenly distributed, as expected. Please click here to view a larger version of this figure.
Figure 4: Example of qPCR amplification curves from sort quality control measuring GAPDH expression. No-cell samples (red) show a background level of expression. 1-cell samples (green) divide into two distinct groups, one with high expression and one with low expression similar to the no-cell samples. The high-expression samples most likely received a real cell during sorting; the low-expression samples most likely did not. 10-cell (blue) and 100-cell (purple) samples show higher expression than the 1-cell samples; the 10-cell samples are close to the highest-expressing 1-cell samples because of sort inefficiency. Standards (black) are evenly distributed, as expected. Please click here to view a larger version of this figure.
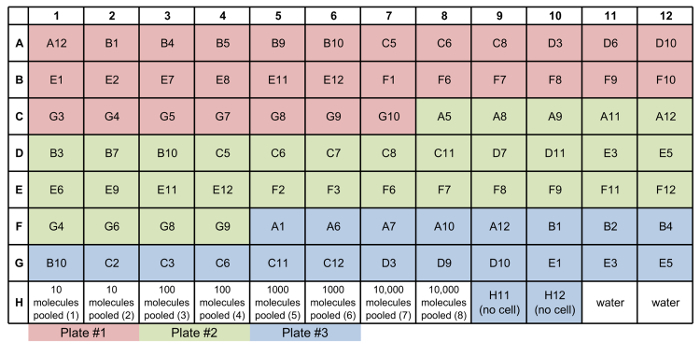 Figure 5: Example of Sample Mixes plate map. This plate map includes 1-cell samples from three different plates (rows A-G), standards mixed from all three plates (H1-H8), no-cell controls (H9-H10), and no-template controls for qPCR (H11-H12). Please click here to view a larger version of this figure.
Figure 5: Example of Sample Mixes plate map. This plate map includes 1-cell samples from three different plates (rows A-G), standards mixed from all three plates (H1-H8), no-cell controls (H9-H10), and no-template controls for qPCR (H11-H12). Please click here to view a larger version of this figure.
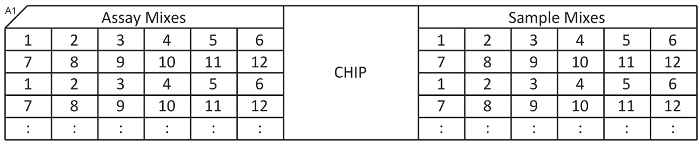 Figure 6:Diagram of microfluidic qPCR chip with the order of loading (1, 2, 3…). The notch (A1) is the top left corner of the plate. Please click here to view a larger version of this figure.
Figure 6:Diagram of microfluidic qPCR chip with the order of loading (1, 2, 3…). The notch (A1) is the top left corner of the plate. Please click here to view a larger version of this figure.
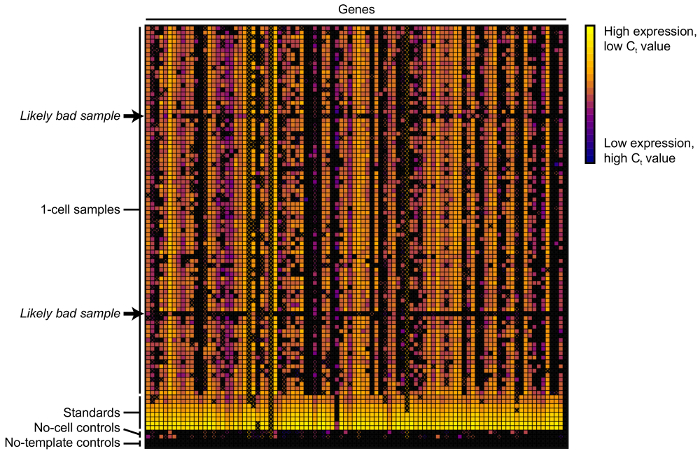 Figure 7:Heat map of the qPCR results. Each row represents a 1-cell sample, standard, or control; each column represents a measured transcript. Rows with an unusual number of black squares or Xs (arrows) likely indicate samples that did not receive an actual cell. Please click here to view a larger version of this figure.
Figure 7:Heat map of the qPCR results. Each row represents a 1-cell sample, standard, or control; each column represents a measured transcript. Rows with an unusual number of black squares or Xs (arrows) likely indicate samples that did not receive an actual cell. Please click here to view a larger version of this figure.
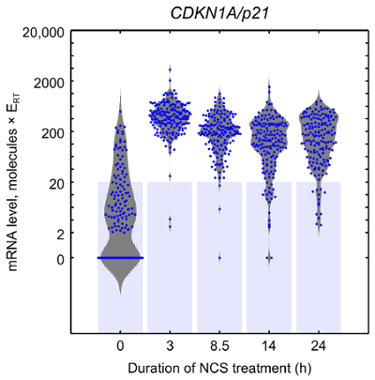 Figure 8: Beeswarm and violin plots. In a beeswarm plot (blue points), each point represents gene expression in a particular cell. In a violin plot (gray shape), the width of the "violin" represents the frequency of gene expression measurements around a particular level in the population of cells analyzed. Light blue bars represent the lowest standard. Measurements above the blue bars are interpolated between standards, while measurements within the blue bars are extrapolated. The data shown here are measurements of CDKN1A expression in MCF-7 cells expressing Venus-tagged p53 treated with neocarzinostatin (NCS) for the indicated times in order to induce DNA double-strand breaks. This figure has been modified from previous publication11. Please click here to view a larger version of this figure.
Figure 8: Beeswarm and violin plots. In a beeswarm plot (blue points), each point represents gene expression in a particular cell. In a violin plot (gray shape), the width of the "violin" represents the frequency of gene expression measurements around a particular level in the population of cells analyzed. Light blue bars represent the lowest standard. Measurements above the blue bars are interpolated between standards, while measurements within the blue bars are extrapolated. The data shown here are measurements of CDKN1A expression in MCF-7 cells expressing Venus-tagged p53 treated with neocarzinostatin (NCS) for the indicated times in order to induce DNA double-strand breaks. This figure has been modified from previous publication11. Please click here to view a larger version of this figure.
| Condition | Temperature | Time |
| Hold | 50 °C | 15 min |
| Hold | 95 °C | 2 min |
| 20 cycles | 95 °C | 15 sec |
| 60 °C | 4 min | |
| Hold | 4 °C | ∞ |
Table 1: Reverse transcription and specific target amplification (RTSTA) thermal cycling program.
| Condition | Temperature | Time |
| Hold | 37 °C | 30 min |
| Hold | 80 °C | 15 min |
| Hold | 4 °C | ∞ |
Table 2: Exonuclease I treatment (EXOI) thermal cycling program.
| Component | Volume/well (µl) | 96 volumes w/ 10% overage (µl) |
| 2x reaction mix | 5.00 | 528.00 |
| Reverse transcriptase/polymerase mix | 0.20 | 21.12 |
| 10x STA Primer Mix | 1.00 | 105.60 |
| 2.64 U/µl (dilute) RNAse inhibitor | 0.02 | 2.00 |
| Nuclease-free water | 0.78 | 82.48 |
| Total | 7.00 | 739.20 |
Table 3: Components of the lysis buffer for the cells.
| Component | Volume/well (µl) | 96 volumes w/ 10% overage (µl) |
| Nuclease-free water | 2.52 | 266.00 |
| Exonuclease I Reaction Buffer (10x) | 0.36 | 38.00 |
| Exonuclease I (20 U/µl) | 0.72 | 76.00 |
| Total | 3.60 | 380.00 |
Table 4: Components of the exonuclease mix to treat the amplified cDNA samples.
Discussion
We have presented a method for isolating individual mammalian cells from a population of adherent cells grown in culture and for assaying the expression of approximately 96 genes in each cell. Good advance preparation is critical for this method to work well. In particular, designing and testing primer pairs specific to the transcripts of interest (steps 1.2-1.3) are time-consuming but important steps, as the primers determine the quality of the single-cell measurements. Once reliable primer pairs have been obtained, they are used to amplify cDNA from the transcripts of interest; the amplicons are then combined together in equimolar amounts to make standards (step 1.4). This step is critical, as the standards are required to estimate the absolute transcript counts; standards should be made once, aliquoted, and used for all subsequent rounds of cell sorting and gene expression measurements. Likewise, on the day that the cells are sorted, it is especially important to dilute the standards carefully using low-binding pipette tips and microcentrifuge tubes when making plates of lysis buffer (steps 3.7 and 3.11). Aiming the cell sorter (step 4.2) is another critical step; this must be done very carefully in order for the cells to successfully hit the target of 9 µl of lysis buffer in a PCR plate.
There are several modifications to the protocol that can be made to adapt it to a variety of research needs. Here, we focused on a DNA binding dye-based qPCR approach, which provides a relatively affordable method for quantifying gene expression. However, this method has the potential for creating a high background signal, since any amplified DNA, even that of off-target sequences, results in a fluorescent signal. Such a background can be minimized by ensuring that the primer pair used to amplify a specific target yields a melt curve with a single peak and a single PCR product of the correct size (Figure 2). If the background is still a concern after using such quality-control methods, a probe-based qPCR approach can be used to minimize off-target detection, albeit at a greater cost of reagents. Another option would be a nested PCR approach, in which one set of primers is used in the RTSTA step to reverse-transcribe and amplify a large region of the transcript and a second set of primers, which amplifies a smaller region contained within that large region, is used to measure gene expression by qPCR. This approach has been shown to improve the specificity of DNA binding dye-based qPCR23.
Several methods are available for isolating individual cells for gene expression analysis. The choice of cell isolation method depends on several factors, including the availability of equipment (such as a fluorescence-activated cell sorter), the source of the cells to be evaluated (such as established cell lines versus primary cells from a tissue), or the speed with which the cells must be isolated. In the example presented in this protocol, we used FACS of a cell line expressing a clearly detectable fluorescent-tagged protein. FACS has the benefits of rare cell detection capabilities and relatively rapid cell isolation. One challenge of the method is that the deposition of the cells of interest into the requisite small volume of lysis buffer in a 96-well PCR plate can be inefficient and may require the optimization of the cell sorting geometry to ensure a successful isolation. Alternative approaches that may lead to greater precision in cell isolation at lower speeds and/or higher costs include the micropipetting of individual cells, the laser capture microdissection of specific cells from a tumor sample, or the use of microfluidic systems (e.g., the Fluidigm C1 system).
One major benefit of the protocol described here is that the internal controls, a dilution series of purified PCR amplicons, enable the estimation of the absolute number of each transcript in each cell. Without these standards, only relative levels of gene expression can be obtained based on calculations derived from the Ct values. However, with these standards, absolute transcript counts can be estimated, ideally through interpolation within the range of precisely quantified amplicon standards (Figure 8). As with any PCR-based method of gene expression quantification, accurate absolute quantification requires knowledge of the efficiency of each process, including reverse transcription.
A current challenge in quantifying transcript levels in single cells is that the limit of detection for many methods is estimated to be 10-100 mRNAs per cell24, preventing the reliable quantification of a significant percentage of the transcriptome. As an alternative approach, one can use smRNA FISH to quantify targets of particular interest25,26, including those with very few transcripts. Advances in smRNA FISH have expanded the number of distinct transcripts that can be detected at once27 and the number of cells that can be profiled at once28, given the appropriate equipment. Although smRNA FISH has its own limitations (potential false-positive and false-negative transcript detection, restrictions to only a few target genes per cell, the cost of equipment and reagents), it may provide a powerful method to complement and validate results from a qPCR-based analysis of a subset of target genes of interest.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank V. Kapoor in the CCR ETIB Flow Cytometry Core for her aid in performing the cell sorting during the development of this protocol. We also thank M. Raffeld and the CCR LP Molecular Diagnostics Unit and J. Zhu and the NHLBI DNA Sequencing and Genomics Core for their aid in performing the qPCR during the development of this protocol. This research was supported by the Intramural Program of the NIH.
References
- Feinerman O, Veiga J, Dorfman JR, Germain RN, Altan-Bonnet G. Variability and Robustness in T Cell Activation from Regulated Heterogeneity in Protein Levels. Science. 2008;321(5892):1081–1084. doi: 10.1126/science.1158013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459(7245):428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Rosenfeld N, et al. Oscillations and variability in the p53 system. Mol. Syst. Biol. 2006;2(1) doi: 10.1038/msb4100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman-Lerner A, Gordon A, et al. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437(7059):699–706. doi: 10.1038/nature03998. [DOI] [PubMed] [Google Scholar]
- Chong S, Chen C, Ge H, Xie XS. Mechanism of Transcriptional Bursting in Bacteria. Cell. 2014;158(2):314–326. doi: 10.1016/j.cell.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA Synthesis in Mammalian Cells. PLoS Biol. 2006;4(10):e309+. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senecal A, Munsky B, et al. Transcription Factors Modulate c-Fos Transcriptional Bursts. Cell Rep. 2014;8(1):75–83. doi: 10.1016/j.celrep.2014.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey SS, Foley JE, Limsirichai P, Schaffer DV, Arkin AP. Orthogonal control of expression mean and variance by epigenetic features at different genomic loci. Mol. Syst. Biol. 2015;11(5):806+. doi: 10.15252/msb.20145704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar RD, Razooky BS, et al. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc. Natl. Acad. Sci. U.S.A. 2012;109(43):17454–17459. doi: 10.1073/pnas.1213530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thattai M, van Oudenaarden A. Stochastic gene expression in fluctuating environments. Genetics. 2004;167(1):523–530. doi: 10.1534/genetics.167.1.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JR, Fisher BE, Batchelor E. p53 Pulses Diversify Target Gene Expression Dynamics in an mRNA Half-Life-Dependent Manner and Delineate Co-regulated Target Gene Subnetworks. Cell Syst. 2016;2(4):272–282. doi: 10.1016/j.cels.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004;36(2):147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer. 2009;9(10):749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008;9(5):402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. 2012;13(1):134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PCR Technologies: A Technical Guide. St. Louis, MO: Sigma-Aldrich; 2014. [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell. 2008;30(3):277–289. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flow Cytometry: Principles and Applications. Totowa, NJ: Humana Press; 2007. [Google Scholar]
- Real-time PCR. Taylor & Francis; 2006. [Google Scholar]
- Song L, Langfelder P, Horvath S. Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinf. 2012;13(1):328. doi: 10.1186/1471-2105-13-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, et al. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinf. 2006;7(Suppl 1) doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haff LA. Improved quantitative PCR using nested primers. PCR Methods Appl. 1994;3(6):332–337. doi: 10.1101/gr.3.6.332. [DOI] [PubMed] [Google Scholar]
- Hashimshony T, Senderovich N, et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016;17:77. doi: 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronander E, Bengtsson DC, Joergensen L, Jensen ATR, Arnot DE. Analysis of Single-cell Gene Transcription by RNA Fluorescent In Situ Hybridization (FISH) J. Vis. Exp. 2012. p. e4073. [DOI] [PMC free article] [PubMed]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008;5(10):877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck E, Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods. 2012;9(7):743–748. doi: 10.1038/nmeth.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battich N, Stoeger T, Pelkmans L. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat. Methods. 2013;10(11):1127–1133. doi: 10.1038/nmeth.2657. [DOI] [PubMed] [Google Scholar]