

Abstract
Detection of genomic changes at single cell resolution is important for characterizing genetic heterogeneity and evolution in normal tissues, cancers, and microbial populations. Traditional methods for assessing genetic heterogeneity have been limited by low resolution, low sensitivity, and/or low specificity. Single cell sequencing has emerged as a powerful tool for detecting genetic heterogeneity with high resolution, high sensitivity and, when appropriately analyzed, high specificity. Here we provide a protocol for the isolation, whole genome amplification, sequencing, and analysis of single cells. Our approach allows for the reliable identification of megabase-scale copy number variants in single cells. However, aspects of this protocol can also be applied to investigate other types of genetic alterations in single cells.
Keywords: Genetics, Issue 120, copy number alteration, aneuploidy, genomic heterogeneity, single cell, whole genome amplification, whole genome sequencing
Introduction
Alterations in DNA copy number can range in size from several base pairs (copy number variants) (CNVs) to entire chromosomes (aneuploidy). Copy number alterations affecting large regions of the genome can have significant phenotypic consequences by altering the expression of up to thousands of genes1,2. CNVs that are present in all cells of a population can be detected by bulk sequencing or microarray-based methods3,4. However, populations can also be genetically heterogeneous, with CNVs existing in a subset of the population or even single cells. Genetic heterogeneity is common in cancer, driving tumor evolution, and also present in normal tissues, with unknown consequence5,6,7,8,9,10.
Traditionally, genetic heterogeneity was assessed either by cytologic approaches or bulk sequencing. Cytologic approaches, such as fluorescence in situ hybridization (FISH), chromosome spreads, and spectral karyotyping (SKY), have the benefit of identifying alterations present in individual cells but have high error rates due to artifacts of hybridization and spreading11. These approaches are also limited in their resolution—only revealing copy number changes spanning several megabases. Sequencing or microarrays of bulk DNA, though higher in accuracy and resolution, is less sensitive. In order to detect genetic heterogeneity by population-based approaches, the variants must be present in a substantial fraction of cells in the population. The emergence of methods to amplify the genomic DNA from single cells has made it possible to sequence the genome of single cells. Single cell sequencing has the advantages of high resolution, high sensitivity, and, when appropriate quality control methods are applied, high accuracy12.
Here, we describe a method for detecting megabase-scale copy number alterations in single cells. We isolate single cells by microaspiration, amplify genomic DNA using linker-adaptor PCR, prepare libraries for next-generation sequencing, and detect copy number variants by both hidden Markov model and circular binary segmentation.
Protocol
1. Isolating Single Cells
- Prepare the microaspirator
- Remove the clear plastic end from the aspirator tube assembly and insert the narrow end into one end of a 1 foot-long PVC tubing with 3/16" inner diameter.
- Insert the outlet of a 0.2 µm syringe filter into the other end of the 1 foot-long PVC tubing with 3/6" inner diameter.
- Insert the inlet of the 0.2 µm syringe filter into one end of a 6 inch-long PVC tubing with 5/16" inner diameter.
- Optional: Cut the 6 inch-long PVC tubing with 5/16" inner diameter in half and insert an in-line water trap between the two halves.
- Break a 5 mL plastic serological pipette at the 1 mL graduation and insert the broken end into the open end of the PVC tubing with 5/16" inner diameter.
- Insert the outlet of the 5 mL plastic serological pipette into the flexible end of an aspirator tube assembly (where the clear plastic end had been removed).
- For storage, cover the red mouthpiece of the aspirator tube assembly with a sterile plastic tube.
- Draw out a glass capillary tube to an inner diameter of 10-30 µm by bringing the middle of the tube near a Bunsen burner flame and applying tension on either end of the tube. Break the drawn tube in the middle to create two potential aspirator needles. Repeat this step with several tubes as only some tubes will end up with an inner diameter that is appropriate for picking single cells.
- For storage, place two strips of modeling clay in a 15 cm Petri dish and secure the aspirator needles across the strips.
- Pick cells
- Prepare a single cell suspension as appropriate for the cell type and experiment. For example, to prepare adherent cells such as human fibroblast cell lines, harvest cells by trypsinization and transfer cells to a conical tube containing appropriate media.
- Before, during, or after preparing the single cell suspension (depending on how long it takes to prepare the single cell suspension), setup the hood for whole genome amplification.
- Spray down the surface of the hood, pipette tip boxes, and pipette tips with 10% bleach and wipe with a paper towel. Repeat this step with 70% ethanol.
- Add 8 µL of water from the whole genome amplification (WGA) kit to individual wells of a 96-well PCR plate, one well for each cell to be sequenced. Cover the 96-well PCR plate with a lid from a 96-well tissue culture plate and place on ice.
- Add 1,000 cells to 10 mL media or phosphate buffered saline (PBS) in a 15 cm Petri dish and place the dish on ice to prevent the cells from adhering to the dish.
- Bring the cells in the Petri dish and the 96-well PCR plate with lid on ice to a light microscope with 10X objective.
- Increase the opening of the aspirator needle by gently tapping the drawn out end of the aspirator needle on a hard surface such that the tip breaks off. Insert the wide end of the aspirator needle into the clear end of the microaspirator.
- Place the Petri dish containing cells on the microscope stage. Place the red mouthpiece of the aspirator in the mouth. Use one hand to move the aspirator needle and the other hand to move the Petri dish containing cells. Identify single cells to be sequenced.
- Using mouth suction, draw a single cell into the aspirator needle along with ~1-2 µL of media or PBS. Transfer the cell into the 8 µL of water inside a single well of the 96-well PCR plate. Avoid introducing bubbles when transferring the cell.
- Repeat step 1.2.7 until the desired number of cells have been isolated. Keep the PCR plate on ice and covered with lid in between picking cells. Mark the wells that have received cells.
- While picking single cells, thaw the 10X single cell lysis and fragmentation buffer from the whole genome amplification kit. After picking the desired number of cells, proceed immediately to whole genome amplification.
2. Whole Genome Amplification
To prevent contamination during whole genome amplification, add all reagents inside a tissue culture hood, use pipette tips with filters, and change the pipette tip in between wells.
Prepare a working lysis and fragmentation buffer solution from the whole genome amplification (WGA) kit. For each set of up to 32 cells, combine 32 µL of 10x single cell lysis and fragmentation buffer and 2 µL of proteinase K solution in a microcentrifuge tube. Vortex the tube to mix the solutions.
Add 1 µL of working lysis and fragmentation buffer solution to each well and pipette up and down to mix.
Cover the plate and seal all wells with a plastic film. Briefly centrifuge the plate in a mini plate spinner.
Thermal cycle as follows: 50 °C, 1 h and 99 °C, 4 min.
Cool the plate on ice and briefly centrifuge the plate in a mini plate spinner.
Prepare a working library preparation buffer solution from the whole genome amplification kit. For each cell, combine 2 µL of 1x single cell library preparation buffer and 1 µL of library stabilization solution in a microcentrifuge tube. Prepare the working solution for several cells in the same microcentrifuge tube.
Remove the plastic film and add 3 µL of working library preparation buffer solution to each well. Pipette the contents of the well up and down to mix. Replace the plastic film. NOTE: The plastic film can be reused throughout the whole genome amplification process until the wells start to bore holes in the film. When this occurs, switch to a new plastic film.
Briefly centrifuge the plate in a mini plate spinner and incubate at 95 °C for 2 min.
Cool the plate on ice and briefly centrifuge the plate in a mini plate spinner.
Remove the plastic film, add 1 µL library preparation enzyme to each well, and pipette the contents of the well up and down to mix. Keep the library preparation enzyme on ice or in a cold block throughout this step. Replace the plastic film.
Briefly centrifuge the plate in mini plate spinner and thermal cycle as follows: 16 °C, 20 min, 24 °C, 20 min, 37 °C, 20 min, 75 °C, 5 min, and hold at 4 °C.
Cool the plate on ice and briefly centrifuge the plate in a mini plate spinner.
Prepare a working amplification mix from the whole genome amplification kit. For each cell, combine 48.5 µL water, 7.5 µL 10x amplification master mix, and 5 µL WGA DNA polymerase in a microcentrifuge tube. Prepare the working mix for several cells in the same microcentrifuge tube. Keep the working mix on ice.
Remove the plastic film, add 61 µL working amplification mix to each well, and pipette contents of well up and down to mix. Keep the working amplification mix on ice or in a cold block throughout this step. Replace the plastic film.
Briefly centrifuge the plate in a mini plate spinner and thermal cycle as follows: 95 °C, 3 min, 25 cycles of 94 °C, 30 sec and 65 °C, 5 min, and hold at 4 °C.
Transfer 60 µL of each sample to a separate microcentrifuge tube and store at - 20 °C for use in step 3.
Add DNA loading dye to the remaining volume of each sample (i.e. 3 µL of 6x DNA loading dye to 15 µL of sample) and run 5 µL from each reaction on a 1% agarose gel. NOTE: Cells that successfully amplified will appear as a smear from 250 to 1,000 bp. Samples that do not show a smear or only show a faint smear are unlikely to yield useful sequencing data.
3. Sequencing
- Purify samples with paramagnetic beads.
- Thaw DNA samples on ice. Vortex paramagnetic beads until the solution is homogenous and incubate the beads at room temperature for at least 30 min.
- Transfer 20 µL of each DNA sample into a clean microcentrifuge tube. The remainder of the sample can be stored at -20 °C.
- Add 30 µL (1.5x) of paramagnetic beads to each DNA sample and vortex to mix. Incubate the samples at room temperature for 10 min. Leave the stock solution of beads at room temperature for use in step 3.4.
- Place the tubes on a magnetic tube strip for 2 min, or until the beads form a pellet and the supernatant is clear.
- Using a P200 pipette, remove as much of the supernatant as possible without disturbing the beads.
- Add 180 µL of 80% ethanol to the DNA-bead mixture. Rotate the tubes several times relative to the magnet to "rinse" the beads through the ethanol solution.
- Using a P200 pipette, remove as much of the ethanol wash as possible without disturbing the beads.
- Repeat 3.1.6 and 3.1.7.
- Air dry the beads for approximately 10 min or until no more ethanol is visible. Proceed to the next step when the beads appear cracked or fall off the wall of the tube.
- Remove the samples from the magnetic strip. Add 40 µL of 10 mM Tris pH 8.0 to elute the DNA from the beads and vortex the samples to resuspend the beads. Incubate for 2 min at room temperature.
- Briefly centrifuge the samples to collect the liquid at the bottom of the tube and place the tubes on a magnetic tube strip for at least two min. Transfer the eluent into clean microcentrifuge tubes without disturbing the beads.
- Sample normalization
- Quantify the concentration of each sample using a spectrophotometer. The concentrations should range from 10 to 30 ng/µL.
- Dilute each sample to 0.2 ng/µL using 10 mM Tris pH 8.0. The following steps will require a minimum input of 5 µL from this dilution.
- Library preparation
- Prepare sequencing libraries by following the instructions of the library preparation kit13.
- Final cleanup
- Clean the samples according to step 3.1. For read lengths of 50 bp, 75 bp, or 150 bp, use a ratio of bead to sample volume of 1.5x, 1x, or 0.6x, respectively. Elute with 15 µL of 10 mM Tris pH 8.0.
- Run the samples on a fragment analyzer to check the size distribution of the library14. The size distribution should be evenly spread from 150 to 900 bp.
- Quantify samples using qPCR
- Using a library quantification kit, set up a qPCR reaction according to the kit instructions15. Leave a column blank to add positive standards (DNA standards from the kit) and negative standards (water or other blank solution).
- Thermal cycle as follows: 95 °C, 5 min (ramp rate of 4.8 °C/sec) and 35 cycles of 95 °C, 30 sec (ramp rate of 4.8 °C/sec) and 60 °C, 45 sec (ramp rate of 2.5 °C/sec).
- Pooling
- Determine how many lanes on the flowcell are needed and divide the samples into groups, with one group per lane.
- For each group of samples, choose the sample with the lowest concentration based on the qPCR data from step 3.5.2 and normalize all samples in that group to that concentration using 10 mM Tris pH 8.0.
- Pool the samples in each group together.
- Sequencing
- Load pools on next-generation sequencing machine following standard procedures16.
4. Data Analysis
NOTE: A Unix-based environment is required to run the programs and scripts in this section. Install the software mentioned in the protocol following their installation guides. All scripts can be found at https://sourceforge.net/projects/singlecellseqcnv/.
Trim the reads to 40-nt using fastx_trimmer from FASTX-Toolkit version 0.0.1317. fastx_trimmer -Q33 -i example.fastq -l 40 -o example.trim.fastq
Align the reads to the appropriate reference genome (mm9 for mouse, hg19 for human) using BWA version 0.6.1 with default options18. bwa aln mm9.fa example.trim.fastq >example.sai bwa samse mm9.fa example.sai example.trim.fastq >example.sam
Remove alignments to chrM and random chromosomes, then sort and index the resulting BAM file using SAMTools version 0.1.1919. grep -v -w chrM example.sam |grep -v random >example.filtered.sam samtools view -uSh example.filtered.sam | samtools sort - example.filtered samtools index example.filtered.bam
- Run HMMcopy to detect CNVs20
- Use gcCounter in HMMcopy to generate a GC percentage reference file for the genome. Use option "-w 500000" to specify window size. Use the same version of the fasta reference file used in step 4.2, but make sure chrM and random chromosomes are removed from the fasta file. gcCounter -w 500000 mm9.fa >mm9_gc.wig
- Use generateMap.pl in HMMcopy to generate a wiggle file for mappability. Use option "-w 40" to specify read length. Use the same fasta reference file used in step 4.4.1. generateMap.pl -b mm9.fa generateMap.pl -w 40 -i mm9.fa mm9.fa -o mm9.bigwig
- Use mapCounter in HMMcopy to generate a mappability reference file for the genome. Use option "-w 500000" to specify window size. mapCounter -w 500000 mm9.bigwig >mm9_map.wig
- Use readCounter in HMMcopy to generate a wiggle file for each BAM file. readCounter -w 500000 example.filtered.bam >input.wig
- Modify the paths to the reference files in the R scripts (run_hmmcopy.mm9.r or run_hmmcopy.hg19.r), use the files generated above in 4.4.1 (e.g., mm9_gc.wig) and 4.4.3 (e.g., mm9_map.wig) for the variables gfile and mfile, which refer to the GC percentage reference file and mappability reference file, respectively. Then run the provided R script "run_hmmcopy.mm9.r" or "run_hmmcopy.hg19.r". R CMD BATCH run_hmmcopy.mm9.r or R CMD BATCH run_hmmcopy.hg19.r
- For batch processing of a set of BAM files, put the BAM files in a single folder and run the provided script "HMMpipe.pl" in the package to call segments and compute variability scores (VS). Follow the format: perl HMMpipe.pl Folder_to_BAMfiles mm9
- Run DNAcopy to detect CNVs21.
- Use SAMtools version 0.1.19 to extract uniquely mapped reads from each BAM file. samtools view -h -F 0x0004 example.filtered.bam | egrep -i "^@|XT:A:U" |samtools view -Shu - >example.mapped.bam
- Use MarkDuplicates in Picard version 1.94 to mark PCR duplicates in the BAM file22. java -jar MarkDuplicates.jar INPUT=example.mapped.bam OUTPUT=example.nondup.bam METRICS_FILE=example.dup REMOVE_DUPLICATES=true
- Use coveragebed in bedtools version 2.17.0 to count mapped reads in each of the predefined dynamic 500kb mappable windows in the provided BED file "mm9.500k.dynamic.win.bed" or "hg19.500k.dynamic.win.bed"23. coverageBed -abam example.nondup.bam -b mm9.500k.dynamic.win.bed -counts | sort -k1,1 -k2,2n >example.nondup.counts
- Use the provided Perl script "normalizeGC.pl" to normalize the read counts based on the provided GC content reference file "mm9.500k.dynamic.win.fa.gc.txt" or "hg19.500k.dynamic.win.fa.gc.txt". perl normalizeGC.pl mm9.500k.dynamic.win.fa.gc.txt example.nondup.counts >example.bam.norm.counts
- Use the provided R script "dnacopy.r" to call the segments. R CMD BATCH dnacopy.r
- Identify CNVs
- Exclude cells for which the VS calculated in 4.4.6 exceeds 0.26.
- Filter the segments called by HMMcopy in 4.4.6 to include only those for which the median log2 ratio is greater than 0.4 (putative gain) or less than -0.35 (putative loss).
- Filter the segments called by DNAcopy in 4.5.5 to include only those for which the segment mean is greater than 1.32 or less than 0.6.
- Overlap the segments from 4.6.2 and 4.6.3, calling CNVs only in regions where both HMMcopy and DNAcopy identify a putative gain or putative loss.
Representative Results
The assembled aspirator should appear similar to the one in Figure 1A. The needle should be drawn such that it is sufficiently wide to accommodate a single cell but not so wide that a large volume is drawn up with the single cell. Aspirating single cells is easiest when there are between one and five cells in a 10X field (Figure 1B).
If whole genome amplification is successful, the sample will appear as a smear on an agarose gel (Figure 2, lanes 1, 2, 4, 5, 6, and 7). A faint or absent smear indicates a failed amplification reaction and the sample should not be sequenced (Figure 2, lanes 3 and 8).
Following library preparation, the fragment size distribution of the samples should be assessed by capillary electrophoresis on a fragment analyzer. Successfully prepared libraries will have a rather even distribution of fragment sizes from 150 to 900 bp (Figure 3, A,B). Failed library preparation will result in a skewed fragment size distribution and such libraries should not be sequenced (Figure 3C).
Processing sequencing data through the hidden Markov model (HMMcopy) and circular binary segmentation (DNAcopy) will parse the genome of each cell into segments of estimated copy number. These segments can then be filtered to identify those with an estimated copy number consistent with gain or loss in a single cell (Table 1). These filtered segments from HMMcopy and DNAcopy should then be overlapped to identify high-confidence CNVs.
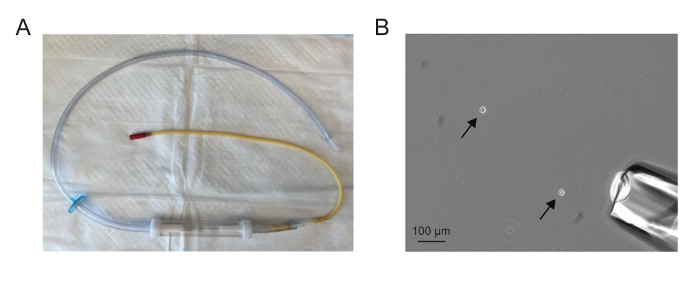 Figure 1. Single cell isolation. (A) An assembled microaspirator. (B) A 10X field showing dissociated cells (arrows) and microaspirator needle (lower right corner). Aspirating single cells is easiest when there are between one and five cells in a 10X field. Please click here to view a larger version of this figure.
Figure 1. Single cell isolation. (A) An assembled microaspirator. (B) A 10X field showing dissociated cells (arrows) and microaspirator needle (lower right corner). Aspirating single cells is easiest when there are between one and five cells in a 10X field. Please click here to view a larger version of this figure.
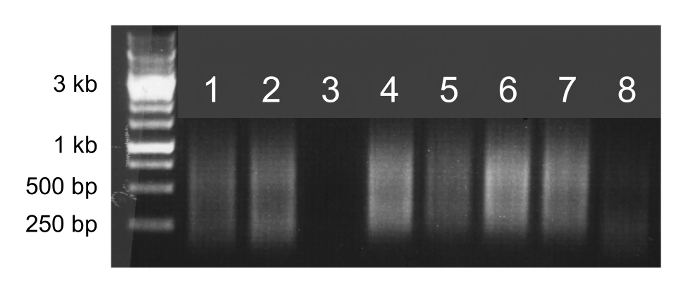 Figure 2. Whole genome amplification. Agarose gel electrophoresis of 5 µl of whole genome amplification products. Samples that successfully amplify will appear as bright smears from 100 bp to 1 kb (lanes 1, 2, 4, 5, 6, and 7) and can be sequenced. Samples that do not successfully amplify will produce faint smears or no smear (lanes 3 and 8) and should not be sequenced. Please click here to view a larger version of this figure.
Figure 2. Whole genome amplification. Agarose gel electrophoresis of 5 µl of whole genome amplification products. Samples that successfully amplify will appear as bright smears from 100 bp to 1 kb (lanes 1, 2, 4, 5, 6, and 7) and can be sequenced. Samples that do not successfully amplify will produce faint smears or no smear (lanes 3 and 8) and should not be sequenced. Please click here to view a larger version of this figure.
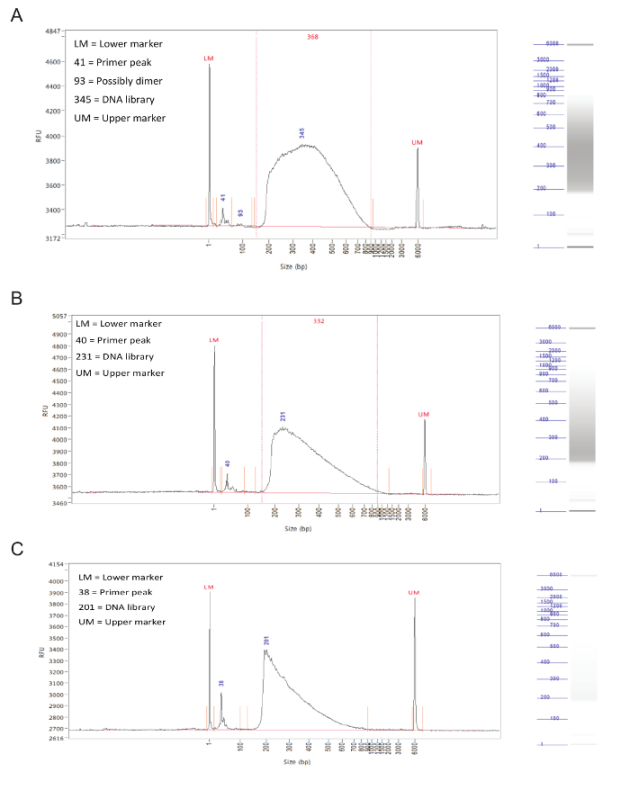 Figure 3. Library preparation. Representative results from a fragment analyzer. The graphs show fragment size (in bp) on X axis and relative fluorescence units (RFU) on Y axis. To the right of each graph is a simulated gel lane. (A) Results for an ideal sample, with an even distribution between 150 and 900 bp and without sharp peaks or bias toward one side. This sample is acceptable for sequencing. (B) Results from an okay sample, with a size distribution skewed toward lower fragment sizes. While this is not optimal, the sample can still be sequenced. (C) Results from a failed sample, with predominantly small fragment sizes. This is likely caused by prolonged incubation during the tagmentation step of library preparation. This sample should not be sequenced. Please click here to view a larger version of this figure.
Figure 3. Library preparation. Representative results from a fragment analyzer. The graphs show fragment size (in bp) on X axis and relative fluorescence units (RFU) on Y axis. To the right of each graph is a simulated gel lane. (A) Results for an ideal sample, with an even distribution between 150 and 900 bp and without sharp peaks or bias toward one side. This sample is acceptable for sequencing. (B) Results from an okay sample, with a size distribution skewed toward lower fragment sizes. While this is not optimal, the sample can still be sequenced. (C) Results from a failed sample, with predominantly small fragment sizes. This is likely caused by prolonged incubation during the tagmentation step of library preparation. This sample should not be sequenced. Please click here to view a larger version of this figure.
| HMMcopy | |||||||
| Sample | Segment | Chr | Start | End | State | Median | |
| D15-4998 | 23 | chr8 | 144,500,001 | 146,500,000 | 6 | 0.5008794 | |
| D15-4998 | 29 | chr10 | 67,000,001 | 134,500,000 | 6 | 0.4031945 | |
| D15-4998 | 52 | chr19 | 1 | 20,000,000 | 6 | 0.4616884 | |
| D15-4998 | 57 | chrY | 1 | 59,500,000 | 2 | -1.506532 | |
| DNAcopy | |||||||
| Sample | Chrom | Start bin | End bin | Start | End | Seg mean | Seg med |
| D15-4998 | chr10 | 88 | 197 | 62,612,945 | 129,971,511 | 1.4688 | 0.157 |
| D15-4998 | chr19 | 0 | 31 | 0 | 28,416,392 | 1.4141 | 0.1674 |
| D15-4998 | chrX | 77 | 126 | 51,659,160 | 95,343,369 | 1.3548 | 0.1874 |
| D15-4998 | chrY | 0 | 14 | 0 | 23,805,358 | -2.7004 | 0.3591 |
Table 1. Data analysis. Filtered segments generated by HMMcopy and DNAcopy from a single cell. Overlapping these two results reveals a gain on chromosome 10 from 67 to 130 Mb as well as a gain on chromosome 19 from 0 to 20 Mb. We have found that gains on the proximal portion of chromosome 19 are an artifact of single cell sequencing 12.
Discussion
Traditionally, identifying CNVs and aneuploidy at the level of single cells required cytologic methods such as FISH and SKY. Now, single cell sequencing has emerged as an alternative approach for such questions. Single cell sequencing has advantages over FISH and SKY as it is both genome-wide and high resolution. Moreover, when appropriate quality control methods are applied, single cell sequencing can provide a more reliable assessment of CNVs and aneuploidy as it is not susceptible to the hybridization and spreading artifacts inherent to FISH and SKY. However, many of the recent applications of single cell sequencing have not been substantiated by thorough assessment of the sensitivity and specificity of the methods and analyses. Indeed, some of the analytic approaches used by other studies are associated with high frequencies (>50%) of false positive CNV calls12. The approach we describe has been rigorously tested using cells of known CNV burden in order to determine true and false discovery rates and optimize the sensitivity and specificity of CNV detection12. Using the quality control and analytic approaches described in this protocol, approximately 20% of 5 Mb gains, 75% of 5 Mb losses, and all CNVs exceeding 10 Mb can be detected. Though determining the false discovery rate of single cell sequencing is difficult, we have estimated it to be less than 25%. This protocol can be applied to cells from a variety of sources, the scripts can be modified to adjust the resolution of CNV detection, and the protocol can be adapted to identify other types of genomic alterations.
There are a variety of means of dissociating fresh tissues into single cells, and many publications describe procedures optimized for specific tissues, such as skin24 and brain25. We prefer to isolate single cells by microaspiration as it allows for visual assessment of each cell to be sequenced. However, it is also possible to isolate single cells by fluorescence-activated cell sorting (FACS)26 and microfluidic devices27. If single cell isolation and whole genome amplification is performed manually, it is reasonable to isolate and amplify up to forty cells in a single sitting. In order to obtain high quality single cell sequencing data, it is crucial that the amplification of single cell genomes is uniform and complete. We find that the quality of single cells isolated as well as the efficiency of lysis and amplification has a significant impact on the quality of sequencing data. As such, the cells should be harvested from their native environment just prior to isolation and whole genome amplification should begin immediately after cells are isolated. Moreover, the lysis and fragmentation step should be followed exactly as described in the steps 2.3-2.6.
The algorithms can be adjusted to change the resolution of CNV detection, with opposing effects on sensitivity and specificity12. It is also possible to adjust the thresholds to detect whole-chromosome aneuploidy in the setting of tetraploidy11. However, we find that our approach is limited to detecting CNVs greater than 5 Mb, as noise introduced during whole genome amplification complicates the detection of smaller variants12. Future improvements in whole genome amplification approaches should ultimately enhance the resolution of CNV detection using single cell sequencing.
Single cell sequencing allows for investigation of not only copy number alterations but also single nucleotide variations28,29 and structural variation30. Our protocol for single cell isolation can be applied to answer these other questions. However, the choice of whole genome amplification method depends on the specific application. The method described in this protocol, which is based on polymerase chain reaction, is best suited for detecting copy number alterations because it is associated with lower levels of amplification bias32. For investigating other types of genomic alterations, such as single nucleotide polymorphisms, other methods of whole genome amplification are believed to be more suitable31,32.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Stuart Levine for comments on this manuscript. This work was supported by the National Institutes of Health Grant GM056800 and the Kathy and Curt Marble Cancer Research Fund to Angelika Amon and in part by the Koch Institute Support Grant P30-CA14051. Angelika Amon is also an investigator of the Howard Hughes Medical Institute and the Glenn Foundation for Biomedical Research. K.A.K. is supported by the NIGMS Training Grant T32GM007753.
References
- Kahlem P. Transcript Level Alterations Reflect Gene Dosage Effects Across Multiple Tissues in a Mouse Model of Down Syndrome. Genome Res. 2004;14(7):1258–1267. doi: 10.1101/gr.1951304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science. 2007;317(5840):916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Itsara A. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Human Gen. 2009;84(2):148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH. Global diversity, population stratification, and selection of human copy-number variation. Science. 2015;349(6253):aab3761. doi: 10.1126/science.aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20(1):68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast cancer res and treat. 2007;102(2):143–155. doi: 10.1007/s10549-006-9317-6. [DOI] [PubMed] [Google Scholar]
- Yates LR. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015;21(7):751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Human Gen. 2012;90(2):217–228. doi: 10.1016/j.ajhg.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Gen. 2012;44(6):642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KB. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Gen. 2012;44(6):651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, Amon A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. 2014;111(37):13409–13414. doi: 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Amon A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 2016;26(3):376–384. doi: 10.1101/gr.198937.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nextera XT DNA Library Preparation Kit. at. 2016. at: http://www.illumina.com/products/nextera_xt_dna_library_prep_kit.html.
- Advanced Analytical Fragment Analyzer. 2016. at: http://aati-us.com/product/fragment-analyzer.
- KAPA Library Quantification Kit. 2016. at: https://www.kapabiosystems.com/product-applications/products/next-generation-sequencing-2/library-quantification/
- Illumina HiSeq 2000. 2016. at: http://support.illumina.com/sequencing/sequencing_instruments/hiseq_2000.html.
- FASTX-Toolkit. 2016. at: http://hannonlab.cshl.edu/fastx_toolkit/
- Burrows-Wheeler Aligner. 2016. at: http://bio-bwa.sourceforge.net/
- SAMTools. 2016. at: http://samtools.sourceforge.net/
- HMMcopy. 2016. at: http://compbio.bccrc.ca/software/hmmcopy/
- DNAcopy. 2016. at: https://bioconductor.org/packages/release/bioc/html/DNAcopy.html.
- Picard. 2016. at: http://broadinstitute.github.io/picard/
- bedtools. 2016. at: http://bedtools.readthedocs.io/en/latest/
- Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat. Protoc. 2008;3(5):799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2007;2(6):1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- Navin N. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, Chen P. Single-Cell Genetic Analysis Using Automated Microfluidics to Resolve Somatic Mosaicism. PLoS ONE. 2015;10(8):e0135007. doi: 10.1371/journal.pone.0135007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, Xie XS. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science. 2012;338(6114):1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522(7555):179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay IC, Voet T. Single Cell Genomics: Advances and Future Perspectives. PLoS Genetics. 2014;10(1):e1004126. doi: 10.1371/journal.pgen.1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat. Rev. Gen. 2016. pp. 1–14. [DOI] [PubMed]