

Abstract
Mesenchymal stem/stromal cells (MSCs) hold great promise in bioengineering and regenerative medicine. MSCs can be isolated from multiple adult tissues via their strong adherence to tissue culture plastic and then further expanded in vitro, most commonly using fetal bovine serum (FBS). Since FBS can cause MSCs to become immunogenic, its presence in MSC cultures limits both clinical and experimental applications of the cells. Therefore, studies employing chemically defined xeno-free (XF) media for MSC cultures are extremely valuable. Many beneficial effects of MSCs have been attributed to their ability to regulate inflammation and immunity, mainly through secretion of immunomodulatory factors such as tumor necrosis factor-stimulated gene 6 (TSG6) and prostaglandin E2 (PGE2). However, MSCs require activation to produce these factors and since the effect of MSCs is often transient, great interest has emerged to discover ways of pre-activating the cells prior to their use, thus eliminating the lag time for activation in vivo. Here we present protocols to efficiently activate or prime MSCs in three-dimensional (3D) culturesunder chemically defined XF conditions and to administer these pre-activated MSCs in vivo. Specifically, we first describe methods to generate spherical MSC micro-tissues or 'spheroids' in hanging drops using XF medium and demonstrate how the spheres and conditioned medium (CM) can be harvested for various applications. Second, we describe gene expression screens and in vitro functional assays to rapidly assess the level of MSC activation in spheroids, emphasizing the anti-inflammatory and anti-cancer potential of the cells. Third, we describe a novel method to inject intact MSC spheroids into the mouse peritoneal cavity for in vivo efficacy testing. Overall, the protocols herein overcome major challenges of obtaining pre-activated MSCs under chemically defined XF conditions and provide a flexible system to administer MSC spheroids for therapies.
Keywords: Developmental Biology, Issue 121, Stem cell therapy, Mesenchymal stem cells, 3D culture, Xeno-free, Spheroids, Cell transplantation, Anti-inflammatory, Immunomodulation, Anti-cancer, Peritoneum
Introduction
Mesenchymal stem/stromal cells (MSCs) have shown great potential for various regenerative medicine approaches. MSCs were initially isolated as a stromal component of bone marrow but have since been obtained from numerous other adult tissues, including adipose tissue1,2,3. Interestingly, the main isolation method embraces the remarkable property of MSCs to adhere tightly onto tissue culture plastic in the presence of fetal bovine serum (FBS). Whilst this traditional isolation technique permits easy and rapid expansion of MSCs in two-dimensional (2D) culture, it is also very artificial and disregards significance of the native three-dimensional (3D) environment leading to potential loss of important cellular characteristics4,5,6. Therefore, the study of MSCs in 3D cultures, which are more physiological than traditional 2D cultures, has emerged in search for "lost/diminished" MSC characteristics. Furthermore, great interest has risen to identify xeno-free (XF) chemically defined conditions for MSC culture and activation, and thus make the cells more amenable for clinical applications.
Many studies have been published demonstrating the 3D culture of MSCs both in biomaterials and as spherical aggregates or spheroids. MSCs in biomaterials were initially designed for tissue engineering approaches to replace damaged tissues with cell-seeded scaffolds, whereas spheroid cultures of MSCs were seen as a way to understand MSC behavior in vivo after administration of the cells for therapies in pre-clinical or clinical trials4,5,7. Interestingly, MSCs form spheroids spontaneously when adherence to tissue culture plastic is not permitted8,9,10. Traditionally, cell aggregation was facilitated by spinner flask methods or liquid overlay techniques, methods used initially in cancer biology in efforts to try to mimic the tumor microenvironment. More recently, additional methods have surfaced that demonstrate cell aggregation in culture dishes pre-coated with specific chemicals to prevent cell-to-plastic adhesion4,5,6. One of the simplest and most economical methods to generate MSC spheroids is to culture them in hanging drops, a technique that was often used to produce embryoid bodies from embryonic stem cells. With hanging drop culture technique, cell adherence to the tissue culture plastic is prevented by suspending the cells in a drop of medium on the underside of a tissue culture dish lid and allowing gravity to facilitate cell aggregation in the apex of the drop. The spheroid size can be readily manipulated by changing the cell concentration or the drop volume, making hanging drop cultures particularly easy to control.
Early studies on the 3D culture of MSCs demonstrated radical differences in the characteristics of the cells in 3D compared to their 2D counterparts6,8,9. At the same time, reports demonstrated that the beneficial effects of MSCs in vivo relied on their ability to become activated by micro-environmental cues and, in response, to produce anti-inflammatory and immunomodulatory factors11. Interestingly, many of these factors such as prostaglandin E2 (PGE2), tumor necrosis factor-stimulated gene 6 (TSG6), and hepatocyte growth factor (HGF) were produced in much larger quantities by MSC spheroids than traditional 2D MSCs paving the way for the idea of using 3D cultures to activate the cells8,12,13. Moreover, gene activation in 3D cultures appeared to recapitulate mechanisms, at least in part, of cell activation after injection into mice12. By activating MSCs prior to their use in experiments, effects of the cells could be prolonged and more prominent as the traditional MSC effect in vivo is often delayed and transient, and can be described as "hit and run". During the past several years, important functional studies using MSC spheroids have demonstrated that they can suppress inflammatory responses and modulate immunity in vivo by influencing effector cells such as macrophages, dendritic cells, neutrophils, and T cells making spheroids an attractive form of primed MSCs2,3. In addition, production of anti-cancer molecules, such as interleukin-24 (IL-24) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), are increased in 3D cultures of MSCs relative to monolayer MSCs, a phenomenon that could be exploited for targeted cancer therapies8,10,14.
As the traditional MSC culture required not only the use of tissue culture plastic but also FBS, another hurdle to make MSC spheroids more amenable for clinical use had to be overcome. To tackle this hurdle, we recently showed formation of MSC spheroids under specific chemically defined XF conditions and established that the resulting MSC spheroids were activated to produce the same anti-inflammatory and anti-cancer molecules as the spheroids generated in conditions with FBS14. Here, these findings are presented in several detailed protocols that demonstrate the generation of pre-activated MSCs in 3D cultures using XF media. In addition, protocols are presented that describe effective ways to assess the activation levels of the MSCs in regards to their anti-inflammatory, immunomodulatory and anti-cancer effects, together with a practical method to deliver the intact spheroids into mice.
Protocol
1. MSC Isolation and Expansion
Obtain early passage MSCs from Center for the Preparation and Distribution of Adult Stem Cells (http://medicine.tamhsc.edu/irm/msc-distribution.html)15 as frozen vials. Alternatively, isolate MSCs from bone marrow aspirates following a routine protocol14 and store as frozen vials.
Prepare complete culture medium (CCM), which is minimal essential medium alpha (αMEM) supplemented with 15-20% premium select FBS, 2 mM L-glutamine, and 1x penicillin-streptomycin. Sterilize the medium by filtration.
Place 30 ml of CCM into a 150 mm cell culture dish and incubate the dish for 30 min at 37 °C in a humidified incubator containing 5% CO2.
Incubate the frozen vial containing approximately 106 MSCs for 2 min in a 37 °C water bath.
Transfer the contents of the vial to the culture dish using a 5 ml serological pipet and wash the vial several times with 1-2 ml CCM from the dish to capture residual cells. Place the dish overnight in an appropriate incubator.
Carefully wash the culture twice with room temperature phosphate buffered saline (PBS) and detach the MSCs with 3 ml of pre-warmed 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA) solution. Detachment usually takes approximately 3-4 min in the incubator.
Neutralize the trypsin in the dish with 6 ml CCM pre-warmed to 37 °C and transfer the cell suspension into a 50 ml conical tube. Combine with washes of PBS to maximize cell yield.
Centrifuge the cells (450 x g) for 10 min at room temperature and aspirate the supernatant.
Re-suspend the cells in a small volume of CCM and count the viable cells using a hemocytometer and trypan blue.
Seed an appropriate number of 150 mm dishes at 100 cells/cm2 in 30 ml of warm CCM and incubate the cultures until approximately 70-80% confluent, usually 7 days with medium changes every 2-3 days and 24-48 hr prior to harvest.
Harvest the cells as above with trypsin/EDTA and combine all the cells into a single conical tube with the appropriate medium. Centrifuge again and re-suspend the cells into a small volume of the same medium for cell counts. Use standard CCM (containing FBS) or various commercially available XF media formulations, here referred to as XF medium-1 (XFM-1) and XF medium-2 (XFM-2), both with and without human serum albumin (HSA, 13 mg/ml) supplementation.
2. Preparation of Activated MSC Spheroids in 3-D Hanging Drop Cultures under XF Conditions
To generate spheroids of approximately 25,000 cells, dilute the harvested MSCs into CCM, XFM-1, XFM-1 + HSA (13 mg/ml), XFM-2, or XFM-2 + HSA (13 mg/ ml) at 714 cells/µl.
Pipette 35 µl/drops of the cells onto the underside of a 150 mm inverted culture dish lid. Multiple drops can be easily pipetted by using a multichannel pipette. NOTE: If larger or smaller spheroids are desired, change the cell concentration or the drop volume (25-45 µl) appropriately.
Transfer 20 ml of room temperature PBS into the base of the dish and in one continuous and steady motion flip the lid, containing the drops, so that the drop apices face downward. Place the lid carefully on the base of the culture dish.
Transfer the dish into the incubator for 3 days without disturbing it to permit appropriate sphere assembly.
After 72 hr, begin the spheroid harvest by removing the lid carefully and inverting it so that the drops, once again, face upward.
Angle the lid to approximately 10-20° and push the drops, containing the spheroids, to the plate edge using a cell lifter.
Transfer the spheres and medium into a 15 ml conical tube with a 1,000 µl pipette. Use room temperature PBS and the pipette with the same tip to wash the plate and ensure maximal recovery of spheroids.
For real-time PCR assays (protocol 3), centrifuge the sphere suspension at 450 x g for 5 min at room temperature and aspirate the supernatant. Wash the spheroids with PBS and repeat both the centrifugation and aspiration steps. For delivery into animals (protocol 8), allow spheres to settle in the bottom of the tube (~3-4 min) without centrifugation.
3. Real-time PCR of Anti-inflammatory and Anti-cancer Markers
Use the RNA extraction kit with homogenizer columns and RNase-Free DNase Set to isolate DNA-free total RNA from monolayer MSCs (Protocol 1) and MSC spheroids (Protocol 2) that were washed with PBS and centrifuged.
Lyse at least 100,000 monolayer MSCs and 20 spheroids from each condition in separate 15 ml conical tubes with RLT buffer containing β-mercaptoethanol (1:100).
Transfer the thoroughly mixed lysates into 1.5 ml RNase-free tubes and place into a freezer (-80 °C) for at least 3 hr to aid in spheroid lysis.
Thaw the cell lysates on ice, vortex briefly, and follow the manufacturer's instructions for RNA isolation using a commercially available mammalian total RNA isolation kit.
Quantify and assess the quality of the isolated RNA using a spectrophotometer.
Use the High Capacity cDNA Reverse Transcription Kit or equivalent according to manufacturer's instructions to generate cDNA for real-time PCR.
Place cDNA samples, commercially designed real-time PCR primers/probes (Gene Expression Assays), and PCR Master Mix on ice. Use primers/probes for GAPDH (control), PTGS2 (COX-2, anti-inflammatory), TNFAIP6 (TSG6, anti-inflammatory), TRAIL (anti-cancer), and IL-24 (anti-cancer).
Prepare the real-time PCR reactions according to manufacturer's instructions for Gene Expression Assays and Fast Universal Master Mix.
Follow the guidelines for the real-time PCR instrument for generating the experiment documents and performing the run.
Analyze the data using the ΔΔCT method by calculating the relative changes (relative quantity, RQ) in gene expression using GAPDH as the endogenous control and monolayer MSCs as a baseline8,14.
4. Collection of CM for Assays of Immunomodulatory and Anti-cancer Potential
Harvest the spheroids and CM as described above into a 15 ml conical tube, using a cell lifter and a pipette, however, do not wash the dish lids with PBS as this will dilute the CM.
Centrifuge at 450 x g for 5 min at room temperature, carefully collect the cell-free supernatant with a pipette, and transfer the supernatant into 1.5 ml tubes.
Centrifuge at 10,000 x g for 5-10 min at room temperature, carefully collect the clarified CM with a pipette, and transfer the CM into new 1.5 ml tubes for storage in a freezer (-80 °C). Prepare aliquots of the CM prior to freezing when using it for multiple assays. NOTE: Prior to performing downstream assays, PGE2 concentration, a good indicator of anti-inflammatory potential of CM, can be determined using a commercial ELISA kit according to manufacturer's instructions. TSG6 concentration can be determined using a published protocol14.
5. Macrophage Assay to Assess the Anti-inflammatory Potential of MSC-CM
Prepare macrophage medium, which consists of Dulbecco's modified Eagle medium (DMEM) containing L-alanyl-L-glutamine and supplemented with 10% premium select FBS and 1x penicillin-streptomycin. Filter-sterilize the medium.
Obtain a frozen vial of approximately 106 J774 mouse macrophages and incubate the vial for 2 min in a 37 °C water bath.
Transfer the contents of the vial into a 15 ml conical tube, add 9 ml of macrophage medium, and centrifuge for 5 min at 200-300 x g and at room temperature.
Aspirate the supernatant, suspend the macrophages into 1 ml of macrophage medium, and transfer the macrophages into a 150 mm petri dish containing 30 ml of macrophage medium.
Change the medium every 2 days and incubate the cells in the incubator until approximately 70-80% confluent. NOTE: The macrophages do not adhere tightly to the petri dish, thus care should be taken not to lose cells with medium change.
Harvest the macrophages by spraying the macrophage medium onto the cells with a pipette. Transfer the cells into a 50 ml conical tube and centrifuge for 5 min at 200-300 x g and room temperature.
Aspirate the supernatant and suspend the cells in a small volume of macrophage medium for cell counts with a hemocytometer. Adjust the concentration to 200,000 cells/ml with macrophage medium.
To start the set up for the macrophage assay, add 480 µl of macrophage medium into wells of a 12-well plate.
Add 20 µl of macrophage medium into non-stimulated control wells and 20 µl of non-conditioned or MSC-conditioned medium, prepared above, in experimental wells. Mix the medium in the wells by tapping the plate.
Transfer 500 µl of the macrophage cell suspension to the non-stimulated macrophage control wells.
Stimulate the remaining macrophages with addition of 1:500-1:1,000 of 0.1 mg/ml lipopolysaccharide (LPS) and incubate 5-10 min at room temperature.
Transfer 500 µl of the stimulated macrophages into the wells with non-conditioned or MSC-conditioned medium. Mix the wells by steadily rocking the plate several times.
Transfer the plate to an incubator for 16-18 hr.
Harvest the macrophage-conditioned medium and centrifuge at 500 x g for 5 min at room temperature.
Transfer the supernatant into new tubes and assay for tumor necrosis factor-alpha (TNF-α) and interleukin-10 (IL-10) content by commercial ELISA kits following the manufacturer's instructions.
6. Splenocyte Assay to Assess the Immunomodulatory Potential of Spheroid-CM
Prepare complete splenocyte medium, which consists of Rosewell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% heat-inactivated FBS, 1x penicillin-streptomycin, and 2-mercaptoethanol. Filter-sterilize the medium.
Harvest spleens from euthanized-male BALB/c mice through an incision prepared on the left side of the abdomen over the spleen, approximately mid-way between the front and hind legs14. Transfer the spleen to a 70 µm cell strainer to isolate the splenocytes14.
Isolate the splenocytes/lymphocytes by pushing the spleens through the 70 µm strainer into a sterile petri dish using the soft rubber/plastic end of a plunger from a 2-3 ml syringe14. Use a grinding circular motion to dissociate the spleens.
Wash the cell strainer several times with cold PBS and transfer cells from the petri dish into a 50 ml conical tube. Fill the tube with cold PBS and centrifuge (500 x g) at 4 °C for 10 min.
Aspirate the supernatant and clear the cells from erythrocytes by incubation with 5-10 ml red blood cell lysis solution (per spleen) for 5-10 min.
Stop the lysis by adding cold PBS to the tube and centrifuge for 5-10 min at 500 x g and at 4°C.
Suspend the isolated cells in complete splenocyte medium, filter through a 40 µm strainer, and count the cells using a hemocytometer. Add splenocyte medium to the cell suspension to achieve a final cell concentration of 106 cells/ml NOTE: This technique typically yields approximately 3 x 107 splenocytes per mouse spleen.
Stimulate the appropriate number of splenocytes with 2 µg/ml anti-CD3e antibody for 5 min. NOTE: The quantity of splenocytes required depends on the number of experimental conditions as determined by the investigator (see step 6.9). For every experimental sample/well, 106 splenocytes are required.
Transfer 106 splenocytes (stimulated or non-stimulated) into wells of a 12-well plate and add non-conditioned (controls) or MSC-CM into wells at final dilution of 1:10-1:20.
Harvest the splenocyte CM after 24 hr and centrifuge at 500 x g for 5 min at room temperature.
Transfer the supernatant into new tubes and assay for interferon gamma (IFN-γ) content by commercial ELISA kit following the manufacturer's instructions.
7. Prostate Cancer Assay to Assess the Anti-cancer Potential of Spheroid-CM
Prepare complete RPMI growth medium that is RPMI-1640 medium supplemented with 10% FBS, 1x penicillin-streptomycin.
Obtain a frozen vial of LNCaP prostate cancer cells and incubate the vial for 2 min in a 37 °C water bath.
Transfer the contents of the vial into a culture dish containing 30 ml of complete RPMI growth medium using a motorized pipettor and a 5 ml serological pipet. Wash the vial several times with the medium from the dish and place the dish into the incubator.
Just before the cells reach confluency, wash the culture twice with room temperature PBS and detach the LNCaP cells with 3 ml of pre-warmed 0.25% trypsin/EDTA solution.
Neutralize the trypsin in the dish with the complete RPMI growth medium and transfer the cells together with washes into a 50 ml conical tube.
Centrifuge the cells at 450 x g for 10 min at room temperature and aspirate the supernatant.
Re-suspend the cells into small volume of the complete RPMI growth medium and count the cancer cells using a hemocytometer.
Transfer LNCaP cells into a 96-well dish at 5,000 cells/well in 120 µl of complete RPMI growth medium containing 25% of non-conditioned or spheroid-CM. Incubate in the incubator for 72 hr.
- To perform a cell growth assay using a cell proliferation assay kit, aspirate the medium from wells and transfer the plate into a freezer (-80 °C) for at least 3 hr.
- Thaw the plate and lyse the cells in each well by adding 100 µl of cell lysis reagent containing 180 mM NaCl, 1 mM EDTA, and 0.2 mg/ml RNase A.
- For a standard curve, lyse known amounts of LNCaP cells as described above.
- After 1 hr, add 100 µl of 1x cell lysis buffer containing 1:100 dilution of cell proliferation assay reagent and measure the fluorescence in each well to determine the cell amount.
8. Intraperitoneal Delivery of Intact Spheroids
Prepare spheroid MSCs as described above (protocol 2) and resuspend the spheres in sterile Hank's Balanced Salt Solution (HBSS) supplemented with 0.2-0.5 % HSA to maintain XF conditions. HSA in the solution helps to prevent sphere adhesion to plastic during injection.
Transfer 40-120 spheroids into 15 ml conical tubes and wash the cells by adding 6 ml HBSS/HSA to the tubes. Allow 3-4 min for spheres to descend. Prepare an independent conical tube of spheroids for each animal. Also, prepare additional tubes of spheroids for assays determining spheroid retention (see 8.18 below) and production of therapeutic factors after transfer (see 8.19 below) if desired.
Aspirate the supernatant, overlay the spheroids with 100-200 µl of sterile HBSS supplemented with 0.2-0.5% HSA, and place the tubes on ice.
Briefly anesthetize the animals with 2% isoflurane in 100% oxygen until the disappearance of pinch-toe reflex for approximately 2 min in the induction chamber.
Place the animal inside the BSL-2 cabinet, dorsal aspect down (ventral side facing up), with the continuous flow of 1.5% isoflurane/oxygen mixture via a nose cone.
Clean the lower mouse abdomen with an antiseptic such as 70% alcohol.
Remove the cap of the 20 G intravenous (iv) catheter and perform the intraperitoneal injection with the catheter-stiletto assembly. Use one hand to hold the mouse skin and another to perform the injection. Locate the point of injection approximately in the middle of an artificial line connecting a flexed knee joint and genital area with the needle tip pointing toward the midline. NOTE: The catheter-stiletto assembly has a higher resistance than a regular needle, therefore care has to be taken as not to inject the needle too deep into the abdomen, which could result in injury to internal organs. Penetration of the skin and then muscles/peritoneal membrane can be felt during catheter placement.
Gently remove the metal stiletto/needle from the catheter-stiletto assembly. Check the stiletto for blood, urine and feces and discard it. Blood on the needle indicates puncture of internal organ(s).
Hold the plastic catheter in an upright position during the injections to permit fluid flow.
Confirm the proper placement of the plastic catheter by injecting approximately 100 µl of sterile HBSS/HSA through the catheter using a pipette with a 100-200 µl tip. Place the end of the pipette tip inside the catheter syringe adapter forming a tight seal. Slowly inject the fluid inside the peritoneal cavity. NOTE: The fluid in a properly positioned catheter will freely flow inside the peritoneal cavity with only minor adjustment of the catheter. Improper placement of the catheter (subcutaneous or intra-intestinal, or if the tip of the needle injured peritoneal organs such as kidneys) will increase resistance and reduce flow. Subcutaneous placement will result in a formation of an obvious "bubble" under the skin.
Collect the MSC spheroids, prepared in 100-200 µl of HBSS/HSA (step 8.3), using a 200 µl pipette tip, and transfer the entire volume of spheres (40-120 spheres) into the peritoneal cavity through the catheter as described above.
Wash the tube containing spheres with 100 µl of HBSS/HSA using the same tip as above to collect any residual spheroids and inject them into the peritoneal cavity.
(Optional) Inject an additional 100 µl of HBSS/HSA into the peritoneal cavity to wash the catheter.
Place the gauze pad soaked in antiseptic over the catheter and slowly remove the catheter from the peritoneal cavity and discard it.
Gently massage the peritoneal cavity with the fingers to ensure even distribution of the spheroids.
Let the animal recover under close supervision and watch for bleeding from the injection site.
Inspect the catheter and 15 ml tube for any remaining spheroids. It is normal to observe a few spheres in the catheter/tube. NOTE: The technique described for MSC spheroid delivery can be adopted for use in normal animals or in a variety of injury models. This technique has been successfully used to inject spheroids in mouse corneal injury and endotoxemia models, as well as in a zymosan-induced peritonitis model8.
- To quantify the number of retained spheroids (optional), use a DNA-based cell number quantification kit/reagent. Hold the used catheter over the 15 ml tube and vigorously dispense HBSS/HSA through the catheter to force any remaining spheres back into the 15 ml tube.
- Centrifuge the 15 ml tube at 500 x g for 5 min at room temperature and aspirate the supernatant. Transfer the tube containing the spheroid pellet into a freezer (-80 °C) for at least 3 hr.
- Thaw the tube and lyse the spheres by adding 500 µl of cell lysis reagent containing 180 mM NaCl, 1 mM EDTA, and 0.2 mg/ml RNase A.
- For a control, freeze and lyse a known amount of spheroids (preferably equivalent to the number of spheroids prepared for a single injection) as described above.
- After 1 hr, transfer 100 µl of the cell lysate, in duplicate, into a 96-well microplate and add to each well 100 µl of 1x cell lysis buffer containing a 1:50 dilution of cell proliferation assay reagent from the kit. After 10 min, measure the fluorescence in each well to determine the number of spheres that were not injected by comparing the experimental sample(s) with the control sample.
- Evaluate the activation level of spheroids prepared for injection (optional) by measuring concentration of PGE2 and TSG6 produced by transferred spheres. Transfer 40-120 spheres through the catheter, as described above, into a 12-well or 6-well plate containing 1 or 2 ml αMEM supplemented with 2% FBS and 1x penicillin/streptomycin. Place the plates in the incubator for 6 hr.
- Transfer the medium from the wells after 6 hr into 1.5 or 15 ml tubes and centrifuge the tubes at 450 x g for 5 min at room temperature.
- Transfer the cell-free supernatant into fresh 1.5 ml tubes using a pipette and centrifuge at 10,000 x g for 5-10 min at room temperature.
- Transfer the clarified CM (supernatant) into new 1.5 ml tubes and assay for PGE2 concentration (good indicator of anti-inflammatory potential of CM) using a commercial ELISA kit according to manufacturer's instructions. TSG6 concentration can be determined using a published protocol14.
Representative Results
In the current work, hanging drop cultures were employed to generate compact spherical micro-tissues or 'spheroids' of activated MSCs under XF conditions. The investigational roadmap in Figure 1 depicts that MSCs are encouraged to self-assemble into spheroids when suspended in hanging drops for 72 hr, after which the spheroids, or the CM loaded with sphere-derived therapeutic factors, can be collected and potentially utilized in both research and clinical applications. The large number of cells needed for sphere production can be acquired within one week by seeding the MSCs at low density, typically 100-200 cells per cm2, and then expanding the cells for 6-7 days until approximately 80% confluent (Figure 2). Cell growth kinetics, determined by counting viable cells daily, indicated that a robust expansion phase (day 3-7) follows a brief lag phase of 1-2 days (Figure 2C). MSCs in passage 2 or 3 typically yield 50-100 million cells from a single vial of 1 million cryopreserved cells when cultured in CCM.
Following expansion and harvest, the MSCs are suspended at high cell concentration to generate spheroids, typically 500-1,000 cells per µl. Hanging drop cultures are prepared by transferring 25-40 µl droplets of medium containing the MSCs onto the underside of a tissue-culture dish lid, which is subsequently flipped and appropriately positioned onto the base of the dish (Figure 3). When suspended in 35 µl drops of CCM at 714 cells per µl (i.e. 25,000 cells per drop), MSCs first assemble into small aggregates that eventually coalesce to generate a single compact sphere at 72 hr in the apex of the drop (Figure 3C). Spheroids also form over 72 hr in several types of commercially available XF media optimized for MSC expansion, here referred to as XFM-1 and XFM-2. However, formation of single compact spheres in XFM-1 requires supplementation with 13 mg/ml human serum albumin (HSA), a concentration that reflects the estimated total protein content in CCM (Figure 3D). Single compact spheres do not readily form in basic XFM-1 without HSA or in protein-free αMEM (Figure 3D). During sphere assembly, MSC phenotype changes radically (Figure 4), and when in the appropriate chemically-defined XF medium (i.e. XFM-1 + HSA), expression of numerous anti-inflammatory factors are upregulated including PGE2 and TSG6 (Figure 4C). However, production of TSG6 and PGE2 is not markedly augmented in MSCs cultured as spheres in XFM-2 or XFM-1 without HSA (Figure 4C). Notably these anti-inflammatory factors, as well as the anti-cancer factors TRAIL and IL-24, were highly upregulated in spheroid MSCs relative to monolayer MSCs, when the spheres were cultured in XFM-1 supplemented with either recombinant HSA (rHSA) or clinical grade HSA (cHSA) prepared from human blood (Figure 4D).
To evaluate the therapeutic potential of MSC spheres prepared in various media formulations, several practical tests are performed (Figure 5). The immune-modulatory properties of spheroids are first determined in vitro by measuring the effects of sphere CM on levels of cytokines produced by LPS-stimulated macrophages and CD3-stimulated splenocytes/lymphocytes (Figure 5). CM from MSC spheres cultured in CCM and XFM-1/HSA markedly suppressed production of macrophage TNFα and splenocyte IFNγ, while at the same time enhanced levels of the anti-inflammatory cytokine IL-10 (Figure 5A and 5B). The anti-cancer properties of spheres are evaluated by measuring the effects of sphere CM on growth and morphology of LNCaP prostate cancer cells. The XF medium XFM-1/HSA effectively reduced LNCaP proliferation similar to FBS-containing CCM (Figure 5C and 5D).
Importantly, intact MSC spheres can be administered into the peritoneal cavity of mice to test the anti-inflammatory activity of the cells in vivo (Figure 6). For these experiments, spheres are prepared for injection in HBSS containing a low concentration of HSA, which minimizes their adhesion to plastic tubing while preserving a XF state. Subsequently, spheres can be easily injected after transfer from the collection tube (Figure 6A), using a pipette (Figure 6B), through a needle/catheter assembly (Figure 6C and 6D) appropriately positioned for a standard intraperitoneal injection. Using this approach, MSC spheroids can be regularly transferred at an efficiency greater than 90% (Figure 6E and 6F) without disrupting their integrity (Figure 6G and 6H). Improper positioning of the catheter tip within the peritoneal cavity leads to poor sphere delivery and high retention (Figure 6F). Importantly, level of sphere retention within the pipette/catheter can be qualitatively determined by visual inspection, or quantitatively by collecting/lysing the retained spheres and measuring cell number with a commercially available DNA-based cell quantification reagent/kit (Figure 6F). Post-injection sphere retention analysis helps create inclusion/exclusion criteria during experimentation. Notably, the ability for CCM and XFM-1/HSA spheroids to secrete high levels of TSG6 and PGE2 are maintained at least 6 hr after transfer of the spheres from hanging drop cultures (Figure 6I). Similar to their in vitro effects, XFM-1/HSA spheres injected into the peritoneal cavity of mice, immediately following induction of systemic inflammation by iv administration of LPS, enhanced PGE2 and IL-10 levels in the peritoneum while decreasing pro-inflammatory TNFα (Figure 6J).
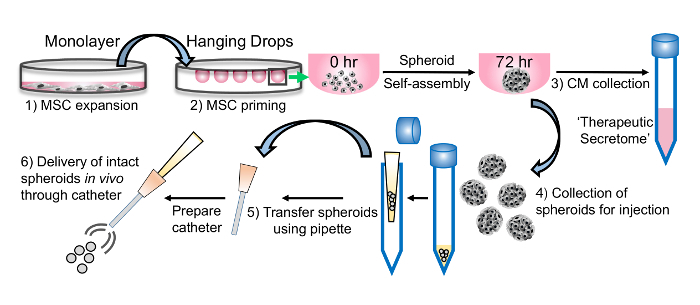 Figure 1: Schematic representation of the platform developed for harnessing the secretome of MSCs primed as spherical micro-tissues under XF conditions. Following expansion as monolayer cultures (1), the MSCs are suspended in hanging drops from the lid of a culture dish (2) at high cell concentration to activate/prime the cells. After 72 hr, both the (3) conditioned medium (CM) and (4) intact spheroids can be harvested from the droplets and utilized in various downstream applications. The CM is rich in immune-modulating factors, as well as other therapeutic components. The compact spheroids that form in hanging drops can be easily transferred using a pipette (5) and effectively administered in vivo through a catheter (6). Please click here to view a larger version of this figure.
Figure 1: Schematic representation of the platform developed for harnessing the secretome of MSCs primed as spherical micro-tissues under XF conditions. Following expansion as monolayer cultures (1), the MSCs are suspended in hanging drops from the lid of a culture dish (2) at high cell concentration to activate/prime the cells. After 72 hr, both the (3) conditioned medium (CM) and (4) intact spheroids can be harvested from the droplets and utilized in various downstream applications. The CM is rich in immune-modulating factors, as well as other therapeutic components. The compact spheroids that form in hanging drops can be easily transferred using a pipette (5) and effectively administered in vivo through a catheter (6). Please click here to view a larger version of this figure.
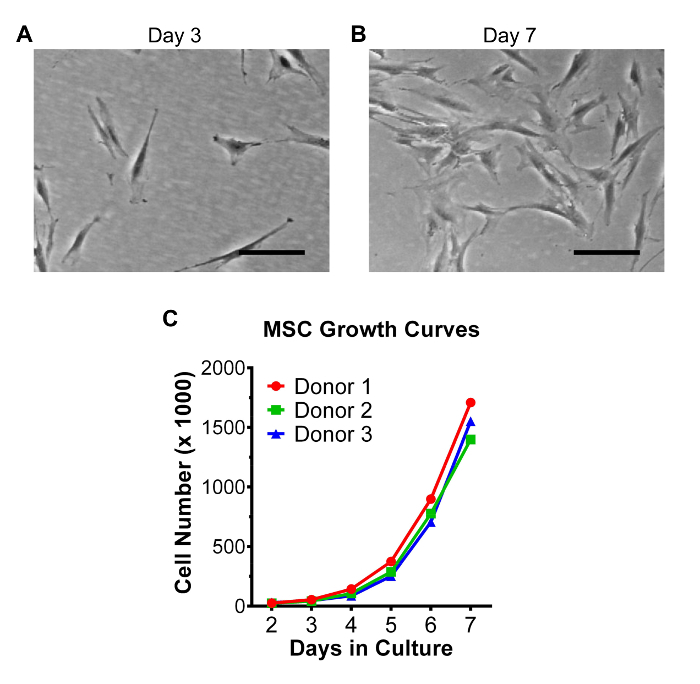 Figure 2: Growth characteristics of bone marrow-derived MSCs in monolayer cultures. MSCs in passage 2 were seeded at low density (100 cells per cm2) in 15 cm dishes (15,000 cells per dish) and cultured for 7 days in CCM. Medium was changed on day 3 and then again on day 5 or 6 (24-48 hr prior to harvest). Representative phase-contrast micrographs of MSCs 3 days (A) and 7 days (B) after plating the cells. Scale bar = 200 µm. (C) Growth kinetics of MSCs obtained from 3 donors were determined by counting the number of viable adherent cells daily from day 2 to day 7. Please click here to view a larger version of this figure.
Figure 2: Growth characteristics of bone marrow-derived MSCs in monolayer cultures. MSCs in passage 2 were seeded at low density (100 cells per cm2) in 15 cm dishes (15,000 cells per dish) and cultured for 7 days in CCM. Medium was changed on day 3 and then again on day 5 or 6 (24-48 hr prior to harvest). Representative phase-contrast micrographs of MSCs 3 days (A) and 7 days (B) after plating the cells. Scale bar = 200 µm. (C) Growth kinetics of MSCs obtained from 3 donors were determined by counting the number of viable adherent cells daily from day 2 to day 7. Please click here to view a larger version of this figure.
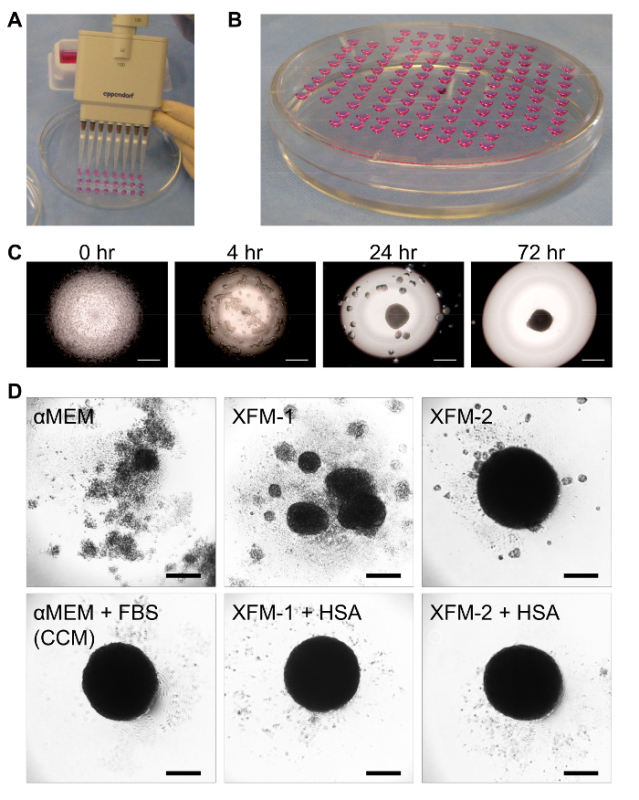 Figure 3: Preparation of hanging drop cultures for priming MSC spheroids in XF medium. (A) Photograph depicting the technique to rapidly layer hanging drops on the underside of a tissue culture dish lid. (B) Photo illustrating hanging drops after inversion and positioning of the lid onto the dish base. (C) Time-dependent changes in the aggregation of 25,000 MSCs in a hanging drop as visualized by phase-contrast microscopy. The objective was focused on the apex of the drop. Scale bar = 500 µm. (D) Images of MSC spheres formed after 3 days in hanging drop cultures using various media formulations including αMEM with and without FBS (i.e. CCM), XFM-1 with and without HSA, and XFM-2 with and without HSA. Scale bar = 200 µm. Data in panel D were obtained from the original article with permission from the publisher14. Please click here to view a larger version of this figure.
Figure 3: Preparation of hanging drop cultures for priming MSC spheroids in XF medium. (A) Photograph depicting the technique to rapidly layer hanging drops on the underside of a tissue culture dish lid. (B) Photo illustrating hanging drops after inversion and positioning of the lid onto the dish base. (C) Time-dependent changes in the aggregation of 25,000 MSCs in a hanging drop as visualized by phase-contrast microscopy. The objective was focused on the apex of the drop. Scale bar = 500 µm. (D) Images of MSC spheres formed after 3 days in hanging drop cultures using various media formulations including αMEM with and without FBS (i.e. CCM), XFM-1 with and without HSA, and XFM-2 with and without HSA. Scale bar = 200 µm. Data in panel D were obtained from the original article with permission from the publisher14. Please click here to view a larger version of this figure.
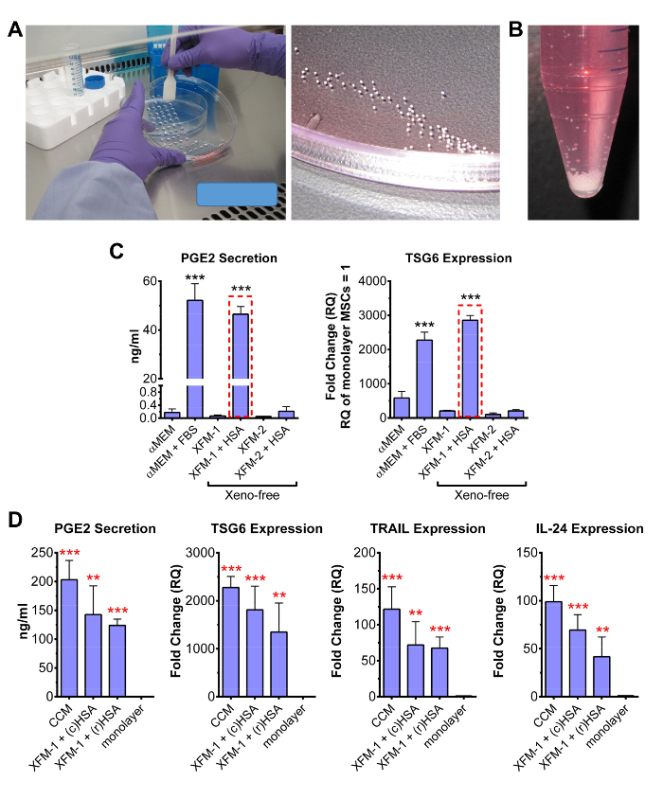 Figure 4: Evaluation of MSC activation in 3-D XF cultures. (A) Spheres/CM are harvested after 72 hr by inverting/tilting the dish lid and forcing the droplets to the edge using a cell lifter. Spheres assembled from 25,000 cells can be easily visualized during collection. (B) Photograph of approximately 500 spheres collected from lids of numerous tissue culture plates. Spheres rapidly descend to the bottom of tube without centrifugation. (C) Level of PGE2 secreted from MSC spheres after 72 hr was measured by ELISA. Real-time RT-PCR was used to measure TSG6 levels. Both TSG6 and PGE2 are valuable markers for screening MSC activation in spheroids. The XF medium XFM-1 + HSA showed high level of PGE2 and TSG6 production (red boxes). Data are shown as mean ± SD and were analyzed by Student's t-test (*** p < 0.001, compared to αMEM sample). (D) PGE2 ELISA and real-time RT-PCR assays on spheres produced in CCM and XFM-1 supplemented specifically with 13 mg/ml recombinant HSA (rHSA), or clinical grade HSA (cHSA) prepared from human venous blood. Fold changes were determined from adherent monolayer MSCs (RQ = 1). Data are shown as mean ± SD and were analyzed by Student's t-test (** p < 0.01, *** p < 0.001, compared to monolayer MSCs). Some data in panels C and D were obtained from the original article with permission from the publisher14. Please click here to view a larger version of this figure.
Figure 4: Evaluation of MSC activation in 3-D XF cultures. (A) Spheres/CM are harvested after 72 hr by inverting/tilting the dish lid and forcing the droplets to the edge using a cell lifter. Spheres assembled from 25,000 cells can be easily visualized during collection. (B) Photograph of approximately 500 spheres collected from lids of numerous tissue culture plates. Spheres rapidly descend to the bottom of tube without centrifugation. (C) Level of PGE2 secreted from MSC spheres after 72 hr was measured by ELISA. Real-time RT-PCR was used to measure TSG6 levels. Both TSG6 and PGE2 are valuable markers for screening MSC activation in spheroids. The XF medium XFM-1 + HSA showed high level of PGE2 and TSG6 production (red boxes). Data are shown as mean ± SD and were analyzed by Student's t-test (*** p < 0.001, compared to αMEM sample). (D) PGE2 ELISA and real-time RT-PCR assays on spheres produced in CCM and XFM-1 supplemented specifically with 13 mg/ml recombinant HSA (rHSA), or clinical grade HSA (cHSA) prepared from human venous blood. Fold changes were determined from adherent monolayer MSCs (RQ = 1). Data are shown as mean ± SD and were analyzed by Student's t-test (** p < 0.01, *** p < 0.001, compared to monolayer MSCs). Some data in panels C and D were obtained from the original article with permission from the publisher14. Please click here to view a larger version of this figure.
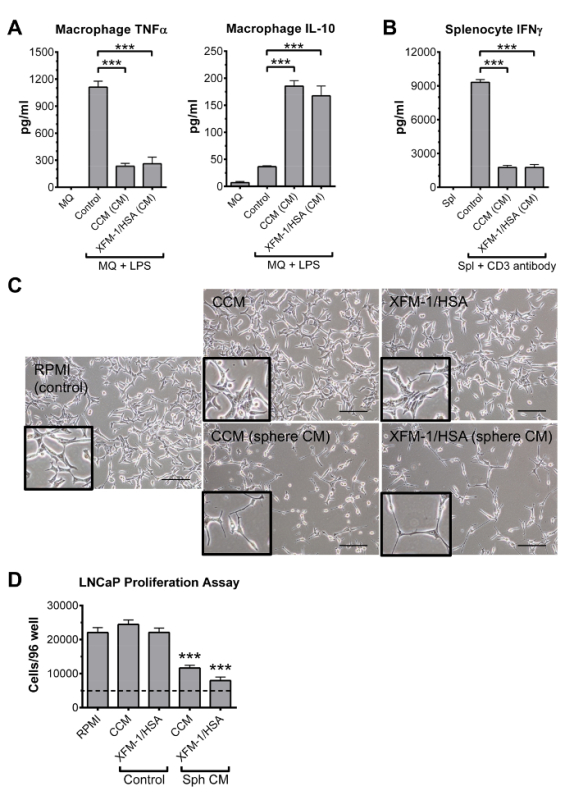 Figure 5: In vitro functional assays for evaluating the anti-inflammatory and anti-cancer properties of MSC-CM collected from hanging drop cultures. (A) Representative results from ELISAs showing the effects of sphere CM (CCM and XFM-1/HSA) on production of TNFα and IL-10 by LPS-stimulated mouse macrophages (MQ). (B) Representative results from ELISA showing the effects of sphere CM (CCM and XFM-1/HSA) on production of IFNγ by mouse splenocytes (Spl) stimulated with an anti-CD3 antibody. (C) Phase contrast micrographs of LNCaP prostate cancer cells treated with basic CCM and XFM-1/HSA or conditioned medium (CM) from MSC spheres prepared in CCM and XFM-1/HSA. LNCaP cells were cultured in RPMI medium (control). Scale bar is 200 µm. (D) Quantitative changes in growth of the LNCaP prostate cancer cells was determined with a commercially available DNA-based cell quantification reagent/kit. Dotted line indicates seeding density of 5,000 cells per well. Data in all panels are shown as mean ± SD, and were analyzed by Student's t-test (*** p < 0.001). Some data in all panels were obtained from the original articles with permission from the publisher14. Please click here to view a larger version of this figure.
Figure 5: In vitro functional assays for evaluating the anti-inflammatory and anti-cancer properties of MSC-CM collected from hanging drop cultures. (A) Representative results from ELISAs showing the effects of sphere CM (CCM and XFM-1/HSA) on production of TNFα and IL-10 by LPS-stimulated mouse macrophages (MQ). (B) Representative results from ELISA showing the effects of sphere CM (CCM and XFM-1/HSA) on production of IFNγ by mouse splenocytes (Spl) stimulated with an anti-CD3 antibody. (C) Phase contrast micrographs of LNCaP prostate cancer cells treated with basic CCM and XFM-1/HSA or conditioned medium (CM) from MSC spheres prepared in CCM and XFM-1/HSA. LNCaP cells were cultured in RPMI medium (control). Scale bar is 200 µm. (D) Quantitative changes in growth of the LNCaP prostate cancer cells was determined with a commercially available DNA-based cell quantification reagent/kit. Dotted line indicates seeding density of 5,000 cells per well. Data in all panels are shown as mean ± SD, and were analyzed by Student's t-test (*** p < 0.001). Some data in all panels were obtained from the original articles with permission from the publisher14. Please click here to view a larger version of this figure.
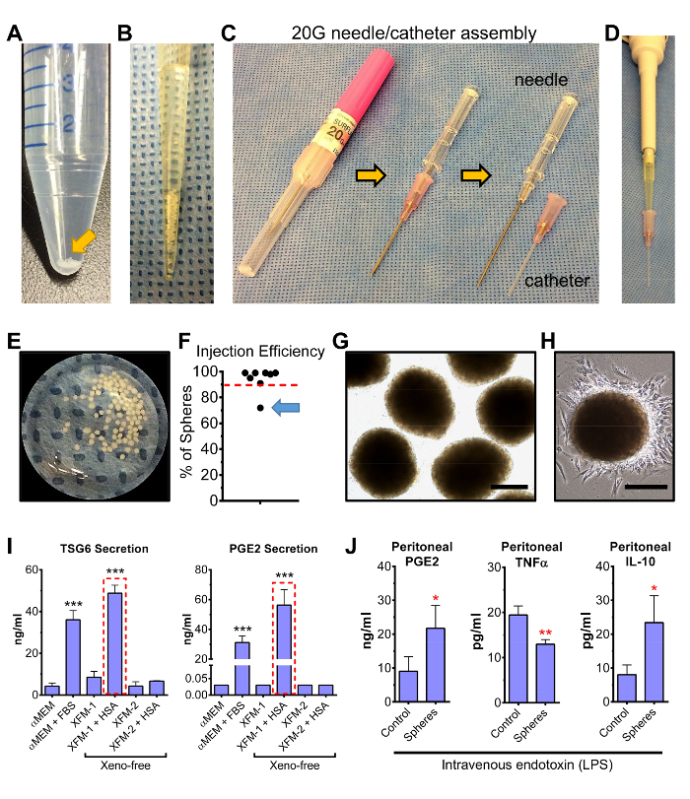 Figure 6: Efficient transfer of intact MSCs spheroids, prepared under XF conditions, using a 20G iv needle/catheter system. (A) Representative photograph of a 15 ml conical tube with XFM-1/HSA MSC spheres prepared for injection in HBSS containing 0.2% HSA. (B) Representative photograph of the MSC spheres after aspiration into a 200 µl pipette tip. (C) Image of the 20 G needle/catheter assembly used to administer intact MSC spheroids into mice. (D) Image demonstrating proper positioning of the pipette tip into the adapter of the polyurethane catheter. (E) Image of the MSC spheres immediately after passing them through the catheter into a culture dish. Approximately 95 spheres out of 100 were transferred. (F) Efficiency of sphere delivery into the peritoneal cavity of mice was determined by collecting/lysing spheres retained in the pipette/catheter and measuring cell numbers, via a commercial DNA-based cell quantification assay, relative to the number of cells obtained from a full complement of lysed spheres. Dotted red line indicates a transfer efficiency of 90%. Poor sphere delivery (71%) was observed with one injection (arrow). (G) Phase-contrast micrographs of spheres in panel E (magnified view). Scale bar = 200 µm. (H) Phase-contrast micrograph of a XFM-1/HSA sphere 16 hr after transfer onto a tissue culture dish. The fibroblastic, spindle-shaped morphology of MSCs is maintained in cells that migrated out of the sphere. Scale bar = 200 µm. (I) ELISA assays for the anti-inflammatory factors TSG6 and PGE2 were performed on CM collected 6 hr after transfer of the spheres into 6-well plates containing 2 ml αMEM/2% FBS. Spheres prepared in the XF medium XFM-1/HSA showed highest level of TSG6 and PGE2 secretion (red boxes). Data, expressed as mean ± SD, were analyzed by Student's t-test (*** p < 0.001, compared to αMEM sample). Some data were obtained from the original article with permission from the publisher14. (J) Representative graphs illustrating the ability for spheroids, injected into the peritoneal cavity, to increase peritoneal PGE2 and IL-10 while decreasing level of TNFα in mice challenged by intravenous injection of endotoxin (LPS). Samples were obtained by peritoneal lavage 6 hr after induction of inflammation and delivery of 80 spheroids. Data, expressed as mean ± SD, were analyzed by Student's t-test (* p < 0.05, ** p < 0.01, compared to control). Please click here to view a larger version of this figure.
Figure 6: Efficient transfer of intact MSCs spheroids, prepared under XF conditions, using a 20G iv needle/catheter system. (A) Representative photograph of a 15 ml conical tube with XFM-1/HSA MSC spheres prepared for injection in HBSS containing 0.2% HSA. (B) Representative photograph of the MSC spheres after aspiration into a 200 µl pipette tip. (C) Image of the 20 G needle/catheter assembly used to administer intact MSC spheroids into mice. (D) Image demonstrating proper positioning of the pipette tip into the adapter of the polyurethane catheter. (E) Image of the MSC spheres immediately after passing them through the catheter into a culture dish. Approximately 95 spheres out of 100 were transferred. (F) Efficiency of sphere delivery into the peritoneal cavity of mice was determined by collecting/lysing spheres retained in the pipette/catheter and measuring cell numbers, via a commercial DNA-based cell quantification assay, relative to the number of cells obtained from a full complement of lysed spheres. Dotted red line indicates a transfer efficiency of 90%. Poor sphere delivery (71%) was observed with one injection (arrow). (G) Phase-contrast micrographs of spheres in panel E (magnified view). Scale bar = 200 µm. (H) Phase-contrast micrograph of a XFM-1/HSA sphere 16 hr after transfer onto a tissue culture dish. The fibroblastic, spindle-shaped morphology of MSCs is maintained in cells that migrated out of the sphere. Scale bar = 200 µm. (I) ELISA assays for the anti-inflammatory factors TSG6 and PGE2 were performed on CM collected 6 hr after transfer of the spheres into 6-well plates containing 2 ml αMEM/2% FBS. Spheres prepared in the XF medium XFM-1/HSA showed highest level of TSG6 and PGE2 secretion (red boxes). Data, expressed as mean ± SD, were analyzed by Student's t-test (*** p < 0.001, compared to αMEM sample). Some data were obtained from the original article with permission from the publisher14. (J) Representative graphs illustrating the ability for spheroids, injected into the peritoneal cavity, to increase peritoneal PGE2 and IL-10 while decreasing level of TNFα in mice challenged by intravenous injection of endotoxin (LPS). Samples were obtained by peritoneal lavage 6 hr after induction of inflammation and delivery of 80 spheroids. Data, expressed as mean ± SD, were analyzed by Student's t-test (* p < 0.05, ** p < 0.01, compared to control). Please click here to view a larger version of this figure.
Discussion
The optimal MSC for use in some research and clinical applications should be highly activated to maximize their benefit, and preferentially prepared under chemically defined XF conditions to minimize the delivery of potential antigens from xenogeneic medium components such as FBS. In the protocols described here, we have shown methods to 1) activate MSCs in 3D culture by formation of spheroids, 2) achieve the 3D activation of MSCs under XF conditions, 3) evaluate the activation levels of spheroid MSCs in regards to their anti-inflammatory, immune-modulatory, and anti-cancer potential, and 4) deliver activated MSCs as intact spheroids into mice.
MSCs are multipotent stem cells that can be isolated via plastic adherence from numerous adult tissues including bone marrow and adipose tissue. MSCs are easily propagated on tissue culture plastic under relatively high (10-20%) FBS concentrations, express a distinct set of surface markers, and can be differentiated at least into adipogenic, osteogenic, and chondrogenic lineages1,16,17,18. MSCs have been demonstrated to exert beneficial effects in various animal models of human diseases, even though they appear to persist only transiently after their administration in vivo. The effect of MSCs has therefore been called "hit and run" and is often mediated by the anti-inflammatory and immunomodulatory paracrine factors, such as PGE2, TSG-6, and indoleamine 2,3-dioxygenase (IDO), secreted by MSCs2,3,11,19,20,21. However, in order to produce these factors, MSCs require activation, either through signals from a damaged tissue or from immune cells of the recipient. Subsequently, there has been an emerging interest to develop MSC pre-activation or priming protocols before delivery of the cells into animals or patients22. Also, the presence of antigenic FBS components in standard MSC preparations has raised concerns. Therefore, studies have been conducted to determine optimal chemically defined XF conditions for MSC cultures. In our recent work, we first showed that MSCs can be activated in 3D by formation of spheroids, and then discovered that cell activation can also be achieved under specific XF conditions8,12,13,14.
3D cell culture techniques have been employed for decades with the goal of providing genuine physiological conditions in cell-based research. Cultures in 2D disregard the 3D nature of tissues and therefore do not always recapitulate the cell-to-cell and cell-to-matrix interactions important in cell signaling. Many 3D culture techniques use rotating vessels, spinner flasks, or various non-adherent surfaces, however, the biggest drawback in many of these methods is the heterogeneity of the generated spheroid size and/or the requirement of specific expensive equipment. Research has also been performed using hanging drop cultures that essentially encourage MSCs to spontaneously aggregate into a single spheroid4,5,6,7,8,9,23. Notably, hanging drop cultures support assembly of relatively uniform micro-tissues/aggregates, with the added benefit of being able to easily manipulate size of the cell aggregate by adjusting cell concentration and/or drop volume. Moreover, mastery of the hanging drop culture technique does not require extensive training. Drops of 25-40 µl can be easily prepared with MSCs at high cell concentrations, typically 500-1,000 cells per µl. Drops smaller than 20 µl tend to evaporate quickly, and those larger than 45 µl often smear across the lid during inversion. However, when flipping a lid containing droplets of any size, speed and directionality are critical parameters to consider for maintaining surface tension and appropriate shape of the drop/spheroid. Uninterrupted culture and proper airflow in the incubator are also important for ensuring the MSCs aggregate into a single sphere within each drop.
A disadvantage of this technique is that plates containing hanging drops should not be stacked in the incubator or moved during sphere assembly, making scale-up for patient therapies difficult. Moreover, changing medium in hanging drop cultures is extremely challenging, thus long-term cultures should be avoided. In addition, the low media-to-cell ratio in hanging drops can cause nutrient depletion and waste accumulation eventually leading to the loss of important cellular functions and/or cell death.
However, with proper hanging drop technique, we have demonstrated that MSCs in spheroids become highly active or primed, in that the cells become factories of the powerful anti-inflammatory molecules PGE2 and TSG6, as well as the anti-cancer molecules IL-24 and TRAIL. We have shown that MSC spheroids are indeed anti-inflammatory and immunomodulatory, and have anti-cancer properties superior to their 2D monolayer counterparts in numerous functional assays using cultured macrophages, splenocytes, and prostate cancer cells8,12,13,14. Furthermore, we have shown that MSC spheroids can exert potent anti-inflammatory effects when administered into the peritoneal cavity of mice with peritonitis8. Specifically, we showed that large spheroids of approximately 400-500 µm in diameter (25,000-30,000 MSCs each) can be efficiently delivered in a small volume of HBSS using a 20G needle/catheter assembly and a standard pipette. With this technique, improper catheter placement, which results in high resistance to fluid flow, can be easily determined prior to spheroid transfer by first injecting a small volume of HBSS/HSA as described in the protocols. The spheroids in a properly positioned catheter will flow freely, provided that the HBSS is supplemented with HSA to minimize sphere adhesion to the plastic tubing, and can be easily visualized. Moreover, this technique prevents shear stress on cells which can occur using a standard needle/syringe for injection, and avoids the need for a surgical incision which carries a higher risk of infection and is more technically challenging. One major disadvantage is that large numbers of spheroids can only be injected into spacious body cavities, such as the peritoneal cavity, as the resistance against sphere transfer through the catheter is high in constricted areas.
We also recently demonstrated that the same level of MSC activation in spheroids could be achieved by using a specific commercially available XF medium, referred to here as XFM-1, as long as it was supplemented with HSA obtained from human blood or prepared through recombinant techniques14. It is important to note here that MSC spheroids do not produce high levels of PGE2 and TSG6 in all types of XF media14, probably because most XF media for MSCs were initially formulated for optimal expansion of the cells in 2D. It is also important to note that different types of HSA vary in their potency14. In addition, the absolute level of PGE2 detected in CM of activated spheroids can vary slightly as the ELISA employed for PGE2 is indeed a competition assay. Thus, it is crucial to include proper controls in every PGE2 ELISA and to avoid directly comparing data between samples assayed at different times or in different plates. Moreover, MSCs must be thoroughly washed in XF medium prior to preparing hanging drops in order to remove residual CCM and, therefore, limit carryover of FBS components. The protocols described here can be easily adopted to evaluate other media formulations on sphere formation and therapeutic gene expression.
Clearly, numerous cell-signaling events that do not normally occur in 2D MSCs are set in motion when MSCs are cultured in 3D. Cell-to-cell and cell-to-matrix interactions, mediated by cadherins and integrins guide spheroid formation and compaction24,25,26 and are likely critical for spheroid therapy. As the cells aggregate and compact into spheroids, various stress signals, including minor apoptosis, aid in the process of MSC activation that results in production of numerous potentially therapeutic factors4,5. We previously showed the critical role of autocrine IL-1 signaling in MSC activation and production of PGE2 and TSG612. While much remains unknown about the various signaling pathways that aid in MSC activation, the hanging drop culture platform affords a practical way to study this phenomenon and improve MSC-based therapies. Overall, we have described, in the protocols here, the means of activating or priming MSCs in 3D cultures, under chemically-defined XF conditions, to produce anti-inflammatory, immunomodulatory, and anti-cancer factors. We have also described how these intact MSC spheroids can be delivered in vivo.
Disclosures
The authors declare they have no competing financial interests.
Acknowledgments
This work was funded in part by grant P40RR17447 from the National Institute of Health and award RP150637 from the Cancer Prevention and Research Institute of Texas. We would like to thank Dr. Darwin J. Prockop for his support on the project.
References
- Keating A. Mesenchymal stromal cells: New directions. Cell Stem Cell. 2012;10(6):709–716. doi: 10.1016/j.stem.2012.05.015. [DOI] [PubMed] [Google Scholar]
- English K, Wood KJ. Mesenchymal stromal cells in transplantation rejection and tolerance. Cold Spring Harb Perspect.Med. 2013;3(5):a015560. doi: 10.1101/cshperspect.a015560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat.Rev.Immunol. 2012;12(5):383–396. doi: 10.1038/nri3209. [DOI] [PubMed] [Google Scholar]
- Follin B, Juhl M, Cohen S, Perdersen AE, Kastrup J, Ekblond A. Increased Paracrine Immunomodulatory Potential of Mesenchymal Stromal Cells in Three-Dimensional Culture. Tissue Eng.Part B.Rev. 2016. [DOI] [PMC free article] [PubMed]
- Cesarz Z, Tamama K. Spheroid Culture of Mesenchymal Stem Cells. Stem Cells Int. 2016. p. 9176357. [DOI] [PMC free article] [PubMed]
- Achilli TM, Meyer J, Morgan JR. Advances in the formation, use and understanding of multi-cellular spheroids. Expert Opin.Biol. Ther. 2012;12(10):1347–1360. doi: 10.1517/14712598.2012.707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sart S, Tsai AC, Li Y, Ma T. Three-dimensional aggregates of mesenchymal stem cells: cellular mechanisms, biological properties, and applications. Tissue Eng.Part B.Rev. 2014;20(5):365–380. doi: 10.1089/ten.teb.2013.0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosh TJ, et al. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc.Natl.Acad.Sci.U.S.A. 2010;107(31):13724–13729. doi: 10.1073/pnas.1008117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova IA, et al. Mesenchymal stem cells support migration, extracellular matrix invasion, proliferation, and survival of endothelial cells in vitro. Stem Cells. 2007;25(7):1761–1768. doi: 10.1634/stemcells.2007-0022. [DOI] [PubMed] [Google Scholar]
- Frith JE, Thomson B, Genever PG. Dynamic three-dimensional culture methods enhance mesenchymal stem cell properties and increase therapeutic potential. Tissue Eng.Part C.Methods. 2010;16(4):735–749. doi: 10.1089/ten.TEC.2009.0432. [DOI] [PubMed] [Google Scholar]
- Lee RH, et al. Intravenous hMSCs Improve Myocardial Infarction in Mice because Cells Embolized in Lung Are Activated to Secrete the Anti-inflammatory Protein TSG-6. Cell Stem Cell. 2009;5(1):54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosh TJ, Ylostalo JH, Bazhanov N, Kuhlman J, Prockop DJ. Dynamic compaction of human mesenchymal stem/precursor cells into spheres self-activates caspase-dependent il1 signaling to enhance secretion of modulators of inflammation and immunity (PGE2, TSG6, and STC1) Stem Cells. 2013;31(11):2443–2456. doi: 10.1002/stem.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylostalo JH, Bartosh TJ, Coble K, Prockop DJ. Human mesenchymal stem/stromal cells cultured as spheroids are self-activated to produce prostaglandin E2 that directs stimulated macrophages into an anti-inflammatory phenotype. Stem Cells. 2012;30(10):2283–2296. doi: 10.1002/stem.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylostalo JH, Bartosh TJ, Tiblow A, Prockop DJ. Unique characteristics of human mesenchymal stromal/progenitor cells pre-activated in 3-dimensional cultures under different conditions. Cytotherapy. 2014;16(11):1486–1500. doi: 10.1016/j.jcyt.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White. 2016. http://www.medicine.tamhsc.edu/irm/msc-distribution.html.
- Pittenger MF, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat.Rev.Immunol. 2008;8(9):726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- Fontaine MJ, Shih H, Schafer R, Pittenger MF. Unraveling the Mesenchymal Stromal Cells' Paracrine Immunomodulatory Effects. Transfus.Med.Rev. 2016;30(1):37–43. doi: 10.1016/j.tmrv.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Nemeth K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat.Med. 2009;15(1):42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prockop DJ. Concise review: two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells. 2013;31(10):2042–2046. doi: 10.1002/stem.1400. [DOI] [PubMed] [Google Scholar]
- Krampera M. Mesenchymal stromal cell 'licensing': a multistep process. Leukemia. 2011;25(9):1408–1414. doi: 10.1038/leu.2011.108. [DOI] [PubMed] [Google Scholar]
- Saleh FA, Genever PG. Turning round: multipotent stromal cells, a three-dimensional revolution? Cytotherapy. 2011;13(8):903–912. doi: 10.3109/14653249.2011.586998. [DOI] [PubMed] [Google Scholar]
- Tsai AC, Liu Y, Yuan X, Ma T. Compaction, fusion, and functional activation of three-dimensional human mesenchymal stem cell aggregate. Tissue Eng.Part A. 2015;21(9-10):1705–1719. doi: 10.1089/ten.tea.2014.0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh HY, Liu BH, Hsu SH. The calcium-dependent regulation of spheroid formation and cardiomyogenic differentiation for MSCs on chitosan membranes. Biomaterials. 2012;33(35):8943–8954. doi: 10.1016/j.biomaterials.2012.08.069. [DOI] [PubMed] [Google Scholar]
- Lee EJ, et al. Spherical bullet formation via E-cadherin promotes therapeutic potency of mesenchymal stem cells derived from human umbilical cord blood for myocardial infarction. Mol.Ther. 2012;20(7):1424–1433. doi: 10.1038/mt.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]