

Abstract
Here we provide protocols for the kinetic examination of lagging-strand DNA synthesis in vitro by the replication proteins of bacteriophage T7. The T7 replisome is one of the simplest replication systems known, composed of only four proteins, which is an attractive feature for biochemical experiments. Special emphasis is placed on the synthesis of ribonucleotide primers by the T7 primase-helicase, which are used by DNA polymerase to initiate DNA synthesis. Because the mechanisms of DNA replication are conserved across evolution, these protocols should be applicable, or useful as a conceptual springboard, to investigators using other model systems. The protocols described here are highly sensitive and an experienced investigator can perform these experiments and obtain data for analysis in about a day. The only specialized piece of equipment required is a rapid-quench flow instrument, but this piece of equipment is relatively common and available from various commercial sources. The major drawbacks of these assays, however, include the use of radioactivity and the relative low throughput.
Keywords: Genetics, Issue 120, DNA synthesis, replisome, kinetics, rapid-quench, primase, polymerase
Introduction
The replication of DNA is a conserved feature of all cellular life. While the identity and number of replication components vary widely, the general mechanisms are shared across evolution1. Here, we describe gel-based, discontinuous kinetic experiments, using radioactive substrates, aimed at understanding the kinetics of lagging-strand DNA synthesis in vitro by components of the T7 replisome. The replication machinery of bacteriophage T7 is very simple, consisting of only four proteins (the DNA polymerase, gp5, and its processivity factor, E. coli thioredoxin, the bifunctional primase-helicase gp4, and the single-stranded DNA binding protein, gp2.5)2. This feature makes it an attractive model system to study the conserved biochemical mechanisms involved in DNA replication.
Special emphasis is placed on the formation of ribonucleotide primers by T7 DNA primase, a critical step in the initiation of DNA synthesis. In addition, one can also examine the usage of these primers by T7 DNA polymerase or other replication proteins by including them in the reaction mixture. The T7 primase-helicase, gp4, catalyzes the formation of primers for DNA synthesis at specific DNA sequences called primase recognition sites, or PRS, (5'-GTC-3'). The cytosine in the PRS is cryptic, it is essential for recognition of the site, but it is not copied into the product3. The first step in primer synthesis by gp4 involves the formation of the dinucleotide pppAC, which is then extended to a trimer, and eventually to functional tetranucleotide primers pppACCC, pppACCA, or pppACAC depending on the sequence in the template4. These primers can then be used by T7 DNA polymerase to initiate DNA synthesis, which is also a gp4-assisted process5,6. In this regard, the primase domain stabilizes the extremely short tetraribonucleotides with the template, preventing their dissociation, engages DNA polymerase in a manner conducive to securing the primer/template into the polymerase active site7. These steps (RNA primer synthesis, primer handoff to polymerase, and extension) are repeated in multiple cycles to replicate the lagging strand and must be coordinated with the replication of the leading strand.
The assays described here are highly sensitive and can be performed in a moderate time frame. However, they are relatively low-throughput and great care must be exercised in the usage and disposal of radioactive materials. Depending on the speed at which the reaction proceeds, one might employ a rapid-quench instrument to attain samples at time scales amenable for meaningful analysis of reaction time courses in either the steady or the pre-steady state. Recently, we used assays described here to provide evidence of the importance of primer release from the primase domain of gp4 in the initiation of Okazaki fragments. Additionally, we found evidence of a regulatory role for the T7 single-stranded DNA binding protein, gp2.5, in promoting efficient primer formation and utilization8.
Protocol
NOTE: Follow all institutional regulations regarding the safe use and disposal of radioactive material, including but not limited to, usage of personal protective equipment, such as gloves, safety glasses, lab coat, and appropriate acrylic shields.
NOTE: The standard buffer consists of 40 mM HEPES-KOH, pH 7.5, 50 mM potassium glutamate, 5 mM DTT, 0.1 mM EDTA (this buffer is pre-made at a 5x concentration) and 0.3 mM of each dNTP. T7 replication proteins are purified as described8,9,10. Single-stranded DNA containing a single T7 primase recognition site (5'-GGGTC-3', 5'-GTGTC-3', 5'-TGGTC-3') can be purchased from a variety of DNA synthesis companies (the length of the ssDNA may depend on the enzyme of choice, for T7 primase, a pentamer is the minimum template length to synthesize a full-length primer11, as the cryptic C is essential, however, if the primer is to be extended by polymerase, additional nucleotides must be present at the 3' end of the template. Reactions are initiated by the addition of a final concentration of 10 mM MgCl2 and incubated at 25 °C. Manual sampling works well for time-points of 5 seconds or greater. If time-points shorter than that are required, it is recommended to use a rapid-quench flow instrument to obtain accurate data.
1. Multiple-turnover Primer Synthesis Reaction by Manual Sampling
Before starting the experiment, aliquot 2 µL of formamide dye buffer (93% (v/v) formamide, 50 mM EDTA, 0.01% xylene cyanol and 0.01% bromophenol blue) into 8 1.5 mL polypropylene tubes. In general, the number of tubes is equal to the number of time-points analyzed.
Prepare a 20 µL mixture consisting 1x buffer, 0.1 mM ATP and CTP, 0.25 mCi/mL [α-32P] CTP, 0.1 µM ssDNA template, and 0.1 µM gp4 (expressed as hexamer concentration — 0.6 µM monomer).
Incubate the mixture at room temperature for 5 min.
Remove 2 µL of the mixture using a pipettor and mix with 2 µL formamide dye buffer in a 1.5 mL tube prepared in step 1.1 for the no reaction control (time zero).
Add 2 µL of 0.1 M MgCl2 to reaction mixture (to a final concentration of 10 mM).
Manually withdraw 2 µL samples of the reaction mixtures using a pipettor at 10 s intervals and mix with formamide dye predispensed in 1.5 mL tubes prepared in step 1.1.
Heat samples at 95 °C for 5 min in a heat block.
Load aliquots of the samples onto a denaturing gel. The small tetraribonucleotide primer products synthesized by the T7 primase are resolved well on a 0.4 mm thick 25% acrylamide:bisacrylamide (19:1), 3 M urea, 1x TBE (100 mM Tris-borate, 1 mM EDTA).
Place entire gel on chromatography paper and cover with plastic wrap. Dry gel using a gel drier.
Prepare a dilution series of the remainder of the reaction mixture and spot the same volume loaded on the gel onto chromatography paper, i.e., spot 4 µL of the dilution if 4 µL of sample were loaded. NOTE: This will serve as the standard curve to determine the concentration of products formed during the reaction. For example, five-fold dilutions using TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) work well, spanning a range of 1:25 to 1:15,625 (52-56), but the dilutions should be adjusted depending on the level of activity.
Expose dry gel and standard using a phosphorimager screen. Quantify the radioactivity in the dilution series and construct a standard curve as described 8,12 NOTE: Inclusion of T7 DNA polymerase to the reaction mixture allows one to examine the usage of tetraribonucleotide primers by this enzyme. Optimally, the polymerase concentration should be saturating to simplify the interpretation of the data. A polymerase concentration of 0.4 µM or higher gives good results. Additionally, the effect of other proteins, such as single-stranded DNA binding proteins, on either primer synthesis or lagging-strand initiation can be examined in this manner.
2. Multiple-turnover Primer Synthesis Reaction Using a Rapid-quench Instrument
- Preparation of reaction mixtures
- Prepare 400 µL of solution A, containing 1x buffer, 0.2 µM gp4 hexamer, 0.2 µM ssDNA template, 0.6 mM dNTPs, 0.2 mM ATP and CTP (including 0.5 mCi/mL [α-32P] CTP.
- Prepare 400 µL of solution B, containing 1x buffer and 20 mM MgCl2.
- Operation of rapid-quench flow instrument
- Turn on rapid-quench flow instrument; after the main menu appears, type 7 on the instrument keyboard to move the syringe driver to the home position.
- Open buffer syringe position to "LOAD".
- Loading buffer syringes
- Attach a 10 mL syringe containing buffer to the drive syringe port. Change syringe knob position to "LOAD".
- Using 10 mL syringes, fill instrument buffer reservoir A and B with 10 mM Tris, pH 7.5, 1 mM EDTA and fill syringe C with quench solution (250 mM EDTA, and 0.2% SDS). Remove air bubbles by pushing plungers in and out.
- Change knob positions to "FIRE". Type 2 to adjust position of the syringe driver bar. Press "ADJUST DOWN" button to lower the syringe until buffer comes out the exit loop: (7-goes down continuously; 8-large step down; 9-small step down). Type "ESC" to return to the main menu.
- Wash tubing
- Attach exit line to vacuum. Change sample knobs to "FLUSH". Close buffer syringe by setting the knobs to the horizontal position. Turn on vacuum and dip the 2 flush loops in H2O for 10 s, then dip in MeOH for 10 s. Dry loops by sucking air for ~15 s, or until dry.
- Loading samples
- Change sample knob position to "LOAD". Place solution A into 1 mL syringe. Plug syringe into one of the sample load ports. Repeat for solution B, and place in opposite sample load port.
- Collection of time-points
- On main menu, type 1 for quench-flow run. Enter reaction time (s) and press, "ENTER". The reaction loop number to use will be displayed. Switch to required reaction loop. Repeat wash procedure in step 2.2.4.
- Turn sample knobs to "LOAD". Load reaction mixes in sample loops until just outside the central mixing compartment. Turn all knobs to "FIRE". Confirm that all knobs are in the correct position.
- Place 1.5 mL tube onto end of exit loop. Press "GO" to run the reaction. Repeat wash procedure in step 2.2.4. Reload buffer syringes A and B until they hit the driver.
- Repeat steps 2.2.6.2-2.2.6.3 for each desired time point. Exception: Don't load solution containing the start reagent (i.e., MgCl2 in this case) when quenching the zero time-point control.
- When finished collecting samples, enter "ESC" to return to main menu. Press "7" to return buffer syringe driver to its home position.
- Instrument clean-up
- Remove reaction mixture syringes. Turn sample loading knob to "LOAD" position. Under vacuum, wash each sample loop with 10 mL 0.2 N NaOH, followed by 10 mL 0.3 N H3PO4 and 10 mL MeOH.
- Turn syringe knobs to "LOAD" position. Press down on all 3 syringe plungers to retrieve reaction and quench buffers. Wash syringes with 5 mL H2O at least twice.
- Fill syringes again with 5 mL H2O and turn knobs to "FIRE" position. Change knob positions and turn on vacuum to remove H2O. Flush as in step 2.2.4. Turn off instrument. NOTE: Products are analyzed as described in steps 1.7-1.12. In addition, T7 DNA polymerase and other replication proteins can be added in the reaction mixture to analyze their effect on primer synthesis or extension.
3. Single-turnover Primer Synthesis Reaction
NOTE: Single-turnover primer synthesis/extension assays are carried out similarly to multiple-turnover reactions, with a few major differences: these assays follow the conversion of radiolabeled ATP into primer or extension products, however the concentration of substrates and proteins are considerably higher. In addition, in order to attain millisecond resolution to allow analysis of the primer synthesis reaction, one must utilize a rapid-quench flow machine.
- Preparation of reaction mixtures.
- Prepare 400 µL of solution A, containing 1x buffer, 3 µM gp4 hexamer, 6 µM ssDNA template, 0.6 mM dNTPs, 1 mM CTP, and 2 nM [γ-32P] ATP. Prepare 400 µL of solution B, containing 1x buffer and 20 mM MgCl2.
- Rapid-quench flow operation
- Carry out reactions as described in step 2.2. If desired, add T7 DNA polymerase or T7 single-stranded-DNA binding protein at a concentration of 15 µM and 20 µM, respectively. In this case, the extension products accumulate at a time regime that allows for manual sampling of the reaction.
4. Data Analysis
Fit product progress curves to non-linear least-squares models using commercially available software capable of least-squares fits and/or numerical integration. The details of the operation will vary with the software employed, but below are generalized forms of equations used for fitting the data.
Determine the rate of product formation of multiple turnover primer synthesis reactions by fitting to progress curves to a linear equation. If there is significant curvature, use the initial rate method13,14 by fitting the data to a single-exponential function: y=A(e-kt ), where y is the product concentration, A is the reaction amplitude, k is the observed rate constant, t is time, in seconds. The slope of the tangent line at time=0 is the initial rate. NOTE: Fit pre-steady state primer synthesis reactions to a linear equation or to the pre-steady state burst equation y=A (e-kburstt )+ratess t, where A is the reaction amplitude, kburst is the observed rate constant for the pre-steady state burst, ratess is the steady-state ratess =kcat [Enzyme]), t is time, in seconds. Fit single-turnover primer synthesis reactions progress curves to a single-exponential function y=A(e-kt). In the case of gp4, fit the data by numerical integration15, to a model with three NTP condensation steps, where each intermediate is in equilibrium with the enzyme, using commercially available software.
Representative Results
The results shown in Figure 1A were obtained as described in step 1 of the protocol, i.e., by manually sampling a primer synthesis reaction catalyzed by gp4 under multiple-turnover conditions. Here, a range of products after gel electrophoresis can be observed by using CTP labeled with 32P at the α-position in primer synthesis reactions (Figure 1A). The labeled precursor, CTP, shows the highest mobility and migrates toward the bottom of the gel, followed by slower migrating species corresponding to the di-, tri-, and tetraribonucleotides synthesized by gp4. Depending on the length of incubation, and template sequence context, additional labeled products corresponding to higher oligomers (pentaribonucleotides, etc.) can be observed. A reaction progress curve is plotted by first determining the concentration of full-length primers (that is, tetra- and pentaribonucleotides) at each timepoint and plotting the concentration of primers synthesized as a function of time.
While obtaining data from steady-state, multiple-turnover experiments is relatively easy, the information content of the data is somewhat limited, as the data is greatly defined by substrate binding and product release events, i.e., these events typically dominate or greatly determine KM and kcat values16. If the reaction is observed at short enough time-points, before the steady-state is established (in other words, the pre-steady state) far greater information content can be derived from kinetic experiments. Examination of reactions in the pre-steady state can provide a trove of information. For example, if a "pre-steady state burst" of product formation is observed, it is good indication that product release is a rate-limiting step in the reaction. Furthermore, one can extract the rate of product formation (or the chemistry step) from the pre-steady state phase of the reaction. In contrast, if a burst of product formation is not observed, then it is evidence that the steady state rate is limited either by the chemical step, or a step preceding it and that the release of product is rapid in comparison.
When a rapid-quench instrument is used to examine the reaction catalyzed by gp4 in time scales in the range of seconds down to a few milliseconds (step 2 of the protocol), it is evident that primer accumulation is not linear, but displays a biphasic profile (Figure 1B, solid line). This is a diagnostic result demonstrating evidence for a pre-steady state burst of product accumulation, which suggests that product formation is rapid and that release is rate-limiting in the steady state. Other enzyme systems may not show this behavior. For example, a hypothetical progress curve is presented which would result if primer formation by gp4 did not proceed with a pre-steady state burst (Figure 1B, dotted line). In cases such as these, the interpretation would be that product formation, or a step preceding it, is the rate-limiting step.
Another embodiment of pre-steady state kinetics is the single-turnover experiment, where a low concentration of labeled substrate is bound by saturating amounts of enzyme (as well as DNA and CTP, in the case of gp4). This type of experiment is uniquely suited to provide information about the formation and decay of intermediate species during the conversion of substrate to product. In the gp4-catalyzed reaction (step 3 of the protocol), primer formation is the result of three nucleotidyl transfer reactions. Recently, we used single-turnover primer synthesis reactions to reveal information about primer intermediates8 (Figure 1C) by fitting the time-dependent accumulation and disappearance of the di-, tri-, and tetraribonucleotide products to a sequential model of three nucleotidyl transfer steps, where the ribonucleotide intermediates are in equilibrium with gp4.
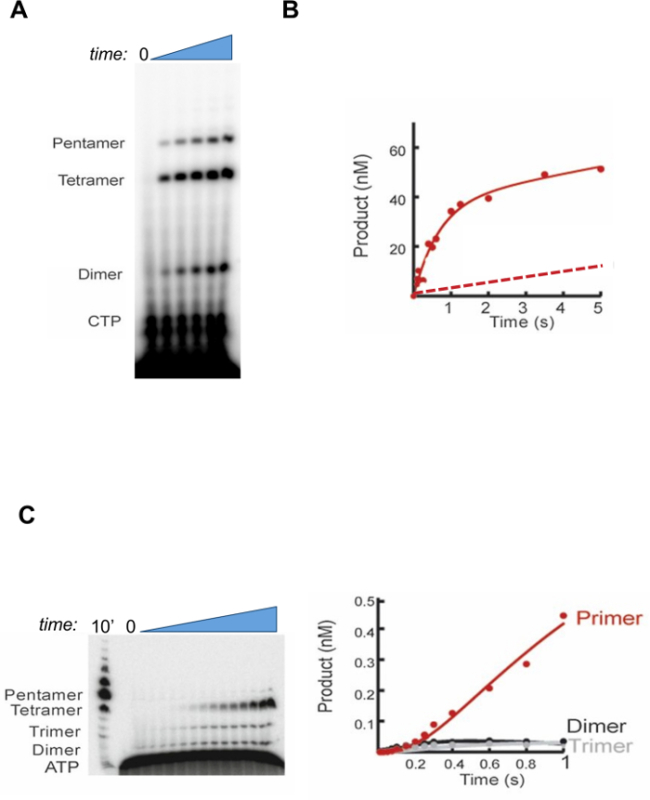 Figure 1:Kinetic analysis of primer synthesis by the T7 primase-helicase. (A) Time-course of primer formation by gp4 on a ssDNA with a single PRS. Samples were removed prior to addition of MgCl2 (time=0) and sampled at intervals following its addition. Products are indicated to the left of the gel image. (B) Solid line: Primer formation by gp4 exhibits a pre-steady state burst. Values are the mean of at least three independent experiments. Dotted line: Representation of a reaction progress curve if the gp4-catalyzed reaction proceeded without a pre-steady state burst. Modified from 8. (C) Single-turnover primer synthesis by gp4. Left: time-course of tetramer formation. Right: plot of intermediate formation versus time. Values are the mean of at least three independent experiments. Data were fit by numerical integration; solid lines show the fit. Modified from 8. Please click here to view a larger version of this figure.
Figure 1:Kinetic analysis of primer synthesis by the T7 primase-helicase. (A) Time-course of primer formation by gp4 on a ssDNA with a single PRS. Samples were removed prior to addition of MgCl2 (time=0) and sampled at intervals following its addition. Products are indicated to the left of the gel image. (B) Solid line: Primer formation by gp4 exhibits a pre-steady state burst. Values are the mean of at least three independent experiments. Dotted line: Representation of a reaction progress curve if the gp4-catalyzed reaction proceeded without a pre-steady state burst. Modified from 8. (C) Single-turnover primer synthesis by gp4. Left: time-course of tetramer formation. Right: plot of intermediate formation versus time. Values are the mean of at least three independent experiments. Data were fit by numerical integration; solid lines show the fit. Modified from 8. Please click here to view a larger version of this figure.
Discussion
The most important factor in performing these experiments is the availability of highly active purified enzyme. During our work with gp4, for example, we found that storage of the purified enzyme in buffer (20 mM potassium phosphate, pH 7.4, 0.1 mM EDTA, 1 mM DTT) containing 50% glycerol at -20 °C leads to a reduction in the specific activity of the preparation over a few months. Therefore we now flash-freeze small aliquots of purified gp4 in 25 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.1 mM EDTA, 1 mM TCEP, and 10% glycerol using liquid nitrogen and store them at -80 °C. While using the rapid-quench flow instrument, especially during acquisition of the first few samples, it is important to allow the reactants to reach the reaction temperature (room temperature) prior to starting the reaction. Also, great care should be taken to thoroughly clean the tubing (step 2.2.4 of the protocol) after each time-point, as any remaining quench reagent in the line will greatly inhibit enzyme activity, leading to erroneous results. Another consideration involves the composition of the polyacrylamide gel and its handling. The products in primer synthesis reactions catalyzed by gp4 do not resolve well in gels with less than at least 20% acrylamide. In fact, in 15% gels, the nucleotide substrate and all reaction products migrate as an indistinct zone of radioactivity. Therefore, it is highly advisable to adjust the acrylamide percentage accordingly if small ribonucleotide products are to be analyzed. A major disadvantage of using gels containing a high percentage of acrylamide is that they do not stick well to paper; therefore great care must be used to transfer them to chromatography paper for drying. Finally, gels containing a high percentage of acrylamide tend to crack in vacuum-powered gel dryers. We find that lowering the drying temperature to 65 °C leads to less cracking of gels. Alternatively, the gels can be exposed to phosphorimager screens without drying.
There are a great number of variations in reaction conditions and reagents that can be tested using this protocol, and which we are pursuing in our studies of gp4, ranging from testing the enzymatic behavior of mutant enzymes, examining the effect of addition of putative small-molecule inhibitors or activators, or alternative substrates, such as nucleotide analogs, as well as alternative DNA templates, or metal ion cofactors. The protocol described above uses room temperature as the reaction temperature. If another reaction temperature is desired, most rapid-quench instruments contain a valve that allows one to connect the instrument to a heat-controlled water bath. In cases where electrophoretic product separation is not satisfactory, particularly if the reaction buffer contains a high concentration (>200 mM) of salt, it is advisable to treat samples after quenching with 1-10 units of alkaline phosphatase and incubate at 37 °C for at least 30 minutes. This will remove the terminal triphosphates from NTPs and ribonucleotide products and enhance their electrophoretic separation. However, this is only practical if the nucleotide is labeled at the α position.
Usage of a rapid-quench instrument is not convenient for high-throughput analysis. An experienced user can perform the rapid-quench flow protocol described here, starting from sample preparation, collecting approximately 18 time-points, and cleaning the instrument in about two hours. An additional time constraint is the time required for gel electrophoresis, gel exposure, and data analysis. Another limitation is that a typical experiment requires 500 to 1,000 µL of reaction volume, and obtaining sufficient enzyme could be challenging in the case of proteins that are not expressed efficiently.
There is great interest in developing assays that enable high-throughput screening of primase activity, particularly to develop novel antibacterial therapeutics17,18. Despite advances in developing non-radioactive, continuous assays for primase activity, these assays suffer from two major limitations and thus preclude their use in rapid kinetic investigations, i.e., lack sufficient time resolution and product sensitivity. Therefore, the radioactive, discontinuous assay described here is currently the gold standard in the kinetic examination of primase activity by gp4, and DNA primases in general. More sophisticated embodiments of the protocol described here can be used in the determination of the kinetic pathway of an enzyme-catalyzed reaction using a rapid-quench flow instrument, for example, in pulse-chase experiments intended to determine the kinetic partitioning of substrates.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank C.C.R. laboratory members for comments and S. Moskowitz for figure preparation. This work was supported by National Institutes of Health Grants F32GM101761 (A.J.H.) and GM54397 (C.C.R.).
References
- Kornberg A, Baker TA. DNA replication. 2nd. W.H. Freeman; 1992. [Google Scholar]
- Hamdan SM, Richardson CC. Motors, switches, and contacts in the replisome. Annu Rev Biochem. 2009;78:205–243. doi: 10.1146/annurev.biochem.78.072407.103248. [DOI] [PubMed] [Google Scholar]
- Mendelman LV, Notarnicola SM, Richardson CC. Roles of bacteriophage T7 gene 4 proteins in providing primase and helicase functions in vivo. Proc Natl Acad Sci U S A. 1992;89(22):10638–10642. doi: 10.1073/pnas.89.22.10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qimron U, Lee SJ, Hamdan SM, Richardson CC. Primer initiation and extension by T7 DNA primase. EMBO J. 2006;25(10):2199–2208. doi: 10.1038/sj.emboj.7601112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Ito T, Wagner G, Richardson CC, Ellenberger T. Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol Cell. 2003;11(5):1349–1360. doi: 10.1016/s1097-2765(03)00195-3. [DOI] [PubMed] [Google Scholar]
- Kusakabe T, Richardson CC. Gene 4 DNA primase of bacteriophage T7 mediates the annealing and extension of ribo-oligonucleotides at primase recognition sites. J Biol Chem. 1997;272(19):12446–12453. doi: 10.1074/jbc.272.19.12446. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Zhu B, Hamdan SM, Richardson CC. Mechanism of sequence-specific template binding by the DNA primase of bacteriophage T7. Nucleic Acids Res. 2010;38(13):4372–4383. doi: 10.1093/nar/gkq205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AJ, Lee SJ, Richardson CC. Primer release is the rate-limiting event in lagging-strand synthesis mediated by the T7 replisome. Proc Natl Acad Sci U S A. 2016;113(21):5916–5921. doi: 10.1073/pnas.1604894113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor S, Richardson CC. A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc Natl Acad Sci U S A. 1995;92(14):6339–6343. doi: 10.1073/pnas.92.14.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YT, Tabor S, Bortner C, Griffith JD, Richardson CC. Purification and characterization of the bacteriophage T7 gene 2.5 protein. A single-stranded DNA-binding protein. J Biol Chem. 1992;267(21):15022–15031. [PubMed] [Google Scholar]
- Frick DN, Richardson CC. DNA primases. Annu Rev Biochem. 2001;70:39–80. doi: 10.1146/annurev.biochem.70.1.39. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Richardson CC. Essential lysine residues in the RNA polymerase domain of the gene 4 primase-helicase of bacteriophage T7. J Biol Chem. 2001;276(52):49419–49426. doi: 10.1074/jbc.M108443200. [DOI] [PubMed] [Google Scholar]
- Cao W, De La Cruz EM. Quantitative full time course analysis of nonlinear enzyme cycling kinetics. Sci Rep. 2013;3:2658. doi: 10.1038/srep02658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MH, Zhao KY. A simple method for determining kinetic constants of complexing inactivation at identical enzyme and inhibitor concentrations. FEBS Lett. 1997;412(3):425–428. doi: 10.1016/s0014-5793(97)00771-0. [DOI] [PubMed] [Google Scholar]
- Johnson KA. Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods Enzymol. 2009;467:601–626. doi: 10.1016/S0076-6879(09)67023-3. [DOI] [PubMed] [Google Scholar]
- Johnson KA. A century of enzyme kinetic analysis, 1913. FEBS Lett. 2013;587(17):2753–2766. doi: 10.1016/j.febslet.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas T, Resto-Roldan E, Sawyer SK, Artsimovitch I, Tsodikov OV. A novel non-radioactive primase-pyrophosphatase activity assay and its application to the discovery of inhibitors of Mycobacterium tuberculosis primase DnaG. Nucleic Acids Res. 2013;41(4):e56. doi: 10.1093/nar/gks1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal G, Doig P. Bacterial DNA replication enzymes as targets for antibacterial drug discovery. Expert Opin Drug Discov. 2012;7(4):327–339. doi: 10.1517/17460441.2012.660478. [DOI] [PubMed] [Google Scholar]