

Abstract
This detailed protocol outlines the implementation of image-guided, laser-based hydrogel degradation for the fabrication of vascular-derived microfluidic networks embedded in PEGDA hydrogels. Here, we describe the creation of virtual masks that allow for image-guided laser control; the photopolymerization of a micromolded PEGDA hydrogel, suitable for microfluidic network fabrication and pressure head-driven flow; the setup and use of a commercially available laser scanning confocal microscope paired with a femtosecond pulsed Ti:S laser to induce hydrogel degradation; and the imaging of fabricated microfluidic networks using fluorescent species and confocal microscopy. Much of the protocol is focused on the proper setup and implementation of the microscope software and microscope macro, as these are crucial steps in using a commercial microscope for microfluidic fabrication purposes that contain a number of intricacies. The image-guided component of this technique allows for the implementation of 3D image stacks or user-generated 3D models, thereby allowing for creative microfluidic design and for the fabrication of complex microfluidic systems of virtually any configuration. With an expected impact in tissue engineering, the methods outlined in this protocol could aid in the fabrication of advanced biomimetic microtissue constructs for organ- and human-on-a-chip devices. By mimicking the complex architecture, tortuosity, size, and density of in vivo vasculature, essential biological transport processes can be replicated in these constructs, leading to more accurate in vitro modeling of drug pharmacokinetics and disease.
Keywords: Bioengineering, Issue 119, biomimetic, tissue engineering, vasculature, biomaterials, microenvironment, microfabrication, biotransport, lymphatic, hydrogel
Introduction
The vascular system, consisting of both lymphatic and cardiovasculature, forms highly dense networks that are essential for the transport of nutrients and oxygen and for the removal of metabolic waste. Accordingly, cells residing in vascularized tissues are never more than 50-100 µm away from a vessel1. The ability to reproduce in vivo vascular architecture in vitro is critical to accurately model in vivo transport processes using engineered constructs. With a recent drive to develop organ-on-a-chip devices2 for high-throughput drug screening3,4 and disease modeling5,6, methods to create microfluidic networks that recapitulate in vivo-like transport in synthetic or natural hydrogels are drawing significant interest. To enhance the biomimicry of microtissues used in these devices, we developed an image-guided, laser-based hydrogel degradation method that utilizes three-dimensional (3D) image stacks of native vasculature as templates to generate vascular-derived microfluidic networks embedded in PEGDA hydrogels7. This protocol outlines the use of a commercially available laser-scanning confocal microscope equipped with a femtosecond pulsed laser to fabricate vascular-derived, biomimetic microfluidic networks in PEGDA hydrogels via image-guided, laser-based degradation.
Current approaches to fluidize hydrogels include the induction of vasculogenic8-10 or angiogenic10-12 endothelial cell self-assembly and microfabrication techniques to create pre-defined channels for endothelialization13-16. While self-assembled networks recapitulate the density and complex architecture of microvasculature, they are often more permeable than in vivo networks11,17,18, which may be problematic in modeling transport for drug screening applications. Self-assembled networks consist of physiologically relevant, capillary-sized vessels, but they can be difficult to integrate with bulk fluid flow due to limitations in generating larger arteriole-sized vessels. As there is no direct control over the assembly of these networks, the final architecture can vary from sample to sample, making it difficult to repeatedly produce networks with the same fluid flow and transport properties.
To generate 3D, hydrogel-embedded microfluidic networks with repeatable geometry and well-defined architecture, a number of microfabrication techniques have been developed, including modular assembly13, 3D printing of sacrificial materials16, direct-write assembly14, and omnidirectional printing15. Applying these methods, microfluidic architecture, and therefore fluid flow and transport properties, can be repeatedly fabricated across many constructs. A major limitation of these approaches, however, is the inability to create microfluidic networks with capillary-sized features, 4 to 10 µm19. Most microfabrication techniques are often limited to features ranging from 150 to 650 µm in diameter13,16. Some existing techniques are capable of generating hierarchical networks with channels across a wide diameter range, 10 to 300 µm for direct-write assembly14, and 18 to 600 µm for omnidirectional printing15, but they are limited in their ability to generate dense networks or to produce multiple microfluidic networks in close proximity within a single construct7.
To overcome some of these limitations, we developed an image-guided, laser-based hydrogel degradation technique that allows repeatable fabrication of biomimetic, hierarchical microfluidic networks that recapitulate the architecture of in vivo microvasculature. To do so, a 790 nm, 140 femtosecond (fs) pulsed laser operating at 80 MHz is raster-scanned in desired 3D locations within a hydrogel, as defined by images of in vivo vasculature. We speculate that the degradation process operates through the laser-induced optical breakdown of water, the resultant plasma formation, the subsequent rapid thermoelastic expansion of water, and the local degradation of the hydrogel as the water expands20. This mechanism varies slightly from laser-based degradation of protein-based hydrogels21-24. Unlike PEGDA, which has a low multiphoton cross-section, proteins often have a large multiphoton cross-section and are therefore degraded via a multiphoton absorption-induced chemical breakage23. To generate image-guided microfluidic networks, the laser shutter is controlled by image-derived virtual masks, which consist of a mosaic of regions of interest9 that define the microfluidic architecture. Using this approach, we have demonstrated the ability to fabricate 3D vascular-derived biomimetic microfluidic networks, which recapitulate the dense and tortuous architecture of in vivo vasculature, in order to locally control the porosity of hydrogels by altering the amount of energy delivered during degradation. We have also been able to generate two independent microfluidic networks that intertwine in close proximity (15 µm) but never directly connect7. We have also demonstrated the ability to endothelialize laser-degraded microchannels through post degradation functionalization with the integrin ligating peptide sequence, Arginine-Glycine-Aspartic Acid-Serine (RGDS), to promote endothelial cell adhesion and lumen formation7.
With this protocol, the generation of complex microfluidic networks in PEGDA hydrogels is made possible via image-guided, laser-based degradation using a commercially available microscope accessible on many university campuses. As the degradation process is guided by digital, virtual masks, this fabrication technique is amenable for the creative design of microfluidic networks, allowing its use in a wide variety of applications. We anticipate that the method described here will be most advantageous in designing biomimetic organ- and human-on-a-chip devices capable of replicating biological transport processes important in modeling drug transport2. This fabrication technique may also be of interest for the generation of in vitro disease models, including cancer metastasis5,6 and blood-brain barrier models25. As laser-based hydrogel degradation has previously been used to create tracks for the guidance of neuronal outgrowth21-23, the image-guided extension of this technique could prove useful in advanced tissue engineering strategies to position cells in user-defined 3D spatial arrangements.
Protocol
1. Generating Virtual Masks
NOTE: A 3D image stack of mouse brain microvasculature was chosen as the microfluidic model for this protocol, having been culled from a much larger dataset containing images of an entire mouse brain microvasculature. Microvascular images were acquired via knife-edged scanning microscopy (KESM)262; similar microvascular data are openly available through the KESM Mouse Brain Atlas28.
Open the image processing software29 and open and crop the 3D TIF image stack of native vasculature so that the desired microfluidic network, to be generated within the hydrogel, fits in a 450 µm x 450 µm x 1.5 mm (x by y by z) window. To do so, draw a square around the desired region and click "Image" → "Crop". Remove the slices in the z-direction by clicking "Image" → "Duplicate" and type in a desired slice range. NOTE: This ensures that the desired features fit within the field of view (x by y) (531.2 x 531.2 µm when using a 20X (NA1.0) water immersion objective with a frame size of 2,884 x 2,884 pixels and a zoom of 0.8 with a laser-scanning confocal microscope) and the working distance (z) of a 20X (NA1.0) water immersion objective. If unknown, the field of view for other systems can be determined by acquiring an image with the desired objective and settings.
Rotate the image stack so that the features are primarily aligned with the direction of raster scanning, or the x-direction, by clicking "Image" → "Transform" → "Rotate". This decreases the time needed for fabrication.
Scale the cropped image stack so that the pixel size matches or exceeds the settings used for degradation on the confocal microscope (0.184 µm/pixel when using a 20X (NA1.0) water immersion objective with a frame size of 2,884 by 2,884 pixels and a zoom of 0.8). To do this, click "Image" → "Adjust" → "Size" and type in "2446" for "Width" (i.e., 450 µm, or the size of the image divided by 0.184 µm/pixel).
Threshold/binarize the image stack by typing "Ctrl + Shift + T". Deselect "Dark Background", and click "apply" → "ok". Finish the process by clicking "Image" → "Lookup Tables" → "Invert LUT".
Using a custom algorithm (source code is available in the supplemental material of the referenced manuscript) in the programming software9, fit the binarized (white) features with a mosaic of single pixel-high quadrilaterals parallel to the direction of raster scanning (the x-direction) to generate regions of interest (ROIs) that guide the laser position and shutter97303132. NOTE: The custom algorithm9 converts each individual plane in the TIF image stack to an RLS file.
Change the file extension of the RLS files to .OVL for use during degradation.
2. Configuring the Laser-scanning Confocal Microscope
NOTE: While other laser scanning microscopes equipped with pulsed lasers can be used, the settings and protocol here describe the use of a laser scanning microscope (LSM) in conjunction with its software.
With the laser-scanning confocal microscope on, open the microscope software, and click "Start System." Select the objective "W Plan-Apochromat 20x/1.0 DIC VIS-IR M27 75mm" in the "Maintain" tab.
- Setting the "Hydrogel Visualization" Configuration
- In the "Acquisition" tab, ensure that the laser line (514 nm Argon laser) is selected and on in the "Laser" window. NOTE: This channel is used to image the hydrogel via eosin Y fluorescence for orientation purposes7.
- In the "Imaging Setup" window, set the "Mode" to "Channel Mode", set "Switch track every" to "Frame", and select "Track1".
- In the "Light Path" window, select only the Ch1 fluorescent detector, with a range of 516 to 735 nm; a main beam splitter (MBS) 458/514 filter on the visible light line; and a plate in the invisible light line. Deselect the bright-field photomultiplier tube detector, "T-PMT".
- In the "Acquisition Mode" window, check that the correct objective is selected and set the "Scan Mode" to "Frame", the "Frame Size" to "2884" in X and Y, the "Line Step" to "1", the "Number Averaging" to "1", the "Averaging Mode" to "Line", the "Averaging Method" to "Mean", the "Bit Depth" to "16 Bit", and the "Direction" to "<->" for bidirectional scanning.
- In the "Acquisition Mode" window, set the "Speed" as desired and set "Corr X" and "Y" to "0.05" or adjust as necessary for the specific microscope being used. Deselect "HDR".
- In the "Acquisition Mode" window, ensure that the "Scan Area" displays an "Image Size" of "531.2 µm x 531.2 µm" and a "Pixel Size" of "0.18", with a "Zoom" set to "0.8"; set all other "Scan Area" values to zero.
- In the "Channels" window, select "Track1" as the Ch1 fluorescent detector. Select laser line "514", with a percent power of "10.0", a "Pinhole" of "1AU", an initial "Gain (Master)" of "800", a "Digital Offset" of "0", and a "Digital Gain" of "1". NOTE: Adjust these settings as necessary for the specific microscope being used.
- Save this configuration as "Hydrogel Visualization".
- Setting the "Channel Formation" Configuration
- In the "Acquisition" tab, ensure that the laser line (790 nm 140 fs pulsed Ti:S laser) is selected and on in the "Laser" window. Set "GDD Correction" to "4000" for the maximum power output at 790 nm.
- Set the "Imaging Setup" window as outlined in step 2.2.2.
- In the "Light Path" window, ensure that no fluorescent detectors are selected, with a MBS 458/514/561/633 filter on the visible light line and a MBS 760+ filter on the invisible light line. Deselect the bright-field photomultiplier tube detector, "T-PMT".
- In the "Acquisition Mode" window, check that the correct objective is listed. Set the "Speed" to "3" and all other settings as outlined in step 2.2.4.
- In the "Channels" window, select "Track1" as the T-PMT detector. Select the laser line "790", with a percent power of "100.0", an initial "Gain (Master)" of "800", a "Digital Offset" of "0", and a "Digital Gain" of "1".
- In the "Stage" window, zero the stage by clicking "set zero". Mark the location by clicking "Mark". This is critical for uploading virtual masks to the microscope macro in step 3.
- In the "Time Series" window, set "Cycles" as "1" and "Interval" as "0".
- In the "Bleach" window, check the "Start Bleaching after # scans" box and set it to a value of "0 of 1". Select the "Different scan speed", with a value of "3" (corresponding to a pixel dwell time of 8.96 µs/pixel). Select the "Safe bleach for GaAsP".
- In the laser line section of the "Bleach" window, select the 790 laser line and set the percent power to "100.0". Leave all other boxes in the bleach window unselected. Save the bleach settings in the "Bleach" window.
- Save this configuration as "Channel Formation". NOTE: The "Z-Stack", "Regions", "Focus", and "Stage" windows are also important to this protocol, but do not need to be set or adjusted during initial configuration.
3. Creating a Recipe and Uploading Virtual Masks to the Microscope Software and Macro
With the laser-scanning confocal microscope still running and the microscope software still on, select only the "Regions" window (next to the "Start Experiment" button) in the "Channel Formation" configuration.
To ensure that the masks load to the microscope macro correctly, set the percent power in the "Channels" window to "0.2" and the "Speed" in the "Acquisition Mode" window to "8", and click "Snap" in the upper left of the screen.
Check that the image size is still "531.2 µm x 531.2 µm", with a pixel size of "0.18", in the "Information" tab of the image. If these values are not correct, check the "Frame Size" and "Zoom" values again.
CRITICAL: Check that the X and Y locations in the "Stage" window are zeroed and that the position is marked before opening the microscope macro. Refer to step 2.3.6.
To open the microscope macro, type "Alt+F8", click "Load", and select the correct version. See details in the List of Specific Materials/Equipment. A long list of sub-macros will open in the "Macro Control" window.
In the "Macro Control" window, click "Run" to open the microscope macro, and then close the "Macro Control" window. NOTE: Alternative versions of the microscope macro may not be compatible with laser-based hydrogel degradation using this protocol.
In the "Saving" tab of the microscope macro, provide an arbitrary "Base File Name", select "Single File Output", and chose a "Temporary File" folder. Set "Open Temp Folder" to location "1", select "Single Temporary Image Folder", and name the recipe in "Store/Apply Recipe" with "Recall Locations List" selected. Click "Store" to initially create the recipe.
In the "Acquisition" tab of the microscope macro, ensure that the "Scan Configuration" matches the name given to the microscope software configuration set in step 2.3. Set the "No of Time Points within Block" to "1", with an "Interval" of "0". Ensure that "Marked Z - Top of the Z Stack" is highlighted green. All other default settings are fine as they are.
In the "Timing" tab of the microscope macro, ensure that "Wait Delay" is highlighted green, set "Experiment Repetitions" and "Group Repetitions" to "1" and "Wait Interval Before Block at First Location Only" to "0". All other default settings are fine as they are. NOTE: The "Group Repetitions" (and not the "Experiment Repetitions") box is where one can set the number of times the laser scans over a region (i.e., repetitions of the recipe).
In the "Z List" tab of the microscope macro, the degradation planes in the z-direction are specified. Set the z-spacing, "dZ", to "1 µm", with the "Number of Positions" set to match the number of virtual masks to be used.
Click the "Create Z Stack" button to make the "Z List" appear in the dropdown "List of Locations" towards the bottom of the window. Check that the number of locations and z-spacing are correct and that the X and Y locations are zeroed. NOTE: If they are not, do not store the recipe; close the microscope macro and repeat step 2.3.6 before re-opening the microscope macro and setting up the recipe again. NOTE: Instead of having 100 masks spaced by 1 µm each, for example, it can be advantageous to have 50 masks, each employed twice and spaced by 1 µm, to get an equivalent microfluidic structure, as loading individual masks to the microscope macro can be time-consuming.
In the "Bleach" tab of the microscope macro, all of the masks must be loaded and matched with a specific numbered location in the "List of Locations". To start, select the "Bleach" box and ensure that "Config." matches the name of the bleach settings in the "Bleach" window in the main microscope software screen (refer to step 2.3.8). Set "Wait Delay After Bleach" to "0", deselect "spot", and leave the "ROI" empty.
Do not alter anything in the "Location", "Tile", "Grid", "Autofocus", "Blocks", and "Options" tabs from the default settings.
To load masks to the microscope macro, select only the "Regions" window in the microscope software and open the "Bleach" tab of the microscope macro. In the "Regions" window, click "Load" and select the .OVL file for the mask at the bottom of the z-stack being degraded. NOTE: The first location in the "Z list" is the bottom of the z-stack, and the microscope macro works its way to the top of the z-stack, or hydrogel.
Once the list of ROIs appears in the "Regions" window, click the arrow button to view the ROIs of the mask within the field of view (on the image snapped in step 3.2). Verify that the ROIs fit within the field of view.
To save the loaded mask in the microscope macro, give the mask a name and number in the "Add Current Region to ROI List" box on the "Bleach" tab, and then click the "Add Current Region to ROI List" button. The name and number will appear in the "ROI" dropdown list with any other masks (including masks from other previously created recipes). Delete the mask in the "Regions" window in the microscope software.
Repeat steps 3.14 and 3.16 to upload and save all masks to the microscope macro.
To assign uploaded masks to each Z-location, highlight position 1 in the dropdown "List of Locations". Select the appropriate ROI from the "ROI" dropdown list. Ensure that the "Bleach" box is selected at the top of the "Bleach" tab in the microscope macro.
Repeat step 3.18 for all positions in the dropdown "List of Locations", and then click "Store" on the "Saving" tab to save the recipe in its final form.
4. Photopolymerizing a PEGDA Hydrogel
- Making Sterile HEPES Buffered Saline (HBS) with Triethanolamine (TEOA)
- In a 1-L beaker, add 500 mL of DI H2O. With a stir bar, stir in 2.922 g of sodium chloride, 1.196 g of HEPES, and 7.5 mL of TEOA in a fume hood.
- Adjust to pH 8.3 with 1 N sodium hydroxide solution by adding dropwise with a Pasteur pipette.
- Sterile filter the solution through a 0.2-µm vacuum filtration cup into a 500-mL autoclaved glass media bottle. Aliquot and store at 4 °C.
Attach a ring of double-sided adhesive with backing to a specialized 60-mm Petri dish with a 20-mm hole cut out of the bottom. Leaving the backing on the adhesive ring, press out any air bubbles from between the Petri dish and the adhesive.
Prepare the materials. Bring the synthesized33 3.4 kDa PEGDA out of the -80 °C freezer and allow it to reach room temperature. Turn on the white light source at least 15 min before photopolymerizing hydrogels.
In a fume hood, add 1 mL of HBS with TEOA to a foil-covered, 2-mL amber centrifuge tube (used to prevent light exposure of the photoinitiator, eosin Y). Add 10 µL of 1 mM eosin Y disodium salt in DI H2O.
In a fume hood, extract a small volume of 1-vinyl-2-pyrrolidinone (NVP) into a glass beaker with a Pasteur pipette. From this volume, pipette 3.5 µL of NVP into the amber centrifuge tube with the HBS, TEOA, and eosin Y.
In a second foil-covered, 2-mL amber centrifuge tube (used to prevent errant photoactivation of the prepolymer solution), add 10 mg of 3.4kDa PEGDA. Add 0.2 mL of the HBS, TEOA, eosin Y, NVP solution for a 5% w/v PEGDA pre-polymer solution. Vortex until the PEGDA is fully dissolved. Using a power meter and detector wand, adjust the intensity of the white light source to 95 mW.
- Building a Poly(dimethylsiloxane) (PDMS) Mold
- Build a PDMS mold7 on a larger surface (glass, polystyrene tissue culture dish) for ease of handling and assemble the molds such that the molded hydrogels fit within a 20-mm diameter circle.
- To generate a rectangular-shaped hydrogel with a thickness of 500 µm, place two 500 µm-thick PDMS spacers onto a PDMS surface to create two defined hydrogel edges.
- Include a 300-µm PDMS spacer to impart a 300 µm-deep well in the surface of the hydrogel, which is helpful for introducing fluids.
Pipette 60 µL of the 5% w/v PEGDA pre-polymer solution onto the PDMS mold. Be careful not to introduce any bubbles during pipetting.
Center and place a [3-(methacryloyloxy)propyl]trimethoxysilane (TMPSA) functionalized7, 40-mm diameter, #1.5 glass coverslip on top of the solution. Ensure that the coverslip rests on the 500-µm PDMS spacers. The hydrogel will bond to the methacrylated coverslip and prevent the hydrogel from floating in the solution.
Place the PDMS, PEGDA pre-polymer solution and coverslip the assembly under the white light source for 3 min.
Flow DI H2O between the mold and the coverslip to separate the hydrogel from the PDMS. Rinse the hydrogel with DI H2O. Dry the edges of the coverslip with lab tissue. Be careful not to touch or wick water from the hydrogel.
After removing the backing from the adhesive, adhere the coverslip to the prepared Petri dish and gently remove air bubbles from between the adhesive and the glass. Submerge the hydrogel in the Petri dish with DI H2O. NOTE: The PDMS mold can be reused to make additional hydrogels if rinsed with DI H2O, dried with nitrogen, and kept free of dust.
5. Fabricating Vascular-derived Microfluidic Networks via Laser-based Degradation
With the laser-scanning confocal microscope and microscope software on, select the objective "W Plan-Apochromat 20x/1.0 DIC VIS-IR M27 75mm" in the "Maintain" tab. In the "Acquisition" tab, make sure the necessary laser lines (514 nm Argon laser and 790 nm 140 fs pulsed Ti:S laser) are on and warmed up.
- Setting up the Hydrogel to Form an Inlet Channel
- Fit the hydrogel and Petri dish on the stage insert and place the stage insert on the microscope stage. Fill the Petri dish with at least 5 mL of DI H2O. Center the hydrogel and well features by eye before lowering the water immersion objective into the DI H2O. NOTE: Do not crush the hydrogel with the objective-the two should never touch!
- To find the hydrogel, switch to the "Hydrogel Visualization" configuration from step 2.2. Click "Live" to turn the laser line on and bring the objective closer to the hydrogel until the eosin Y signal (detected by the Ch1 fluorescent detector) of the top of the hydrogel can be seen. NOTE: To find the hydrogel easily, the percent power can be increased or the pinhole can be opened. Once the objective is focused on the hydrogel, however, drop the percent power to protect the fluorescent detector and decrease the pinhole back to 1AU to avoid oversaturation of the detector and to accurately find the hydrogel surface.
- In the "Stage" window, mark the locations of the top and bottom of the hydrogel and of the bottom of the well. NOTE: The z-values here will help in determining the thickness of the hydrogel and the depth of the well.
- Use the joystick to move in X and Y to find the edge of a well. Place the hydrogel at the right of the screen, with the well at the left, or vice versa. Allow the hydrogel to fill no more than two thirds of the field of view.
- Drop the plane of focus down 150 µm into the hydrogel by setting the "Step Size" to "150" in the "Focus" window and clicking the down arrow next to the "Z-Position" box. Repeat step 5.2.4, if necessary. NOTE: The x, y, and z position of the inlet depends solely on the design of the virtual masks.
- CRITICAL: Zero the X and Y location in the "Stage" window, delete all other marked locations, and mark only this location. NOTE: If this step is not performed, the microscope macro will not properly restore the locations in the previously saved recipe, and the hydrogel will not be degraded in the desired location.
- Forming an Inlet Channel
- "Snap" an image and draw a rectangular region that will be the inlet to the vascular network using the rectangle button in the "Regions" window. NOTE: Make sure part of the region extends out into the well to ensure that the inlet will connect the well to the vascular network.
- Next to the ROI generated in the "Regions" window, select "Acquisition" and deselect "Bleach" and "Analysis".
- Save the "Hydrogel Visualization" configuration, deselect "Regions", and change to the "Channel Formation" configuration, set up in step 2.3. In the "Channel Formation" configuration, select "Regions" again. NOTE: When switching between the "Hydrogel Visualization" and the "Channel Formation" configurations, be sure to deselect regions in the top left of the screen. Sometimes the scaling of regions can change unexpectedly between configurations if this is not done.
- In the "Channel Formation" configuration, make sure that only "Regions" and "Z-Stack" are selected. In the "Z-Stack" window, click the "Center" button at the top and then the "Center" button above "Offset" to set the center of the z-stack as the current Z-position. Depending on how large the inlet needs to be, set the number of "Slices" with an "Interval" of "1". The size of the Z-stack will be displayed underneath "Range".
- Check that "Speed" in the "Acquisition Mode" window is set to "3" and that the "Percent Power" in the "Channels" window is set to "100". Save the configuration and click the "Start Experiment" button. NOTE: The microscope macro is not required for degrading the same simple shape on multiple planes, as performed here to generate an inlet channel. The macro is only required when degrading different regions on each individual plane, and it is used to store the virtual masks within a recipe.
- To visualize the hydrogel during degradation, either increase the "Gain (Master)" in the "Channels" window if the region in the field of view is black, or decrease the "Gain (Master)" if the region is white. NOTE: Bubble formation here is an indication of laser-induced hydrogel degradation.
- To degrade the inlet multiple times instead of just once, adjust the "Cycles" in the "Time Series" window.
- In the "Hydrogel Visualization" configuration, click "live" to ensure that the inlet was formed.
- Setting Up the Hydrogel to Generate the Microfluidic Network
- In the "Regions" window, load any .OVL file from the z-stack of virtual masks and click the arrow button to see the ROIs in the field of view.
- Click "Live" to live scan the "Hydrogel Visualization" configuration and use the joystick or the "Stage" window to position the hydrogel in X and Y, such that the inlet connects to the correct location within the vascular network. NOTE: Make sure the ROIs of the virtual mask overlap slightly with the previously formed inlet.
- Drop the plane of focus down 50 µm from the previous centered Z-location, or 200 µm from the surface of the hydrogel, using the "Step Size" in the "Focus" window and clicking the down arrow next to the "Z-Position" box. This will be the first position in the microscope macro. NOTE: The actual distance to move in the z-direction depends on the network design. Ensure that the desired network does not extend beyond the top or bottom surface of the hydrogel in the z-direction.
- CRITICAL: Zero the X and Y locations in the "Stage" window, delete all other marked locations, and mark only this location. NOTE: This will be the starting point in the microscope macro. The recipe will work its way back towards the surface of the hydrogel.
- "Snap" an image in the "Hydrogel Visualization" configuration, delete and deselect "Regions", and save the configuration.
- Generating the Microfluidic Network
- Change to the "Channel Formation" configuration, and only select the "Time Series" and "Bleaching" windows. Verify that the settings for the "Time Series" and the "Bleaching" windows are as they were initially set in steps 2.3.7 and 2.3.8.
- Set the "Speed" in the "Acquisition Mode" window to "8" and the percent power of the laser line to "0.2" in the "Channels" window. Save the configuration.
- Open the microscope macro following step 3.5. On the "Saving" tab of the macro, select the previously made recipe from step 3 and click "Apply". NOTE: Do not click "Store" or the recipe will be overwritten!
- Check the settings on all tabs and verify that the virtual masks (.OVL files) are still correctly associated with the Z-locations in the dropdown list. Also check that the first Z-location matches the marked location in the "Stage" window of the microscope software. NOTE: Counterintuitively, the second location in the dropdown list will be physically above the first location, as the microscope macro will work its way from the bottom of the hydrogel (farthest away from the objective) to the top of the hydrogel (closest to the objective).
- Save the "Channel Formation" configuration again, and then click "Update" in the microscope macro. Click "No" to "Overwrite Current Location List?", and then click "Start" in the microscope macro to begin the recipe.
6. Visualizing the Microfluidic Network
To view the hydrogel and the formed microfluidic network after degradation, close the microscope macro. Switch to the "Hydrogel Visualization" configuration to view the eosin Y signal of the hydrogel; the degraded network will appear dark. NOTE: The hydrogel degradation cannot be visualized while the microscope macro is running.
To view the microfluidic network specifically, use the "Hydrogel Visualization" configuration at a lower laser power to enable the imaging of introduced fluorescein (FITC)-labeled dextran, which has a brighter signal than the native eosin Y signal of the hydrogel.
Before introducing fluorescently labeled dextran, wick water from the well in the hydrogel using lab tissue. Pipette in 10 µL of 5 mg/mL 2,000 kDa FITC-labeled dextran in DI H2O (pre-filtered through a 0.2-µm syringe filter). NOTE: Use any dextran sized 2,000 kDa or larger to prevent diffusion into the gel. If imaging for longer than 30 min, use an incubated stage with 60% humidity (or higher) to prevent hydrogel drying and evaporation of water from the FITC-labeled dextran solution.
Remove the hydrogel and Petri dish from the stage and mark the Petri dish to enable the easy location of the formed vascular network during later experimentation.
Representative Results
To demonstrate the implementation of image-guided, laser-based hydrogel degradation to generate a 3D, vascular-derived microfluidic network, we utilized a 3D image stack of mouse brain vasculature that was acquired via knife-edged scanning microscopy (KESM)2627. A selected 3D region of the larger microvascular dataset was converted into a series of virtual masks for image-guided microfluidic network formation, as described above. After fabrication, the resulting microfluidic network was perfused with a solution of FITC-labeled, 2,000 kDa dextran and imaged via confocal microscopy. The image stack of the in vivo vasculature that defined the network architecture (Figure 1A and C) and the fabricated microfluidic network (Figure 1B and D) were used to generate Z-projections (Figure 1A and B) and 3D renderings (Figure 1C and D). The comparison of the images demonstrates that image-guided, laser-based hydrogel degradation enables the fabrication of 3D biomimetic microfluidic networks that recapitulate the size, tortuosity, and complex architecture of in vivo vasculature. The technique is amenable to fabrication of networks containing a wide range of diameters; within Figure 1, channels ranging from 3.3 to 28.8 µm in diameter were generated. Note that the larger rectangular structure in the top left of Figure 1B and in the left corner of Figure 1D is the inlet channel to introduce flow into the network.
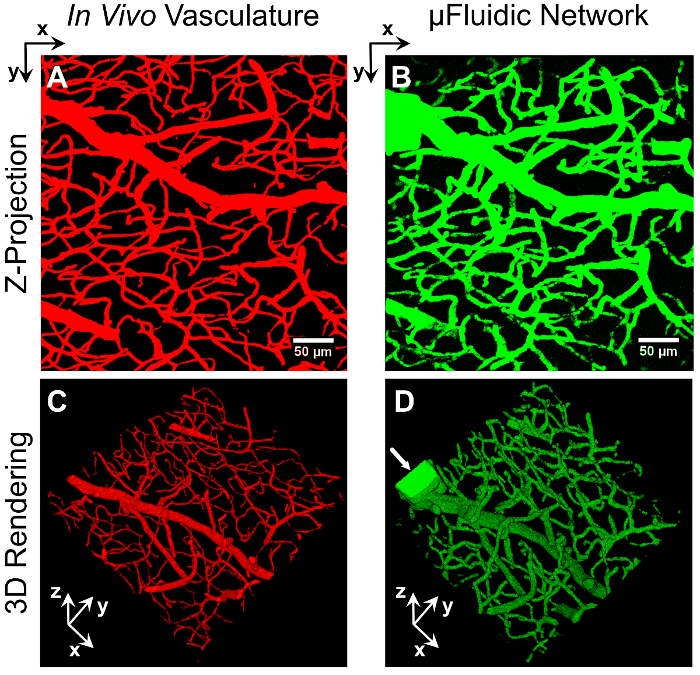 Figure 1: Vascular-Derived Microfluidic Network. A series of virtual masks, derived from an image stack of mouse brain vasculature (A and C), were used to fabricate a 3D biomimetic microfluidic network embedded in a PEGDA hydrogel (B and D) via image-guided, laser-based degradation. The microfluidic network was perfused with 2,000 kDa FITC-labeled dextran and imaged via confocal microscopy. Z-projections (A and B) and 3D renderings (C and D) of the two networks demonstrate the ability to generate microfluidic networks that recapitulate the density, organization, and overall architecture of in vivo vasculature. The arrow (D) marks the rectangular inlet created to introduce the dextran. The scale bar represents 50 µm. Please click here to view a larger version of this figure.
Figure 1: Vascular-Derived Microfluidic Network. A series of virtual masks, derived from an image stack of mouse brain vasculature (A and C), were used to fabricate a 3D biomimetic microfluidic network embedded in a PEGDA hydrogel (B and D) via image-guided, laser-based degradation. The microfluidic network was perfused with 2,000 kDa FITC-labeled dextran and imaged via confocal microscopy. Z-projections (A and B) and 3D renderings (C and D) of the two networks demonstrate the ability to generate microfluidic networks that recapitulate the density, organization, and overall architecture of in vivo vasculature. The arrow (D) marks the rectangular inlet created to introduce the dextran. The scale bar represents 50 µm. Please click here to view a larger version of this figure.
We have implemented two methods, pressure heads and syringe pumps, to initiate fluid flow in these embedded microfluidic networks. For example, a 30 by 30 µm rectangular channel was fabricated to connect two wells in a micromolded PEGDA hydrogel (Figure 2A), with fluid flow driven via a pressure head7. Generated in the left well, the pressure head induced flow through the microchannel and into the well on the right. In Figure 2A, an initial front of tetramethylrhodamine (TRITC)-labeled, 70 kDa dextran was observed moving through the channel using time-lapse microscopy. Later, in Figure 2B, a 10-µm diameter fluorescent polystyrene sphere flowed from the left well, through the channel, to the well on the right. With the duration of flow controlled by the volume of fluid present in the micromolded well at the inlet, and the flow-rate controlled by the hydrostatic gradient generated between the inlet and outlet well, pressure head-driven flow in micromolded hydrogels provides a simple method for flow induction, without the need for an external housing or a syringe pump.
 Figure 2: Pressure Head-Driven Flow in a Micromolded PEGDA Hydrogel. PEGDA was micromolded to form a hydrogel containing two wells connected by an embedded microchannel. A pressure head was generated in the left well to initiate flow of TRITC-labeled, 70 kDa dextran (A1-A3) and a 10-µm polystyrene sphere (B1-B3) through a 30 by 30 µm rectangular channel from one well to the other. The scale bar represents 50 µm. Please click here to view a larger version of this figure.
Figure 2: Pressure Head-Driven Flow in a Micromolded PEGDA Hydrogel. PEGDA was micromolded to form a hydrogel containing two wells connected by an embedded microchannel. A pressure head was generated in the left well to initiate flow of TRITC-labeled, 70 kDa dextran (A1-A3) and a 10-µm polystyrene sphere (B1-B3) through a 30 by 30 µm rectangular channel from one well to the other. The scale bar represents 50 µm. Please click here to view a larger version of this figure.
Alternatively, to generate constant fluid flow within microfluidic networks, syringe pump-driven flow can be initiated by photopolymerizing the hydrogel inside of a secondary microfluidic device with inlet and outlet ports for syringe pump interfacing. In Figure 3A, a commercially available, pre-fabricated microfluidic device is shown with a 1-mm band of hydrogel photopolymerized across the width of the device7. Laser-based degradation was implemented to fabricate microfluidic channels and networks in housed hydrogels using the same protocol described here for freestanding hydrogels. Syringe pump-generated flow can be seen in Figure 3B, where a 20-µm diameter cylindrical channel was fabricated in a hydrogel housed in a microfluidic device. In this 20-µm diameter channel, individual 2-µm fluorescent polystyrene spheres are easily resolved without fluid flow (Figure 3B1), whereas when a 5 µL/s flowrate is applied, the spheres move through the field of view, as evidenced by streaking (Figure 3B2). This same approach was used to induce flow through a 2D, vascular-derived network to monitor flow of TRITC-labeled, 65 kDa dextran through the network and diffusion into the surrounding hydrogel (Figure 3C).
 Figure 3: Syringe Pump-Driven Flow in PEGDA Hydrogels Polymerized in Microfluidic Housings. A commercially available, pre-fabricated microfluidic device (A) with a 17 x 3.8 x 0.53 mm (x,y,z) microchannel houses a photopolymerized 1 x 3.8 x 0.53 mm (x,y,z) PEGDA hydrogel, indicated by the black arrow. A 20-µm diameter cylindrical channel was degraded through the hydrogel, and 2-µm polystyrene spheres were pumped through the device (B). The spheres are clearly resolved without fluid flow (B1), but they appear as streaks at a flowrate of 5 µL/s (B2). In another housed hydrogel, a 2D vascular-derived network was fabricated and TRITC-labeled, 65 kDa dextran was pumped through the network (C). Time-lapse microscopy was used to monitor the fluorescent dextran as it filled the channels and diffused into the surrounding hydrogel. White arrows in (B) and (C) indicate the direction of fluid flow. The calibration bar in (C) indicates the intensity of the fluorescent dextran. The scale bars represent 20 µm in (B1) and (B2) and 50 µm in (C). Please click here to view a larger version of this figure.
Figure 3: Syringe Pump-Driven Flow in PEGDA Hydrogels Polymerized in Microfluidic Housings. A commercially available, pre-fabricated microfluidic device (A) with a 17 x 3.8 x 0.53 mm (x,y,z) microchannel houses a photopolymerized 1 x 3.8 x 0.53 mm (x,y,z) PEGDA hydrogel, indicated by the black arrow. A 20-µm diameter cylindrical channel was degraded through the hydrogel, and 2-µm polystyrene spheres were pumped through the device (B). The spheres are clearly resolved without fluid flow (B1), but they appear as streaks at a flowrate of 5 µL/s (B2). In another housed hydrogel, a 2D vascular-derived network was fabricated and TRITC-labeled, 65 kDa dextran was pumped through the network (C). Time-lapse microscopy was used to monitor the fluorescent dextran as it filled the channels and diffused into the surrounding hydrogel. White arrows in (B) and (C) indicate the direction of fluid flow. The calibration bar in (C) indicates the intensity of the fluorescent dextran. The scale bars represent 20 µm in (B1) and (B2) and 50 µm in (C). Please click here to view a larger version of this figure.
Discussion
Critical in overriding the standard functions of the microscope software to enable the use of virtual masks for image-guided, laser-based degradation, the microscope macro must be setup and manipulated carefully. How the microscope software interfaces with the microscope macro can sometimes be counterintuitive, and much effort was invested in determining the optimal settings in both programs to develop this method. A general understanding of basic laser-scanning confocal microscope use is recommended, especially with the "Stage" and "Focus" windows for finding and correctly positioning the hydrogel in the x, y, and z dimensions, before attempting laser-based degradation. Additionally, the fabrication of an inlet channel is critical to the protocol; suspended fluorescent species must be able to enter the microfluidic systems for successful imaging and network characterization. It is important to note that this protocol applies to a specific laser-scanning confocal microscope configured with a specific femtosecond pulsed laser, running specific versions of the microscope software and the microscope macro (see details in the List of Specific Materials/Equipment). We have discovered that newer versions of the microscope macro lack the necessary control and functionality needed for image-guided laser control. While other microscope platforms and pulsed laser sources can be used, this protocol applies specifically to this system and will need modification according to the platform, laser source, and software implemented. The underlying principles throughout this protocol still apply, however.
A potential modification to this protocol involves changing the hydrogel composition. Here, we utilized 5 wt%, 3.4 kDa PEGDA hydrogels, but laser-based degradation (for purposes aside from microfluidic network generation) has been implemented to degrade both synthetic and natural hydrogels, including PEGylated fibrinogen21,22, silk protein hydrogels23, and collagen24. Adjustment of the laser power, scan speed, spacing between Z-slices, and number of repetitions will aid in determining the optimal parameters to fabricate microfluidic networks in other hydrogel formulations.
One current limitation of the technique is the overall volume of hydrogel that can be degraded in a feasible amount of time. To create open voids or microfluidic features within the hydrogel, the laser dwell time must be at or above 8.96 µs/pixel (or a laser scan speed of 0.021 µm/µs) when using a laser fluence of 37.7 nJ/µm2. With these settings, it takes 1.4 h to degrade vessels within a 0.014 mm3 volume (as seen in Figure 1). Using a laser dwell time of 4.48 µs/pixel or below, the energy delivered is not enough for full degradation of the hydrogel formulation used here. Implementing a different hydrogel composition could overcome this limitation. The use of photolabile gels that contain light-sensitive components34-36 or hydrogels that have large multiphoton cross-sections21-24 are good options that would enable the use of less energy and would result in faster degradation. With respect to dimensional limitations, features have been fabricated up to 1.5 mm deep in the hydrogel7. The depth achievable is a function of the working distance of the objective and can be increased using ultra-long working distance objectives optimized for in vivo imaging. The smallest measured channel7 created using a 20X (NA1.0) water immersion objective had a width of 3.3 µm and depth of 8.9 µm, which is on par with the size of the smallest channels generated in Figure 1. While other labs have used lower NA objectives21,23,24, we anticipate that microfluidic networks with smaller features can be generated using higher NA objectives at the expense of a reduced working distance. Ultimately, however, the resolution of the technique is a function of the point spread function of the focused laser beam, the laser scanning properties, the amount of energy delivered by the laser, and the laser absorption properties of the material being degraded.
Furthermore, laser properties (speed, fluence, spacing between virtual masks, and number of repetitions) must be optimized based on hydrogel properties (crosslink density, macromer/monomer molecular weight, weight percent, and type of hydrogel: protein-based vs. synthetic), as these will inherently alter interactions with the laser and thus the ultimate resolution of the process. As the energy delivered is also influenced by the objective used, additional optimization is required when switching between objectives. With respect to the costs involved, access to a microscope with a pulsed laser is required, and while many different objectives can be used, those with a high NA and long working distance (for in vivo imaging) can be expensive.
Above and beyond the protocol detailed here, microfluidic networks generated using this technique can also be functionalized with cell-adhesive peptides post-channel fabrication to induce endothelial cell adhesion and lumen formation7. Additionally, the incorporation of cell-degradable peptide sequences in the bulk material9 prior to channel degradation could allow for the study of cell migration into and through the hydrogel. For continuous fluidization of the microfluidic network within the hydrogel, hydrogels can be photopolymerized in larger fluidic devices or housings, as demonstrated in Figures 2 and 37.
The generation of vascular networks via vasculogenic or angiogenic self-assembly8-12 provides a straightforward approach to induce vascularization throughout a relatively large hydrogel volume. While this approach results in perfusable fluidic networks, it is difficult to directly control the size, tortuosity, density, and overall network architecture. Due to this limitation, the flow profile and shear rates in the vasculature may differ between experiments. Alternatively, microfabrication techniques13-16 allow for direct control over network architecture but are often limited by their inability to generate small, capillary-sized channels or the fabrication of dense networks that mimic in vivo vascular architecture. While the laser-based hydrogel degradation technique outlined here has been newly repurposed for microfluidic generation, it simultaneously overcomes both the architectural control and size-based limitations of existing microfabrication methods by enabling the creation of 3D biomimetic microfluidic networks that recapitulate the density, tortuosity, size range, and overall architecture of in vivo vasculature. Furthermore, multiple fluidic networks can be generated in a single hydrogel, allowing the study of inter-network transport7. For tissue engineering applications that strive to more closely replicate in vivo transport processes in vitro, the highly resolved hydrogel-embedded microfluidic networks outlined here are well suited. We anticipate that this protocol will be useful in developing tissue constructs that more accurately mimic in vivo transport for use as drug screening devices and in vitro disease models.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank Dr. Jeff Caplan and Mr. Michael Moore at the Delaware Biotechnology Institute Bio-Imaging Center for their support with confocal microscopy. Access to microscopy equipment was supported by the National Institutes of Health (NIH) shared instrumentation grants (S10 RR0272773 and S10 OD016361) and the State of Delaware Federal Research and Development Grant Program (16A00471). This research was supported by grants from the Institutional Development Award (IDeA) from the NIH National Institute of General Medical Sciences (P20GM103446), the American Cancer Society (14-251-07-IRG), the University of Delaware Research Foundation (14A00778), the Cancer Prevention and Research Institute of Texas (CPRIT) (RR140013), and the NIH National Library of Medicine (4 R00 LM011390-02).
References
- Baish JW, Stylianopoulos T, et al. Scaling rules for diffusive drug delivery in tumor and normal tissues. Proc. Natl. Acad. Sci. USA. 2011;108(5):1799–1803. doi: 10.1073/pnas.1018154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nature Biotechnol. 2014;32(8):760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015;14(4):248–260. doi: 10.1038/nrd4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami AM, Hancock MJ, Harrington H, Kaji H, Khademhosseini A. Biomimetic tissues on a chip for drug discovery. Drug Discov. Today. 2012;17(3-4):173–181. doi: 10.1016/j.drudis.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JS, Bersini S, et al. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. USA. 2015;112(1):214–219. doi: 10.1073/pnas.1417115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisano M, Triacca V, Barbee KA, Swartz MA. An in vitro model of the tumor-lymphatic microenvironment with simultaneous transendothelial and luminal flows reveals mechanisms of flow enhanced invasion. Integr. Biol. 2015;7(5):525–533. doi: 10.1039/c5ib00085h. [DOI] [PubMed] [Google Scholar]
- Heintz KA, Bregenzer ME, Mantle JL, Lee KH, West JL, Slater JH. Fabrication of 3D Biomimetic Microfluidic Networks in Hydrogels. Adv. Healthc. Mater. 2016. [DOI] [PMC free article] [PubMed]
- Cuchiara MP, Gould DJ, McHale MK, Dickinson ME, West JL. Integration of Self-Assembled Microvascular Networks with Microfabricated PEG-Based Hydrogels. Adv. Funct. Mater. 2012;22(21):4511–4518. doi: 10.1002/adfm.201200976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver JC, Hoffmann JC, Poché RA, Slater JH, West JL, Dickinson ME. Three-Dimensional Biomimetic Patterning in Hydrogels to Guide Cellular Organization. Adv. Mater. 2012;24(17):2344–2348. doi: 10.1002/adma.201200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee H, Chung M, Jeon NL. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip. 2013;13(8):1489. doi: 10.1039/c3lc41320a. [DOI] [PubMed] [Google Scholar]
- Saik JE, Gould DJ, Keswani AH, Dickinson ME, West JL. Biomimetic Hydrogels with Immobilized EphrinA1 for Therapeutic Angiogenesis. Biomacromolecules. 2011;12(7):2715–2722. doi: 10.1021/bm200492h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisch AH, Lutolf MP, et al. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB J. 2003. [DOI] [PubMed]
- He J, Zhu L, Liu Y, Li D, Jin Z. Sequential assembly of 3D perfusable microfluidic hydrogels. J. Mater. Sci. Mater. Med. 2014;25(11):2491–2500. doi: 10.1007/s10856-014-5270-9. [DOI] [PubMed] [Google Scholar]
- Therriault D, White SR, Lewis JA. Chaotic mixing in three-dimensional microvascular networks fabricated by direct-write assembly. Nat. Mater. 2003;2(4):265–271. doi: 10.1038/nmat863. [DOI] [PubMed] [Google Scholar]
- Wu W, DeConinck A, Lewis JA. Omnidirectional Printing of 3D Microvascular Networks. Adv. Mater. 2011;23(24):H178–H183. doi: 10.1002/adma.201004625. [DOI] [PubMed] [Google Scholar]
- Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA, Lewis JA. 3D Bioprinting of Vascularized, Heterogeneous Cell-Laden Tissue Constructs. Adv. Mater. 2014;26(19):3124–3130. doi: 10.1002/adma.201305506. [DOI] [PubMed] [Google Scholar]
- Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA. 2012;109(34):13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudsari LC, West JL. Studying the influence of angiogenesis in in vitro cancer model systems. Adv. Drug Deliv. Rev. 2015. [DOI] [PubMed]
- Dawson TH. Scaling laws for capillary vessels of mammals at rest and in exercise. Proc. R. Soc. Long. [Biol.] 2003;270(1516):755–763. doi: 10.1098/rspb.2002.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel A, Noack J, Hüttman G, Paltauf G. Mechanisms of femtosecond laser nanosurgery of cells and tissues. Appl. Phys. B. 2005;81(8):1015–1047. [Google Scholar]
- Sarig-Nadir O, Livnat N, Zajdman R, Shoham S, Seliktar D. Laser Photoablation of Guidance Microchannels into Hydrogels Directs Cell Growth in Three Dimensions. Biophys. J. 2009;96(11):4743–4752. doi: 10.1016/j.bpj.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livnat N, Sarig-Nadir O, Seliktar D, Shoham S, editors. Three-dimensional guidance of DRG neurite outgrowth using multi-photon photo-ablation. 2009 4th International IEEE/EMBS Conference on Neural Engineering; 2009. pp. 116–119. [Google Scholar]
- Applegate MB, Coburn J, et al. Laser-based three-dimensional multiscale micropatterning of biocompatible hydrogels for customized tissue engineering scaffolds. Proc. Natl. Acad. Sci. USA. 2015;112(39):12052–12057. doi: 10.1073/pnas.1509405112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilina O, Bakker GJ, Vasaturo A, Hoffman RM, Friedl P. Two-photon laser-generated microtracks in 3D collagen lattices: principles of MMP-dependent and -independent collective cancer cell invasion. PB. 2011;8(2) doi: 10.1088/1478-3975/8/1/015010. [DOI] [PubMed] [Google Scholar]
- Naik P, Cucullo L. In Vitro Blood-Brain Barrier Models: Current and Perspective Technologies. J. Pharm. Sci. 2012;101(4):1337–1354. doi: 10.1002/jps.23022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe Y, Mayerich D, et al. Specimen Preparation, Imaging, and Analysis Protocols for Knife-edge Scanning Microscopy. JoVE. 2011. [DOI] [PMC free article] [PubMed]
- Mayerich D, Kwon J, Sung C, Abbott L, Keyser J, Choe Y. Fast macro-scale transmission imaging of microvascular networks using KESM. Biomed. Opt. Express. 2011;2(10):2888–2896. doi: 10.1364/BOE.2.002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JR, Sung C, et al. Multiscale Exploration of Mouse Brain Microstructures Using the Knife-Edge Scanning Microscope Brain Atlas. Front. Neuroinform. 2011;5 doi: 10.3389/fninf.2011.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Slater JH, Culver JC, Dickinson ME, West JL. Biomimetic Surface Patterning Promotes Mesenchymal Stem Cell Differentiation. ACS Appl. Mater. Interfaces. 2015. [DOI] [PMC free article] [PubMed]
- Slater JH, Culver JC, et al. Recapitulation and Modulation of the Cellular Architecture of a User-Chosen Cell of Interest Using Cell-Derived, Biomimetic Patterning. ACS Nano. 2015;9(6):6128–6138. doi: 10.1021/acsnano.5b01366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater JH, West JL. Fabrication of Multifaceted, Micropatterned Surfaces and Image-Guided Patterning Using Laser Scanning Lithography. Methods Cell Biol. 2014;119:193–217. doi: 10.1016/B978-0-12-416742-1.00011-1. [DOI] [PubMed] [Google Scholar]
- DeLong SA, Gobin AS, West JL. Covalent immobilization of RGDS on hydrogel surfaces to direct cell alignment and migration. J. Control. Release. 2005;109(1-3):139–148. doi: 10.1016/j.jconrel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- DeForest CA, Anseth KS. Cytocompatible click-based hydrogels with dynamically-tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem. 2011;3(12):925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science. 2009;324(5923):59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbitt MW, Kloxin AM, Dyamenahalli KU, Anseth KS. Controlled two-photon photodegradation of PEG hydrogels to study and manipulate subcellular interactions on soft materials. Soft Matter. 2010;6(20):5100–5108. doi: 10.1039/C0SM00174K. [DOI] [PMC free article] [PubMed] [Google Scholar]