

Abstract
The Golgi/secretory pathway Ca2+/Mn2+-transport ATPase (SPCA1a) is implicated in breast cancer and Hailey-Hailey disease. Here, we purified recombinant human SPCA1a from Saccharomyces cerevisiae and measured Ca2+-dependent ATPase activity following reconstitution in proteoliposomes. The purified SPCA1a displays a higher apparent Ca2+ affinity and a lower maximal turnover rate than the purified sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA1a). The lipids cholesteryl hemisuccinate, linoleamide/oleamide, and phosphatidylethanolamine inhibit and phosphatidic acid and sphingomyelin enhance SPCA1a activity. Moreover, SPCA1a is blocked by micromolar concentrations of the commonly used SERCA1a inhibitors thapsigargin (Tg), cyclopiazonic acid, and 2,5-di-tert-butylhydroquinone. Because tissue-specific targeting of SERCA2b by Tg analogues is considered for prostate cancer therapy, the inhibition of SPCA1a by Tg might represent an off-target risk. We assessed the structure-activity relationship (SAR) of Tg for SPCA1a by in silico modeling, site-directed mutagenesis, and measuring the potency of a series of Tg analogues. These indicate that Tg and the analogues are bound via the Tg scaffold but with lower affinity to the same homologous cavity as on the membrane surface of SERCA1a. The lower Tg affinity may depend on a more flexible binding cavity in SPCA1a, with low contributions of the Tg O-3, O-8, and O-10 chains to the binding energy. Conversely, the protein interaction of the Tg O-2 side chain with SPCA1a appears comparable with that of SERCA1a. These differences define a SAR of Tg for SPCA1a distinct from that of SERCA1a, indicating that Tg analogues with a higher specificity for SPCA1a can probably be developed.
Keywords: calcium transport, cholesterol, Golgi, membrane transporter reconstitution, molecular modeling, prostate cancer, protein purification, cyclopiazonic acid, thapsigargin
Introduction
As an important secondary messenger, the partitioning of intracellular Ca2+ controls a wide range of cellular responses, including contraction, secretion, cell growth, differentiation, proliferation, and cell death (1, 2). A sub-micromolar basal cytosolic Ca2+ concentration range is strictly maintained by intracellular Ca2+ pumps, whereas Ca2+ channel activity produces confined spatial and temporal cytosolic Ca2+ signals up to 10–100 μm that control various cellular activities. Ca2+ transporters belonging to the P2-type Ca2+ transport ATPases maintain the low housekeeping cytosolic Ca2+ levels and reduce the cytosolic Ca2+ concentration when a Ca2+-signaling event ceases. The P2-type ATPases encompass the plasma membrane Ca2+-ATPase, SERCA,2 and SPCA Ca2+ pumps that perform active Ca2+ transport across the plasma membrane, the intracellular membranes of the sarco(endo)plasmic reticulum, and the Golgi/secretory pathway, respectively (3). SERCA and SPCA belong to the subfamily P2A and share around 30% sequence identity and over 40% similarity (4).
In humans, the SPCA proteins are encoded by two genes (ATP2C1 and ATP2C2) (4–7). SPCA1 is the housekeeping isoform, which by transporting Ca2+ and Mn2+ into the Golgi lumen supports essential Golgi/secretory pathway functions. Ca2+/Mn2+ transport is required for proper protein folding, enzymatic activities (e.g. glycosylation), sorting, and/or trafficking of proteins (8). In conjunction with SERCA, this ATPase also provides Ca2+-storage pools for subsequent Ca2+-release events (4, 9, 10). A total of four SPCA1 splice variants exist, SPCA1a–d, differing only at the C terminus. Although SPCA1a is considered to be the housekeeping variant, the differential tissue/subcellular distributions of the SPCA1 isoforms remain largely unexplored (11). The more restricted expression pattern of SPCA2 in cultured hippocampal neurons, colon, secretory acini, and luminal epithelial cells of mouse mammary tissue suggests that the second isoform may play a more specialized function, for instance in secretion (6, 12–14).
In humans, SPCA1 haploinsufficiency, due to heterozygous loss-of-function mutations, causes Hailey-Hailey disease (OMIM169600), a heritable autosomal-dominant skin disorder characterized by recurrent skin vesicles and erosions that typically arise in adulthood (15, 16). In addition, SPCA1a and/or SPCA2 proteins are up-regulated in mammary gland during lactation and in various breast cancer sub-types thereby contributing to a pathological Ca2+ dyshomeostasis (17, 18). Independent of its transport activity, SPCA2 interacts with and activates the plasma membrane Ca2+ channel Orai1 via the N and C termini (18), leading to store-independent Ca2+ entry (18) and subsequent Ca2+ transfer into the secretory pathway (19). During lactation, this system contributes to the cellular uptake of Ca2+ in mammary gland epithelial cells for the subsequent release of Ca2+ into the milk (20).
In general, SPCA1 isoforms display a higher Ca2+ affinity but a lower maximal turnover rate than SERCA isoforms (21), but so far, a detailed functional analysis of SPCA isoforms has remained difficult due to the confounding presence of other abundant housekeeping Ca2+-ATPases in cellular membrane fractions. In addition, although several potent SERCA inhibitors (thapsigargin (Tg), cyclopiazonic acid (CPA), and 2,5-di-tert-butylhydroquinone (BHQ)) are widely used to selectively inhibit endoplasmic reticulum Ca2+ transport (4, 22), selective pharmacological tools for SPCA inhibition are lacking. Because of the close sequence homology between SERCA and SPCA isoforms, several of the so-called highly selective SERCA inhibitors are also reported to inhibit SPCA in isolated membrane fractions of various organisms, albeit at much higher concentrations (4, 23–25).
The inhibition of SPCA1a by Tg may give rise to some concerns in connection with the clinical use of Tg analogues that target SERCA for prostate cancer therapy. In this therapeutic approach, a non-toxic, cell-impermeable pro-drug is administered that consists of a Tg analogue, which is coupled to a peptide that is specifically cleaved by the prostate-specific antigen (PSA) protease (26, 27). Extracellular cleavage of the peptide by local PSA in or near the prostate cancer releases the toxic Tg analogue Leu-8ADT, which is taken up by neighboring PSA-excreting cancer cells inducing targeted cell death (27). The estimated 3 orders of magnitude difference in the binding affinity of Tg for SERCA and SPCA may be considered sufficiently discriminatory to enable the use of Tg analogues for selective SERCA inhibition in vitro and in vivo. However, it remains unclear whether this is also true for other Tg analogues.
Toward the design of Tg analogues for clinical therapy related to SPCA dysfunction (see “Discussion”), we wanted to understand the off-target effect of Tg on SPCA1a, the closest relative of SERCA, by determining the structure-activation relationship (SAR) of Tg for SPCA1a. Moreover, due to the reported micromolar affinity of Tg to SPCA1 (24), one might consider Tg as a possible lead compound for the development of an SPCA1a inhibitor. Insights in the SAR of Tg for SPCA1a may aid the design of specific SPCA1a inhibitors as powerful pharmacological tools.
In this study, we purified the human SPCA1a isoform from a yeast recombinant expression system and reconstituted it into lipid vesicles. This purified system eliminates the influence of other membrane-bound contaminants or Ca2+-ATPases, allowing the biochemical characterization of SPCA1a relative to purified and reconstituted SERCA1a. In particular, we demonstrate that SPCA1a activity is sensitive to the lipid environment, and we show that Tg and cholesteric compound sensitivity of SPCA1a may depend on the same or the overlapping binding pocket formed by transmembrane helices M3, M5, and M7. Finally, we have established the SAR of Tg for SPCA1a to facilitate the design of more selective SPCA inhibitors.
Results
Affinity purification of recombinant human SPCA1a from Saccharomyces cerevisiae
The relatively low expression level of SPCA1a in native tissues, typically in a background of high SERCA expression, hampers biochemical analysis of SPCA1a. As an alternative approach, we purified His8-tagged human SPCA1a via Ni-NTA affinity chromatography from S. cerevisiae, which serves as an excellent model system to produce and purify P-type transport ATPases (8). SDS-PAGE analysis reveals that SPCA1a monomer and dimer bands were highly enriched in the elution fractions (Fig. 1A). Using mass spectrometry, we observed a molecular mass of 101.8 kDa, close to the predicted molecular mass of 101.7 kDa and high purity (>95%) of the human SPCA1a (Fig. 1B). The concentrated His-tagged SPCA1a in the elution fraction was further demonstrated by immunoblotting with SPCA1 (Fig. 1C) and His tag (Fig. 1D)-specific antibodies.
Figure 1.

Purification of the human SPCA1a isoform by affinity chromatography. Gel stained for an SDS-PAGE (A), mass spectrometry analysis (B), and immunoblotting (C and D) of hSPCA1a fractions collected during affinity purification. The yeast membrane fractions (m), the flow-through after binding to Ni-NTA beads (F), the flow-through of each washing step (W1–3), each elution fraction (E1–5), and the beads (B) after elution were collected. Equal volumes of each fraction were separated via SDS-PAGE. Immunoblotting was performed with antibodies against SPCA1a (C) or the C-terminal His tag (D). Images are representative for a minimum of n = 3 experiments. L, ladder.
SPCA1a reconstitution into proteoliposomes restores Ca2+-dependent ATPase activity
We found that purified and lipid-free SPCA1a became reversibly inactivated in detergent solution (Fig. 2E). This we considered to be a result of either a removal of specific lipid component(s) essential for activity or loss of physical membrane constraints originally imposed by the membrane lipids. To restore a functional lipid environment, we reconstituted as described under “Experimental procedures” the proteoliposomes from detergent-solubilized SPCA1a by addition of defined lipid components, followed by removal of detergent with Bio-beads. For this, 600 μg of phosphatidylcholine (PC) was first mixed with 200 μg of purified and detergent-solubilized SPCA1a (i.e. the protein was solubilized with detergent at a lipid/protein ratio of 3:1, w/w). The efficiency of SPCA1a incorporation into liposomes was compared for three types of detergents, i.e. Triton X-100 (PC/Triton X-100 = 8:1, 6:1, 5:1, 4:1 (w/w)), n-dodecyl β-d-maltoside (DDM) (PC/DDM = 1:1 (mol/mol)), and octaethylene glycol monododecyl ether (C12E8) (PC/C12E8 = 1:2 (w/w)), in line with already established protocols for SERCA1a (28, 29). Reconstitution with Triton X-100 rendered the most efficient incorporation of SPCA1a into proteoliposomes, especially at a high lipid to detergent ratio (Fig. 2A). Importantly, we were able to recover the Ca2+-dependent ATPase activity of the purified SPCA1a reconstituted with Triton X-100 or DDM but not with C12E8. Moreover, we determined that a lipid/detergent ratio of 20:1 generated the most active SPCA1a in proteoliposomes (Fig. 2B). Therefore, all further reconstitutions were carried out with a lipid/protein/detergent weight ratio of 60:20:3.
Figure 2.

Optimization of SPCA1a reconstitution into proteoliposomes. A, SDS-polyacrylamide gel stained with SPCA1a proteoliposomes generated with the detergents Triton X-100, DDM, or C12E8. Same volume of samples was loaded on the gel after reconstitution. The lipid/Triton X-100 ratios (w/w) are indicated above the lanes. B, comparison of the SPCA1a ATPase activity following reconstitution with a different (w/w) ratio of lipids/Triton X-100. C and D, 20% weight percent of the indicated lipid(s) were supplemented to PC for SPCA1a reconstitution, and the maximal Ca2+-dependent ATPase activity was determined at 1 μm free Ca2+ concentration (n ≥ 3). The ATPase activities were normalized to the activity when only PC was used. Comparison of the specific SPCA1a ATPase activities in the presence of indicated phospholipids (C), CL, SM, or the combination of CL and SM (1:1 molar ratio) (D). E, comparison of the specific ATPase activities between SERCA1a and SPCA1a in proteoliposomes, and SPCA1a in lipid-free, DDM-solubilized state (n = 3). The DDM-solubilized SPCA1a was purified without supplement of PC in the buffers. F, structure of CHEMS. G, dose-responses of CHEMS on reconstituted SERCA1a and SPCA1a ATPase activities. P, purified SPCA1a; L, ladder; PA, phosphatidic acid; PE, phosphatidylethanolamine; PS, phosphatidylserine; PI, phosphatidylinositol; CL, cholesterol; SM, sphingomyelin; CHEMS, cholesteryl hemisuccinate. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Lipids phosphatidic acid, sphingomyelin, cholesterol, and linoleamide/oleamide modulate SPCA1a activity
Membrane proteins are functionally adapted to the local lipid environment of their cellular membranes. We therefore analyzed the impact of different lipid types on the Ca2+-dependent ATPase activity of SPCA1a, which in human colon adenocarcinoma cells is sensitive to cholesterol (CL) and is preferentially associated with lipid rafts (30). For reconstitution, various phospholipids (Fig. 2C), CL, sphingomyelin (SM), or a combination of the latter two (SM + CL) (Fig. 2D) were supplied together with PC, and SPCA1a ATPase activity was assessed. Compared with PC alone, the presence of phosphatidic acid (PA), SM, or SM/CL slightly but significantly enhanced activity, whereas phosphatidylethanolamine (PE) lowered the SPCA1a ATPase activity. In further experiments, PA was routinely added during SPCA1a reconstitution, as has been done previously for SERCA1a reconstitution (29), although we cannot exclude that PA may be a low-abundance lipid in the native Golgi membrane (31, 32).
Next, we directly compared the biochemical properties of purified and reconstituted SPCA1a (4:1 PC/PA ratio) and SERCA1a, the skeletal muscle Ca2+-transport ATPase, which was purified and reconstituted according to a well established protocol (29) (with a 9:1 PC/PA ratio as in Ref. 33) (Fig. 2E). Compared with SERCA1a, SPCA1a displays a higher apparent Ca2+ affinity (Km of 0.125 ± 0.004 and 0.088 ± 0.005 μm, respectively) and a 5-fold lower turnover rate (Vmax of 12.29 ± 0.13 and 2.54 ± 0.04 nmol of Pi/μg/min), in line with previous reports (21).
Although we previously reported that SPCA1a and SERCA1a respond differently to CL (30), we observed here little functional effects of incorporating 20% CL upon reconstitution. However, by replacing CL with cholesteryl hemisuccinate (CHEMS), a more soluble cholesterol derivative (Fig. 2F), we found that it affected both the reconstituted SPCA1a or SERCA1a activities (Fig. 2G). SERCA1a displayed a concentration-dependent inhibition by CHEMS with an IC50 (defined as the concentration that leads to 50% inhibition of activity) of 8.8 ± 0.66 μm and a maximal inhibition of 80% at around 100 μm. For SPCA1a, a slight and not significant activation up to 106% was observed at lower concentrations, whereas higher concentrations above 6 μm impaired SPCA1a to a maximal inhibition of 47% at 300 μm, in line with the previous observation that SPCA1a and SERCA1a respond differently to CL (30).
Because two endogenous lipids oleamide and linoleamide have recently been reported to be inhibitors of SPCA2 (34), we tested here whether these lipids also modify SPCA1a or SERCA1a activity (Fig. 3, A and B). Up to 1 mm, oleamide only partially inhibited SPCA1a and SERCA1a (Fig. 3A), whereas complete inhibition was previously reported for SPCA2 (34). In contrast to SERCA1a, linoleamide fully inhibited SPCA1a with an IC50 of 30 μm (Fig. 3B), which is 8-fold higher than the value that has been reported for SPCA2 (34), although a side by side SPCA1a/2 comparison could not be performed.
Figure 3.
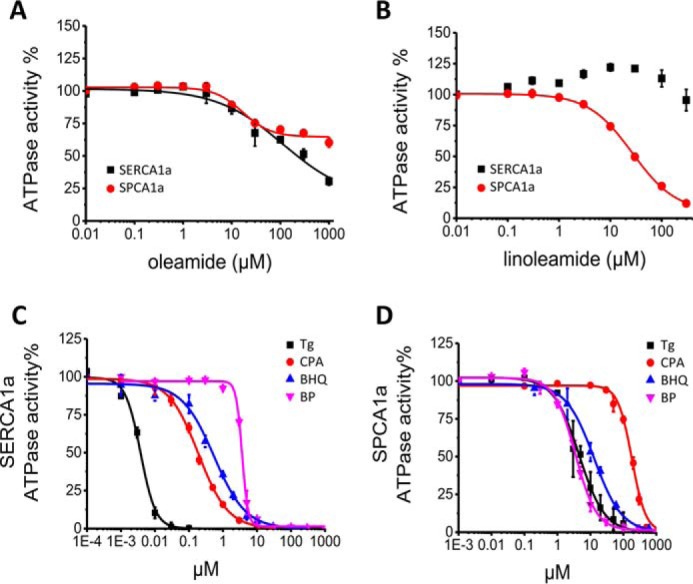
Dose-response effects of SPCA1a and SERCA1a inhibitors. Dose-response curves of oleamide (A) and linoleamide (B) on SERCA1a and SPCA1a. Dose-response curves for the inhibition of SERCA1a (C) and SPCA1a (D) by Tg, CPA, BHQ, and BP. n = 3 for all experiments.
Several SERCA inhibitors block SPCA1a activity at micromolar concentration
To further validate the reconstituted SPCA1a system, we verified whether bisphenol (35, 36) and the SERCA inhibitors Tg, CPA, and BHQ also inhibit SPCA1a (24, 25). We confirmed that all compounds completely inhibit SERCA1a and SPCA1a, but we show that the compounds are effective in different concentration ranges for SERCA1a and SPCA1a (Table 1 and Fig. 3, C and D). For SERCA1a, the IC50 value of Tg falls below 10 nm, whereas higher (sub-μm) IC50 values are observed for CPA and BHQ. 2,2′-Methylenebis(6-tert-butyl-4-methylphenol) (bisphenol, BP) displays the lowest potency with an IC50 above 1 μm. For SPCA1a, the IC50 values of Tg and CPA are at least 3 orders of magnitude higher than for SERCA1a, whereas the IC50 value of BHQ is only 20 times higher. The IC50 value of BP is comparable for the two pumps, indicating that unlike what was suggested by a previous study (35), BP cannot be considered as a more selective inhibitor of SPCA1a than of SERCA1a. Remarkably, the slopes of the dose-response curves for BP (SERCA1a) and CPA (SPCA1a) are higher than 2 (2.9 ± 0.1 and 2.3 ± 0.1, respectively). This was also observed in earlier studies (25, 35) and may reflect differences in the cooperativity (37). In conclusion, the SERCA inhibitors Tg, CPA, and BHQ also inhibit SPCA1a, although at much higher concentrations.
Table 1.
IC50 values for SPCA1a and SERCA1a measured for Tg, CPA, BHQ, and BP
Tg is thapsigargin; CPA is cyclopiazonic acid; BHQ is 2,5-di-tert-butylhydroquinone; BP is bisphenol.
| SPCA1a IC50 | SERCA1a IC50 | |
|---|---|---|
| μm | μm | |
| Tg | 7.7 ± 1.5 | < 0.01 |
| CPA | 182 ± 10 | 0.19 ± 0.01 |
| BHQ | 13 ± 2 | 0.54 ± 0.06 |
| BP | 3.5 ± 0.2 | 3.63 ± 0.09 |
Tg scaffold together with the O-2 moiety are mainly responsible for the inhibition of SPCA1a
Of the so-called specific SERCA inhibitors, Tg has the highest affinity for SPCA1a. Insights into the SAR of Tg on SPCA1a might be helpful to aid the development of Tg analogues with either low or high SPCA1a affinity. We therefore explored the SAR of Tg for SPCA1a and compared it with the well established SAR of Tg for SERCA1a (38).
First, we compared the Tg-binding pocket in the SERCA1a-Tg structure and in an SPCA1a homology model in the same E2 conformation. In SERCA1a, the non-flexible tricyclic guaianolide nucleus of Tg provides a scaffold for the optimal position of side chains within the Tg-binding pocket formed by transmembrane helices M3, M5, and M7 (Fig. 4B). The lipophilic groups at O-3, C-4, O-8, and O-10 of Tg (Fig. 4A) critically determine the hydrophobic interaction and provide complementarity to the binding cavity in SERCA1a (39, 40). In the SERCA1a-Tg structure (41), the O-8 moiety of Tg orients to the interior of M3 and M5, whereas the O-2 chain is located in the interface between the transmembrane region and the membrane (42). By comparing the residues in the Tg-binding pocket of SERCA1a (Fig. 4B) and the homology model of SPCA1a (Fig. 4C), we anticipate that the Tg-binding affinity, mechanism of inhibition, and/or positioning of Tg are affected by several amino acid substitutions, including the homologous SERCA1a/SPCA1a substitutions labeled in Fig. 4, B and C (Phe-256/Leu-265, Val-263/Tyr-272, Ile-765/Leu-707, Ile-829/Leu-771, and Phe-834/Ile-776), which might result in an altered SAR of Tg for SPCA1a as compared with SERCA1a.
Figure 4.

Establishing the structure-activity relationship of Tg in SPCA1a. A, structure of Tg with the numbering of the carbon atoms and side chains indicated. B, Tg pocket in SERCA1a in the E2 conformation with bound Tg (1IWO (41)) with important residues shown as blue sticks. The same view of the SPCA1a homology model is shown in C, and the residues that are different between SPCA1a and SERCA1a are labeled.
To establish the SAR of Tg for SPCA1a, we measured the IC50 values of a series of Tg analogues with various side chain substitutions. First, the acyl groups O-2, O-3, O-8, and O-10 (Fig. 4A) were one by one replaced by –OH or –H, which significantly increases the IC50 of Tg in SERCA1a (39, 40, 43). Remarkably, only the removal of the Tg O-2 chain led to a significantly higher IC50 value in SPCA1a, suggesting that of all tested side groups O-2 contributes the most to the inhibitory effect of Tg in SPCA1a (Table 2). Based on the calculation of the free energy change for binding caused by the removal of a side chain, the O-2 contribution is comparable in SPCA1a and SERCA1a, whereas O-3, O-8, and O-10 contribute much less in SPCA1a (Table 2). The absence of O-3, O-8, and O-10 contributions at least partially explains the lower Tg affinity in SPCA1a than in SERCA1a.
Table 2.
Contributions of the Tg side chain and scaffold modalities in SPCA1a inhibition
IC50 values of Tg analogues with indicated modifications were measured for SPCA1a and compared with the IC50 of Tg by Student′s t test (***, P < 0.001 versus Tg). Changes in free energy of SPCA1a and SERCA1a were calculated from ΔΔG° – RTlnKd Tg − RTlnKd compound. The Kd is based on the concentration of compound required for 50% inhibition of SPCA1a or SERCA1a.
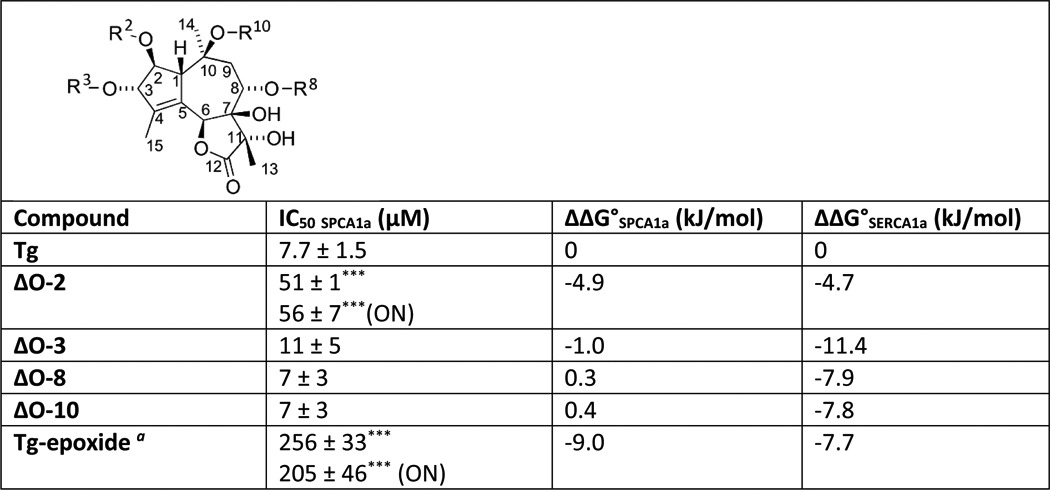
a Tg-epoxide has a β-epoxide group formed between C7 and C11; ON is overnight incubation with compound. ΔO-3, −8, and −10 indicate that the acyl group at O-X has been replaced with a proton, and ΔO-2 means that the octanoyloxy group has been replaced with a proton to give nortrilobolide.
Besides the side-chain contribution by O-2, the Tg scaffold might be mainly responsible for the inhibitory effect of Tg in SPCA1a. Indeed, like in SERCA1a, the conversion of the 7,11- gem diol in Tg into an epoxide led to a dramatic reduction of the inhibitory effect in SPCA1a (Table 2), presumably by the loss of hydrogen donors in the molecule or by the changed conformation of the tricyclic nucleus due to the conversion of the stereochemistry at C-11. Of note, some Tg analogues require a longer pre-incubation time with SERCA1a to obtain the full inhibitory potential, which can be explained by a time-dependent induced fit of Tg into the SERCA1a pocket (44). However, the weaker inhibitory potential of the Tg analogues in which the octanoyloxy group has been substituted with a hydrogen atom (ΔO-2), and Tg-epoxide did not significantly increase after a longer pre-incubation time with SPCA1a (Table 2).
Only the O-2 moiety of Tg binds to a spatially restricted pocket in SPCA1a
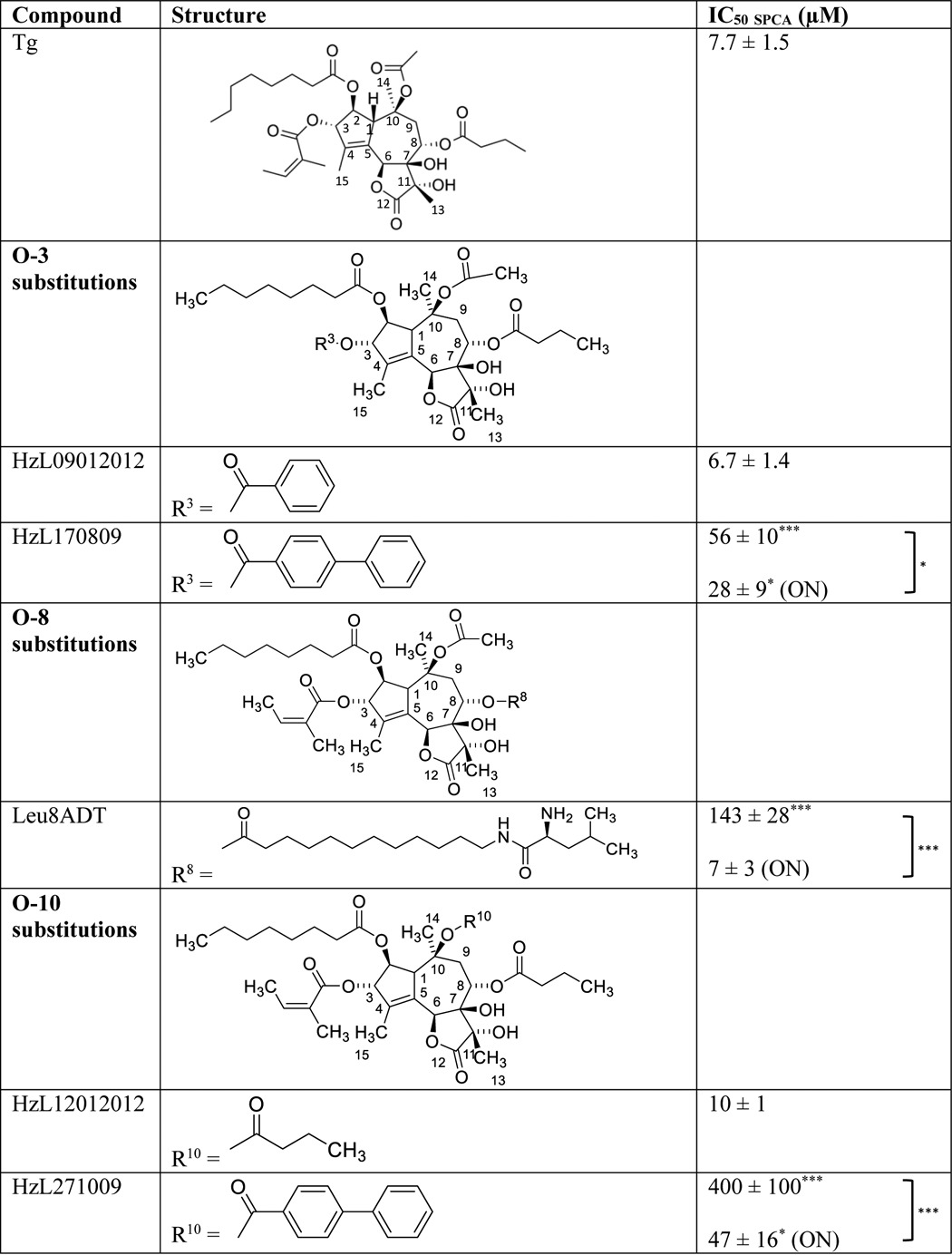
In the further exploration of the poor contribution of the side chains at O-3, O-8, and O-10 to the affinity of Tg to SPCA1a, we considered the possibility that it might arise from a more spacious Tg-binding cavity than in SERCA, which in a more flexible way would accommodate these Tg side groups. This hypothesis was tested by replacing the O-2, O-3, O-8, and O-10 moieties with more bulky groups (Table 3). Replacing O-3 and O-10 with a slightly bulkier chain (HzL09012012 and HzL12012012, respectively) is well tolerated, whereas a more bulky biphenyl group on O-3 (HzL170809) or O-10 (HzL271009) led to a 7- and 49-fold higher IC50, respectively (Table 3). However, the higher IC50 was significantly reduced after a longer pre-incubation time, indicating that a sufficiently large three-dimensional space is potentially available in the Tg pocket near O-3 and O-10 to allow accommodation of biphenyl groups (Table 3). The large but flexible Leu-8ADT substitution on O-8 led to a 19-fold reduction of the potency in SPCA1a most likely as a result of steric hindrance. However, overnight incubation of Leu-8ADT led to a lower IC50, indicative of an induced fit effect and sufficient space to accommodate a larger O-8 chain (Table 3).
Table 3.
IC50 values of Tg analogues with modified O-3, O-8, or O-10 side chains on SPCA1a
Columns provide information on compound name, the structure of the side-chain modification, and the IC50 value on SPCA1a. Student′s t test were performed to compare between IC50 of each analogue and IC50 Tg, and before/after overnight (ON) incubation. * P < 0.05, *** P < 0.001.
Of all tested Tg side groups, O-2 contributes most to the inhibition of SPCA1a. To determine the SAR of Tg in SPCA1a, we therefore focused on the role of O-2 by testing the functional impact of various substitutions of the O-2 group (Table 4). The IC50 remains unaffected when the O-2 octanoyl group was substituted with a benzoyl (HzL20072015) or naphthalenecarboxyl group (HzL18072015). Thus, the difference between HzL20072015, HzL18072015, and the −H substitution of nortrilobolide (ΔO-2) indicates that the O-2 chain facilitates a hydrophobic interaction in the SPCA1a-binding pocket. However, a substitution of the O-2 moiety with a phenylbenzoyl group (HzL17072015) led to a significantly higher IC50 than with smaller substituents (HzL20072015 and HzL18072015), which was independent of the pre-incubation time. This reveals that the local three-dimensional space to accommodate a bulky O-2 group is restricted in SPCA1a, although the O-2 acyl group is larger in one dimension.
Table 4.
IC50 values of Tg analogues with modified O-2 side chain on SPCA1a
Columns provide information on compound name, the structure of the side-chain modification and the IC50 value on SPCA1a. Student′s t test were performed to compare between IC50 of each analogue and IC50 Tg, and before/after overnight (ON) incubation. * P < 0.05, *** P < 0.001.
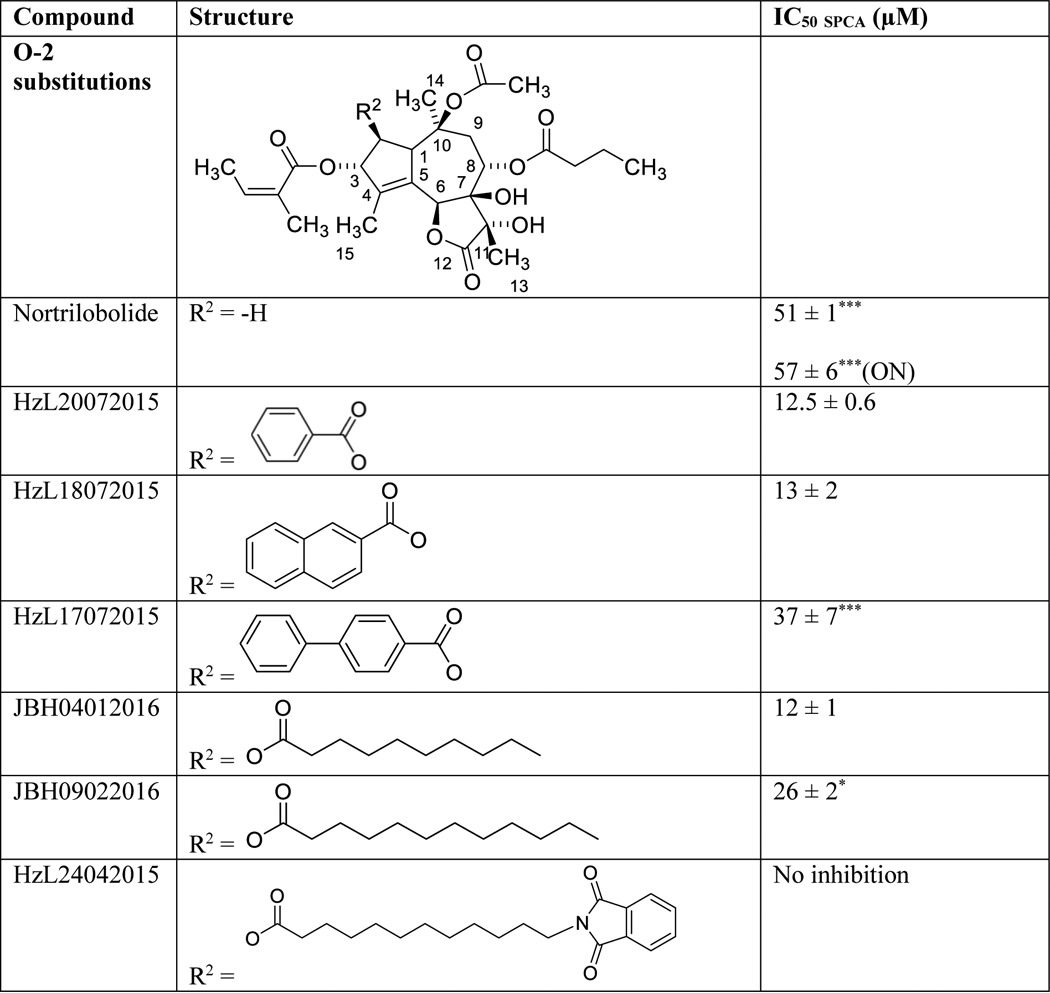
Therefore, we explored the replacement of the O-2 chain with several longer but flexible acyl groups (Table 4). We observed little effect on the IC50 by elongating the O-2 chain with two carbons (JBH04012016), whereas addition of four carbons (JBH09022016) increased the IC50 3.4-fold (Table 4). Of interest, the inhibition of SPCA1a was completely abolished by introducing a more bulky end group (HzL24042015). The inhibitory effect of this compound is unaffected by the pre-incubation time.
Thus, strict spatial restrictions around O-2 are likely present, because only thin flexible O-2 chains are allowed, whereas more voluminous groups lower or prohibit the affinity for SPCA1a.
SPCA1a inhibition by Tg and cholesteric compounds involves the same or overlapping binding pocket
To explore the functional role of amino acid replacements in SPCA1a on Tg binding and confirm that Tg binds to the same pocket in SPCA1a and SERCA1a, we generated and purified three SPCA1a mutants in the Tg-binding pocket in which we substituted one or more of the SPCA1a-specific residues into the corresponding SERCA1a amino acids (L265F, Y272V, and L265F/Y272V/L776F) (Fig. 5A). Phe-256 is a critical residue for Tg central scaffold binding in SERCA1a (45) and is replaced in SPCA1a by Leu-265. The replacement of Val-263 in SERCA1a by Tyr-272 in SPCA1a introduces a bulky aromatic side chain in the Tg pocket possibly hindering Tg binding to SPCA1a. The Leu-776 in SPCA1a corresponds to Phe-834 in SERCA1a, which exerts unfavorable interaction with the O-10 group of Tg.
Figure 5.

Modeling Tg into the binding pocket of SPCA1a. SDS-polyacrylamide gel staining (A), IC50 values of Tg (B), and CHEMS dose-response curves (C) of the reconstituted SPCA1a WT and L265F, Y272V, and L265F/Y272V/I776F mutants. D, crystal structure of SERCA1a-Tg complex (1IWO (41)). E and F, Tg docking simulation performed on the SERCA1a E2 crystal structure without Tg (3W5C (46)) (side-chain torsion rotations were assigned to Glu-255, Phe-256, Gln-259, Leu-260, Val-263, Ile-765, Val-769, Ile-829, Phe-834, and Met-838), rendering two conformational populations: model 1 (E) and model 2 (F). G–I, Tg docking simulation was performed on the SPCA1a homology model (side-chain torsion rotations were assigned to Leu-264, Leu-265, Leu-269, Tyr-272, Leu-707, Ile-711, Ile-770, Leu-771, Ile-776, and Ile-779), rendering three conformational populations (G–I), which are corresponding to two major models according to the orientation of the Tg O-2 and O-8 side chains: model 1 (G) and model 2 (H and I). The O-2 and O-8 chains are indicated in red text boxes. Top (J) and side (K) view of the superimposed 10 Tg scaffolds from the docking result of G–I. The residue Leu-265 is highlighted. TM, transmembrane helix. One-way ANOVA was performed followed by a post hoc Fisher test. *, p < 0.05; **, p < 0.01.
Compared with wild-type SPCA1a (WT), the more SERCA-like L265F and L265F/Y272V/L776F mutants are more sensitive to Tg, whereas Y272V behaves similarly as WT (Fig. 5B). This is consistent with the view that as in SERCA1a, Tg binds to a conserved pocket in the transmembrane region formed by M3, M5, and M7. Furthermore, the L265F substitution in SPCA1a significantly enhanced the binding affinity of Tg, possibly by promoting a direct interaction between Phe-265 and the Tg scaffold as is the case for SERCA1a (39). This result is also consistent with the view that binding of the Tg scaffold in SPCA1a may closely resemble the interaction in SERCA1a. Finally, the three SPCA1a mutants also displayed a more potent and stronger inhibition by CHEMS, a cholesterol analogue (Fig. 5C), suggesting that cholesteric compounds may bind at the same or overlapping site as Tg.
Modeling confirms the importance of the Tg scaffold in the binding to SPCA1a
To further explore how Tg might be accommodated in SPCA1a, we generated a homology model of SPCA1a and performed an in silico docking of Tg into the M3, M5, and M7 pocket, while allowing flexibility of 10 amino acid side chains in the pocket. To validate our modeling approach, we first docked Tg into an E2 crystal structure of SERCA1a without Tg (PDB code 3W5C (46)) and compared the results with the 1IWO crystal structure of SERCA1a with bound Tg (Fig. 5D) (41). Among the best 10 docking poses (Chemscore scoring function of GOLD), two major populations were recognized (Fig. 5, E and F, and Table 5). Model 1 (Fig. 5E) corresponds to a Tg position that is highly similar to the Tg position in the 1IWO crystal structure (Fig. 5D), with the O-8 group that is oriented toward the space flanked by M3 and M5 helices. Of the 10 highest ranked scores, six docking poses, including the five best belong to model 1 (Fig. 5E), validating the docking approach. The other four solutions belong to model 2 (Fig. 5F), which contains docking poses displaying a remarkable flipped orientation of Tg into the pocket, i.e. with the O-2 group of Tg facing M3 and M5.
Table 5.
Scores of the best Tg dockings in SPCA1a and SERCA1a
Poses obtained by in silico docking of Tg in SERCA1a or SPCA1a are divided into different models (1, 2′, and 2″) that show a closely resembling docking position.
| SPCA1a |
SERCA1a |
||
|---|---|---|---|
| Docking score by Gold.Chemscore.Fitness | Population | Docking score by Gold.Chemscore.Fitness | Population |
| 30.22 | 1 | 31.68 | 1 |
| 27.92 | 2′ | 30.25 | 1 |
| 27.80 | 2″ | 30.21 | 1 |
| 27.57 | 2′ | 29.81 | 1 |
| 26.96 | 1 | 29.68 | 1 |
| 26.06 | 1 | 28.61 | 2 |
| 25.55 | 2″ | 28.58 | 2 |
| 24.99 | 2″ | 28.30 | 2 |
| 24.95 | 2″ | 28.19 | 1 |
| 24.87 | 2″ | 28.06 | 2 |
Next, we docked Tg onto the SPCA1a homology model, and again two major populations were recognized with opposite orientations of O-2 and O-8 (Fig. 5, G–I). Model 1 (Fig. 5G) displays a similar Tg orientation as in the SERCA1a crystal structure, except that the O-2 chain extends to the outside of the pump. Model 2 (Fig. 5, H and I) corresponds to docking poses with O-8 pointing away from the protein, whereas O-2 is directed toward the interior of the pump with two main variations in the path of the O-2 chain as displayed in Fig. 5, H and I). Our experimental results highlighted that the inhibition of SPCA1a by Tg is mainly explained by the Tg scaffold and the O-2 moiety, whereas the O-3, O-8, and O-10 side chains contribute little. Of interest, the position of the Tg scaffold in the 10 best docking results is always aligned into the same plane flanked by M3 and M7, irrespective of the opposite O-2/O-8 directions in models 1 (Fig. 5J) or 2 (Fig. 5K). The strict positioning of the Tg scaffold in SPCA1a may indicate the presence of spatial restrictions and/or critical interactions between the scaffold and the protein. The scaffold is in close proximity of Leu-265 on M3, which might explain the affinity-enhancing effect of the L265F substitution (Fig. 5B).
Thus, our docking analysis confirms the contribution of the Tg scaffold to the SPCA1a, and it points to two possible but opposite orientations of Tg inside the pocket formed by M3, M5, and M7.
Discussion
In this study, we purified for the first time the human Golgi/secretory pathway Ca2+/Mn2+-ATPase SPCA1a from a yeast overexpression model. The one-step affinity purification protocol generates hSPCA1a with over 95% purity offering opportunities for structure/function analysis and inhibitor screening. We demonstrate that SPCA1a is highly sensitive to the lipid environment and that several SERCA inhibitors, including Tg, also block SPCA1a activity, although at higher concentrations only. Because of the wide use of Tg in cellular studies and prospects for the use of Tg analogues in prostate cancer therapy, we focused on deciphering the SAR of Tg for SPCA1a. We observed differences in the relative contribution of Tg side chains in the inhibition of SERCA1a versus SPCA1a, which might be exploited for the future design of more selective SPCA1a or SERCA1a inhibitors.
Activity of SPCA1a depends on the membrane environment
Ca2+ transporters are tightly regulated by their lipid environment. For instance, the plasma membrane Ca2+-ATPase contains two lipid-binding sites and can be stimulated by PS, PA, phosphatidylinositol bisphosphate, and cardiolipin (47). SERCA pumps are inhibited by sphingosine (48) and CL (49), whereas SPCA1a associates with CL-rich microdomains of human colon adenocarcinoma cells (30). Also, SPCA2 is inhibited by two endogenous signaling lipids oleamide and linoleamide (34). Here, we demonstrate that SPCA1a is inactive in a lipid-free detergent-solubilized state, but the activity restores upon reconstitution into a PC-rich lipid environment. In proteoliposomes, SPCA1a displays a higher apparent Ca2+ affinity and lower maximal ATPase activity than SERCA1a. The Km value of the purified and reconstituted SPCA1a falls within the reported range for SPCA1 in cell membrane fractions (0.04–0.26 μm (8, 21, 50)). The variation in the reported Km values presumably relates to slight differences in the free Ca2+ concentrations, the choice of the SPCA1 splice variant, and the selected cellular expression system, which may affect the regulation of SPCA1.
Moreover, SPCA1a is sensitive to the SERCA inhibitors Tg, CPA, and BHQ but also to bisphenol and CHEMS. SPCA1a was previously considered as Tg-insensitive, whereas a few studies reported a Tg sensitivity in the range between 0.1 and 30 μm (13, 24, 36). Here, we report that purified SPCA1a in reconstituted proteoliposomes displays an IC50 for Tg of 7.7 μm, which is four times lower than the reported IC50 value of 28 μm in microsomal fractions (24). The variations in the reported Tg sensitivities of SPCA1 may be due to differences in the experimental systems (membrane lipid composition and presence of other proteins or detergent). Thus, overall, the properties of the purified protein correspond well with SPCA1a in a cellular membrane context.
The inclusion of specific lipids in the proteoliposomes impacts on the activity of SPCA1a, with PA and SM leading to stimulation, whereas PE is inhibitory. PA is typically added during the reconstitution of SERCA1a to prevent aggregation (51). Because the stimulatory effect of PA was not observed with other charged lipids, it is also possible that PA serves as a regulatory lipid of SPCA1a. Although PA is a low-abundant lipid in the Golgi/secretory pathway, it can be locally formed by phospholipase D as a signaling lipid that may regulate SPCA1a activity (52, 53). Moreover, we showed that SPCA1a preferentially accumulates in lipid rafts (30), which is in line with the stimulatory effect of SM on the ATPase activity of SPCA1a. The cholesteric compound CHEMS only moderately inhibits SPCA1a, while exhibiting a more potent inhibitory effect on SERCA1a, in line with a different impact of CL on SPCA1a and SERCA1a as suggested previously (30). Although our reconstitution conditions were selected to promote the proper orientation of the protein in the membrane (i.e. the large hydrophilic cytosolic domain facing the outside of the vesicles) (51), we cannot fully rule out the possibility that the functional effects of the lipids may relate to an impact on the distribution between inward-facing and outward-facing enzymes in the proteoliposomes.
Of interest, the potency and efficacy of the CHEMS inhibition is higher in SPCA1a mutants with a more SERCA1a-like Tg pocket. The changes between the Tg-binding site of SERCA1a and SPCA1a might also explain the different sensitivity of SPCA1a and SERCA1a to cholesteric compounds. Although we cannot rule out that these mutations might exert an allosteric effect via an unrelated binding site for cholesteric compounds, it is reasonable to propose that the cholesteric compound binds and overlaps the Tg pocket. Indeed, it was already previously proposed that the SERCA1a Tg pocket might be a CL regulation site (39, 54). In the SERCA1a crystal structure 2EAU, the Tg-binding site was occupied by a lipid (55). Moreover, a fluorescence quenching study suggested the existence of hydrophobic non-annular binding sites for CL in SERCA1a (56), whereas a computational docking analysis modeled the steroid compound Alisol B, which targets SERCA pumps and enhances autophagy, in the Tg site (57). However, a more recent molecular dynamics simulation rather indicates that little CL may occupy the Tg-binding site (58), suggesting that the CL effect on SERCA1a may also be indirect via the impact on the membrane properties.
Inhibition of SPCA1a and SERCA1a by Tg is largely determined by the Tg scaffold
Of all tested pharmacological compounds, we observed the most potent inhibition of SPCA1a with Tg, a plant-derived sesquiterpene lactone that is generally accepted as a highly selective SERCA1a inhibitor. Over the years, structures of the Tg-SERCA1a complex (59–62), supported by extensive functional studies (38–40, 42, 43, 63–66), rendered a comprehensive pharmacophore model that explains the sub-nanomolar affinity of SERCA1a for Tg. The availability of purified SPCA1a and a library of Tg analogues now offered an opportunity to determine the SAR of Tg on SPCA1a, which might be helpful for the design of more specific SERCA1a or SPCA1a inhibitors.
In accordance with the view that Tg targets the same binding pocket in SPCA1a as in SERCA1a, amino acid substitutions in the predicted M3/M5 pocket of SPCA1a impact the affinity for Tg. According to our in silico docking, the Tg scaffold is aligned on the same plane irrespective of the side chain orientations. It was previously demonstrated that an F256L mutation in SERCA1a led to a 4-fold reduction in the Tg sensitivity (67), whereas our complementary experiment shows that the L265F substitution in SPCA1a significantly enhanced SPCA1a's affinity for Tg. Thus, the Phe-256/Leu-265 substitution at least in part explains the lower Tg affinity for SPCA1a than SERCA1a. The Tg scaffold might possibly undergo π-stacking with the L265F residue in the SPCA1a mutant. The importance of the Tg scaffold for inhibition is supported by findings with the 7,11-epoxide, which show a major reduction in the binding energy for both SPCA1a (Table 2) and SERCA1a (38, 39), pointing to possible similarities in the mechanism of inhibition in SPCA1a and SERCA1a.
Tg side groups differently contribute to the inhibition of SPCA1a versus SERCA1a
We further approached the SAR of Tg for SPCA1a by analyzing the IC50 values of a series of Tg analogues with modified side chains. Strikingly, removal of the O-3, O-8, and O-10 acyl groups have little effect on the binding energy of Tg in SPCA1a, which is different for SERCA1a (Table 2) (40, 43). Moreover, substitution by slightly bulkier groups at these sites on SPCA1a is well tolerated, indicating that the Tg pocket in SPCA1a is spacious near O-3, O-8, and O-10 and that only few functional contacts exist between the side chains of Tg and residues of the protein. The removal of O-2, the longest side chain of Tg, leads to a higher IC50 value in SPCA1a, suggesting that besides the relevance of the Tg scaffold, O-2 also contributes to the inhibition of SPCA1a. The analysis of Tg analogues with various O-2 substitutions further indicates that the O-2 chain is accommodated in a narrow hydrophobic cleft, which is restricted in volume. Noteworthy, the change in binding energy of the ΔO-2 compound is remarkably similar for SPCA1a and SERCA1a possibly pointing to a comparable binding mechanism.
By in silico docking analysis, two major docking solutions of Tg O-2 in SPCA1a were predicted. Of interest, the docking model 1 predicts a similar Tg orientation in SPCA1a as observed in the solved SERCA1a-Tg complex, which also might point to a similar inhibitory mechanism in SPCA1a and SERCA1a. This model may explain why a prolonged incubation of Leu-8ADT shows a more potent inhibition in SPCA1a (Table 3), because a similar effect of Leu-8ADT was observed on SERCA1a (44). Alternatively, in line with O-2 being accommodated into a tight hydrophobic cleft, the Tg O-2 chain in the SPCA1a model 2 protrudes into the M3/M5 region. Such a flipped orientation of Tg is also predicted for SERCA1a, although this is not experimentally observed in the solved SERCA1a-Tg complex, which may question the validity of such an orientation.
Toward the design of more specific SPCA1a inhibitors
We demonstrate that the SAR of Tg differs for SPCA1a and SERCA1a, which is mainly attributed to amino acid changes in the Tg pocket that prevent most interactions of the Tg side chains in SPCA1a, but it still allows the Tg scaffold to be accommodated in the pocket. The different SAR of Tg for SERCA1a and SPCA1a provides a strong rationale to consider the Tg scaffold as a lead compound for the development of more specific SPCA1a inhibitors. Solving the crystal structure of SPCA1a in the presence of Tg will greatly facilitate a rational design approach for a Tg-based SPCA1a inhibitor with higher potency and specificity, but so far, crystallization of SPCA1a turns out to be difficult in our hands.
In the absence of detailed structural information, we need to test additional Tg analogues with substitutions of the O-2, O-3, O-8, and O-10 chains to develop Tg analogues with a higher SPCA1a/SERCA1a specificity. Tg analogues with O-2 acyl chains that are slightly shorter would be interesting for establishing the optimal fit of O-2 within the SPCA1a Tg pocket. Moreover, our results indicate that single or combined bulky substitutions at O-3, O-8, and O-10 might lower the potency in SERCA1a, while having little impact on SPCA1a inhibition, favoring a higher SPCA1a/SERCA1a specificity. The more spacious environment near O-3 and O-10 indicates that bulkier side groups on both positions might lead to a tighter fit of Tg within the pocket.
A specific SPCA1a inhibitor might not only be a powerful tool to dissect the cellular and pathological role of SPCA1a in the Ca2+ (dys)homeostasis of various diseases, it might also be of therapeutic interest. Gain of SPCA1a and SPCA2 function is observed in breast cancer, because SPCA1a and/or SPCA2 proteins are up-regulated in various breast cancer sub-types contributing to a pathological Ca2+ dyshomeostasis (17, 18). Knockdown of endogenous SPCA2 prevents Orai1 activation, which reduces the transforming phenotype of MCF-7 cells in soft agar and impairs tumor generation upon injection in nude mice (18). Although this mainly depends on the direct interaction of SPCA2 with Orai1 leading to constitutive cytosolic Ca2+ influx (18), SPCA2 and Orai1 are likely mechanistically coupled, suggesting that also a higher Golgi Ca2+ uptake is contributing to the pathological Ca2+ dyshomeostasis (19). In line with this view, knockdown of SPCA1a in the basal-type breast cancer cell line MDA-MB-231 reduces, independent of Orai1, the processing of insulin-like growth factor receptor IGF1R (17), which might counteract its mitogenic and anti-apoptotic actions. Of further interest, the absence of PMR1, the SPCA1a orthologue in lower eukaryotes such as yeast, flies, and Caenorhabditis elegans, prevents the elevation of cytosolic Ca2+ and cell death caused by α-synuclein overexpression, a major toxic effector in Parkinson disease (68). Finally, SPCA1a mutations lead to a dominant skin disorder, Hailey-Hailey disease, which is generally considered to be caused by loss-of-function mutations that cause haploinsufficiency (15, 16). However, it was recently proposed that dominant disease mutations in ion pumps might introduce a gating defect resulting in ion leakage, which would represent a gain-of-function effect (69). In mice, heterozygous loss of SPCA1a does not trigger a similar skin disease, but it increases the risk of developing squamous cell tumors (70), further highlighting the importance of a balanced SPCA1a activity.
In conclusion, we demonstrate that Tg and cholesteric compounds in SPCA target an overlapping binding site on SPCA1a, and the differences in the SAR of Tg for SPCA1a and SERCA1a might allow the development of more specific inhibitors of SPCA1a.
Experimental procedures
Human SPCA1a expression in yeast
The recombinant human SPCA1a was cloned in the vector pTV001E with a C-terminal His8 tag (CATCATCACCATCACCATCACCAT) via Gateway cloning. Mutations were introduced via Q5 site-directed mutagenesis (New England Biolabs). pTV001-SPCA1a wild-type (WT) or mutant vectors were transformed into the S. cerevisiae strain BY4741a (his3Δ1;leu2Δ0;met15Δ0;ura3Δ0;MATa) as described previously (71). The yeast clone was inoculated in 10 ml of minimal medium (MM) deprived of Leu for overnight growth at 30 °C and gradually expanded into 2 liters MM-Leu culture over 3 days. On day 4, the yeast pellet was transferred into 25 liters of yeast peptone dextrose medium in a 50-liter Cell Bag (GE Healthcare), for 30 h of growth on a shaking system supplied with 30% O2, 70% N2 air mixture. Yeast cells were harvested by centrifugation (5,000 × g, 5 min, 4 °C) and washed with milli-Q water, and the pellets were weighted, flash-frozen, and stored at −20 °C.
Yeast membrane isolation
The yeast membrane was isolated based on the protocol described in Ref. 72. Briefly, the yeast pellet was suspended in lysis buffer (50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 0.6 m sorbitol, 250 μm PMSF, 1 mm DTT, SIGMAFASTTM protease inhibitor mixture) and subjected twice to high pressure homogenization at 140,000 kPa (High Pressure Homogenizer, Avestin). The membrane fractions were collected by differential centrifugation at 1,000 and 16,000 × g, and they were suspended in solubilization buffer (10 mm imidazole, pH 7.5, 50 mm NaH2PO4, pH 7.5, 500 mm NaCl, 20% glycerol, 250 μm PMSF, 1 mm DTT, SIGMAFASTTM). Protein concentration was determined by the Bradford assay. The yeast membrane fraction was aliquoted, flash-frozen in liquid N2, and stored at −80 °C.
Purification of SPCA1a and SERCA1a by affinity chromatography
SERCA1a was purified from rabbit fast twitch skeletal muscle as described before (29), whereas a purification protocol for SPCA1a was optimized as detailed below. The His8-labeled SPCA1a was purified via nickel-affinity chromatography using nickel-charged agarose beads coupled to Ni-NTA. One column volume (CV) of nickel-Sepharose high performance beads (GE Healthcare) was pre-equilibrated with 6 CV of equilibration buffer (50 mm NaH2PO4, pH 7.5, 75 mm imidazole, pH 7.5, 500 mm NaCl, 20% glycerol, 5 mm 2-mercaptoethanol (2-ME), 250 μm PMSF, SIGMAFASTTM, 20 mg/ml DDM (Isogen Life Science, SOL-grade)). The yeast membrane proteins were solubilized in a triple weight amount of DDM detergent solution (200 mg/ml DDM, 250 μm PMSF, SIGMAFASTTM protease inhibitor mixture, 5 mm 2-ME), and non-soluble material was removed by ultracentrifugation (160,000 × g, 45 min, 4 °C). After binding of the supernatant to the column for 1 h at 4 °C, the liquid was drained off. The beads were washed with 6 CV of three consecutive washing buffers composed of binding buffer with a stepwise reduction in the NaCl and DDM concentrations: from 500 to 50 and 50 mm NaCl combined with 5, 1, and 0.25 mg/ml DDM, respectively. SPCA1a became eluted in the final equilibration buffer with 50 mm NaCl and 0.25 mg/ml DDM. The washing and elution buffers were supplemented with PC (Avanti Polar Lipids) at the ratio 1:0.67 (DDM/PC). Afterward, the buffer component of purified SPCA1a was exchanged into 20 mm imidazole, pH 7.5, 100 mm KCl, 20% glycerol, 0.25 mg/ml DDM, 0.167 mg/ml PC, 5 mm 2-ME, SIGMAFASTTM via PD Mantrap G-25 (GE Healthcare) column. Aggregates were removed by centrifugation at 20,000 × g for 20 min at 4 °C. The supernatant was collected and flash-frozen in liquid N2 and stored at −80 °C. The purified SPCA1a was analyzed by MALDI-TOF mass spectrometry (4800 Proteomics Analyzer, AB Sciex) in the linear ion mode in the presence of α-cyano-4-hydroxycinnamic acid (5 mg/ml).
Reconstitution of the SPCA1a and SERCA1a Ca2+-ATPases
The 550-μg lipid mixture dissolved with chloroform in a glass tube was dried in a vacuum desiccator under a N2 stream and overnight in a vacuum desiccator. Liposome buffer (150 μl of 20 mm imidazole, pH 7, 100 mm NaCl, 10 mm MgCl2, and 1 mm DTT) was added and incubated at 37 °C water bath for 1 h with short vortex periods in between. To this (unless otherwise specified) 30 μg of Triton X-100 was added at room temperature, and the mixture was vortexed for 3 min followed by addition of purified SPCA1a (200 μg of protein at a concentration 0.667 mg/ml). Detergent was extracted to form proteoliposomes by treatment with excess Bio-Beads SM-2 (Bio-Rad) for 2.5 h.
The proteoliposomes were purified by sucrose gradient centrifugation (20 and 50% sucrose in 20 mm imidazole, pH 7, 0.2% azide, 100 mm NaCl) at 150,000 × g, 75 min, and 4 °C from which a 450-μl proteoliposome fraction was collected from the turbid layer present between the two sucrose fractions. This sample was flash-frozen in liquid N2 and stored at −80 °C. The reconstitution of SERCA1a was performed according to Ref. 29. PC, PA, and PE from chicken egg, PS and SM from porcine brain, and phosphatidylinositol from bovine liver were purchased from Avanti Polar Lipids, whereas CL was purchased from Sigma.
Protein electrophoresis and immunoblotting
Purified or reconstituted SPCA1a or SERCA1a was separated on NuPAGE 4–12% BisTris polyacrylamide gel (200 V, 35 min, MES buffer) (Thermo Fisher Scientific) and visualized with Imperial Protein Stain (Thermo Fisher Scientific). SeeBlue Plus2 pre-stained protein standard (Thermo Fisher Scientific) was used as a molecular weight marker. The concentration of purified protein was determined on a denaturing gel by comparison with 0.125 to 2 μg/μl bovine serum albumin (BSA) standards (BSA Standard Ampules, Pierce). For immunoblotting, primary antibodies against the His tag (1:2,000 dilution, penta-His, Qiagen) or SPCA1a (1:50,000 dilution, Frodo, homemade) were used. Horseradish peroxidase-linked secondary antibodies (Cell Signaling Technology) were used for enhanced chemiluminescent detection (Bio-Rad ChemiDoc). MagicMark (Thermo Fisher Scientific) was used as the molecular weight marker.
ATPase assay
The Ca2+-dependent ATPase activity assay was performed on reconstituted SPCA1a or SERCA1a samples according to the Baginsky protocol as described previously (73). The reactions were performed for 15 min (SERCA1a, 150 ng in 50 μl) or 30 min SPCA1a (300 ng in 50 μl). The results were fitted with a Hill function.
The dose response of the ATPase to various compounds was performed by measuring the ATPase activity at 1 μm free Ca2+ concentration with increasing concentrations of compounds to be tested in the reaction mix. Reactions with Tg (200 nm for SERCA1a and 500 μm for SPCA1a) samples were used as blanks to correct for irrelevant color development. The data were normalized as the percentage of inhibition compared with that obtained in the absence of inhibitor and fitted with a logistic curve. The CHEMS, CPA, BHQ, BP, and oleamide were purchased from Sigma. Linoleamide was purchased from Enzo Life Sciences, Tg was purchased from Alomone Labs. All compounds were dissolved in dimethyl sulfoxide (DMSO, Sigma).
In silico modeling
The SPCA1a homology model was generated with the homology model module in MOE 2014.09 (74) based on the SERCA1a E2 crystal structure with Tg bound (PDB code 1IWO (41)). Subsequently, the structure of the homology model was energy-minimized using Amber99 (75) force field in the energy minimization module of MOE. Tg was docked in the SPCA1a homology model or SERCA1a crystal structure via GOLD version 5.2 (Genetic Optimization for Ligand Docking (76, 77)). Tg was docked in the SERCA1a E2 crystal structure without any inhibitors (PDB code 3W5C (46)) and compared with the crystal structure 1IWO with Tg bound. We specified the center of the Tg-binding site (x, 33.422; y, −18.503; and z, 82.4683) and selected all atoms in a radius of 11 Å. 10 residues in the Tg-binding site (Glu-255, Phe-256, Gln-259, Leu-260, Val-263, Ile-765, Val-769, Ile-829, Phe-834, and Met-838) were selected to allow for receptor flexibility through side-chain torsion rotations. For each of these residues, flexibility was limited to a library of selected side chain conformers extracted from published crystal structures. Docking conformations for Tg were generated by performing 20 runs using standard Genetic Algorithm parameters. The docking results were scored and sorted using the Chemscore scoring function of GOLD.
The docking poses for Tg or the analogues were generated in the SPCA1a E2 homology model using the GOLD version 5.2 docking software. Here, we specified the center of the binding site (x, −2.8375; y, −22.0812; and z, 12.2) and selected all atoms in a radius of 10 Å. 10 residues in the binding site (Leu-264, Leu-265, Leu-269, Tyr-272, Leu-707, Ile-711, Ile-770, Leu-771, Ile-776, and Ile-779) were selected to allow for receptor flexibility through side-chain torsion rotations. To generate docking conformations, 100 runs were performed for each ligand using Genetic Algorithm parameters. The docking results were scored and sorted using the Chemscore scoring function of GOLD.
Synthesis of Tg analogues
The synthesis of the Tg analogues is described in supplemental Materials and methods.
Statistical analysis
Values are provided as mean ± S.E. Hill or logistic fitting was performed via OriginPro 9 software. Statistical significance was calculated by one-way t test and indicated by asterisks: *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Author contributions
P. V. conceived the idea for the project and coordinated the study. J. C. conducted most of the experiments and analyzed the data. J. D. R., M. D. M., J. B. H., S. S., and I. V. conducted experiments on in silico modeling, Tg analogue synthesis, and SPCA1a purification. J. V. M., J. E., S. B. C., P. V., and F. W. analyzed the data and critically reviewed the manuscript. P. V. and J. C., wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by Flanders Research Foundation FWO Grants G044212N and G0B1115N, the Inter-University Attraction Poles Program P7/13, and by the Danish Council for Strategic Research. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Materials and methods and additional references.
- SERCA
- sarco(endo)plasmic reticulum Ca2+-ATPase
- Tg
- thapsigargin
- PSA
- prostate-specific antigen
- SAR
- structure-activity relationship
- CPA
- cyclopiazonic acid
- BHQ
- 2,5-di-tert-butyl-hydroquinone
- BP (bisphenol)
- 2,2′-methylenebis(6-tert-butyl-4-methylphenol)
- ANOVA
- analysis of variance
- MOE
- molecular operating environment
- PDB
- Protein Data Bank
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- 2-ME
- 2-mercaptoethanol
- DDM
- n-dodecyl β-d-maltoside
- Ni-NTA
- nickel-nitrilotriacetic acid
- PC
- phosphatidylcholine
- CL
- cholesterol
- SM
- sphingomyelin
- PA
- phosphatidic acid
- PE
- phosphatidylethanolamine
- CHEMS
- cholesteryl hemisuccinate
- CV
- column volume
- PS
- phosphatidylserine
- SPCA
- Golgi/secretory pathway Ca2+-ATPase.
References
- 1. Berridge M. J., Bootman M. D., and Roderick H. L. (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 2. Monteith G. R., McAndrew D., Faddy H. M., and Roberts-Thomson S. J. (2007) Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer 7, 519–530 [DOI] [PubMed] [Google Scholar]
- 3. Palmgren M. G., and Nissen P. (2011) P-type ATPases. Annu. Rev. Biophys. 40, 243–266 [DOI] [PubMed] [Google Scholar]
- 4. Vangheluwe P., Sepúlveda M. R., Missiaen L., Raeymaekers L., Wuytack F., and Vanoevelen J. (2009) Intracellular Ca2+- and Mn2+-transport ATPases. Chem. Rev. 109, 4733–4759 [DOI] [PubMed] [Google Scholar]
- 5. Behne M. J., Tu C. L., Aronchik I., Epstein E., Bench G., Bikle D. D., Pozzan T., and Mauro T. M. (2003) Human keratinocyte ATP2C1 localizes to the Golgi and controls Golgi Ca2+ stores. J. Invest. Dermatol. 121, 688–694 [DOI] [PubMed] [Google Scholar]
- 6. Xiang M., Mohamalawari D., and Rao R. (2005) A novel isoform of the secretory pathway Ca2+,Mn2+-ATPase, hSPCA2, has unusual properties and is expressed in the brain. J. Biol. Chem. 280, 11608–11614 [DOI] [PubMed] [Google Scholar]
- 7. Wuytack F., Raeymaekers L., and Missiaen L. (2002) Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 32, 279–305 [DOI] [PubMed] [Google Scholar]
- 8. Ton V. K., Mandal D., Vahadji C., and Rao R. (2002) Functional expression in yeast of the human secretory pathway Ca2+, Mn2+-ATPase defective in Hailey-Hailey disease. J. Biol. Chem. 277, 6422–6427 [DOI] [PubMed] [Google Scholar]
- 9. Mitchell K. J., Tsuboi T., and Rutter G. A. (2004) Role for plasma membrane-related Ca2+-ATPase-1 (ATP2C1) in pancreatic beta-cell 2+ homeostasis revealed by RNA silencing. Diabetes 53, 393–400 [DOI] [PubMed] [Google Scholar]
- 10. Van Baelen K., Vanoevelen J., Callewaert G., Parys J. B., De Smedt H., Raeymaekers L., Rizzuto R., Missiaen L., and Wuytack F. (2003) The contribution of the SPCA1 Ca2+ pump to the Ca2+ accumulation in the Golgi apparatus of HeLa cells assessed via RNA-mediated interference. Biochem. Biophys. Res. Commun. 306, 430–436 [DOI] [PubMed] [Google Scholar]
- 11. Micaroni M., Giacchetti G., Plebani R., Xiao G. G., and Federici L. (2016) ATP2C1 gene mutations in Hailey-Hailey disease and possible roles of SPCA1 isoforms in membrane trafficking. Cell Death Dis. 7, e2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vanoevelen J., Dode L., Van Baelen K., Fairclough R. J., Missiaen L., Raeymaekers L., and Wuytack F. (2005) The secretory pathway Ca2+/Mn2+-ATPase 2 is a Golgi-localized pump with high affinity for Ca2+ ions. J. Biol. Chem. 280, 22800–22808 [DOI] [PubMed] [Google Scholar]
- 13. Baron S., Struyf S., Wuytack F., Van Damme J., Missiaen L., Raeymaekers L., and Vanoevelen J. (2009) Contribution of intracellular Ca2+ stores to Ca2+ signaling during chemokinesis of human neutrophil granulocytes. Biochim. Biophys. Acta 1793, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 14. Faddy H. M., Smart C. E., Xu R., Lee G. Y., Kenny P. A., Feng M., Rao R., Brown M. A., Bissell M. J., Roberts-Thomson S. J., and Monteith G. R. (2008) Localization of plasma membrane and secretory calcium pumps in the mammary gland. Biochem. Biophys. Res. Commun. 369, 977–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Missiaen L., Raeymaekers L., Dode L., Vanoevelen J., Van Baelen K., Parys J. B., Callewaert G., De Smedt H., Segaert S., and Wuytack F. (2004) SPCA1 pumps and Hailey-Hailey disease. Biochem. Biophys. Res. Commun. 322, 1204–1213 [DOI] [PubMed] [Google Scholar]
- 16. Burge S. M. (1992) Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br. J. Dermatol. 126, 275–282 [DOI] [PubMed] [Google Scholar]
- 17. Grice D. M., Vetter I., Faddy H. M., Kenny P. A., Roberts-Thomson S. J., and Monteith G. R. (2010) Golgi calcium pump secretory pathway calcium ATPase 1 (SPCA1) is a key regulator of insulin-like growth factor receptor (IGF1R) processing in the basal-like breast cancer cell line MDA-MB-231. J. Biol. Chem. 285, 37458–37466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feng M., Grice D. M., Faddy H. M., Nguyen N., Leitch S., Wang Y., Muend S., Kenny P. A., Sukumar S., Roberts-Thomson S. J., Monteith G. R., and Rao R. (2010) Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 143, 84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smaardijk S., Chen J., Wuytack F., and Vangheluwe P. (2016) SPCA2 couples Ca2+ influx via Orai1 to Ca2+ uptake into the Golgi/secretory pathway. Tissue Cell 2016, S0040. [DOI] [PubMed] [Google Scholar]
- 20. Cross B. M., Hack A., Reinhardt T. A., and Rao R. (2013) SPCA2 regulates Orai1 trafficking and store independent Ca2+ entry in a model of lactation. PLoS ONE 8, e67348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dode L., Andersen J. P., Raeymaekers L., Missiaen L., Vilsen B., and Wuytack F. (2005) Functional comparison between secretory pathway Ca2+/Mn2+-ATPase (SPCA) 1 and sarcoplasmic reticulum Ca2+-ATPase (SERCA) 1 isoforms by steady-state and transient kinetic analyses. J. Biol. Chem. 280, 39124–39134 [DOI] [PubMed] [Google Scholar]
- 22. Michelangeli F., and East J. M. (2011) A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 39, 789–797 [DOI] [PubMed] [Google Scholar]
- 23. Sorin A., Rosas G., and Rao R. (1997) PMR1, a Ca2+-ATPase in yeast Golgi, has properties distinct from sarco/endoplasmic reticulum and plasma membrane calcium pumps. J. Biol. Chem. 272, 9895–9901 [DOI] [PubMed] [Google Scholar]
- 24. Dode L., Andersen J. P., Vanoevelen J., Raeymaekers L., Missiaen L., Vilsen B., and Wuytack F. (2006) Dissection of the functional differences between human secretory pathway Ca2+/Mn2+-ATPase (SPCA) 1 and 2 isoenzymes by steady-state and transient kinetic analyses. J. Biol. Chem. 281, 3182–3189 [DOI] [PubMed] [Google Scholar]
- 25. Missiaen L., Vanoevelen J., Parys J. B., Raeymaekers L., De Smedt H., Callewaert G., Erneux C., and Wuytack F. (2002) Ca2+ uptake and release properties of a thapsigargin-insensitive nonmitochondrial Ca2+ store in A7r5 and 16HBE14o− cells. J. Biol. Chem. 277, 6898–6902 [DOI] [PubMed] [Google Scholar]
- 26. Denmeade S. R., Mhaka A. M., Rosen D. M., Brennen W. N., Dalrymple S., Dach I., Olesen C., Gurel B., Demarzo A. M., Wilding G., Carducci M. A., Dionne C. A., Moller J. V., Nissen P., Christensen S. B., and Isaacs J. T. (2012) Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 4, 140ra186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doan N. T., Paulsen E. S., Sehgal P., Møller J. V., Nissen P., Denmeade S. R., Isaacs J. T., Dionne C. A., and Christensen S. B. (2015) Targeting thapsigargin toward tumors. Steroids 97, 2–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rigaud J. L. (2002) Membrane proteins: functional and structural studies using reconstituted proteoliposomes and 2-D crystals. Braz. J. Med. Biol. Res. 35, 753–766 [DOI] [PubMed] [Google Scholar]
- 29. Young H. S., Rigaud J. L., Lacapère J. J., Reddy L. G., and Stokes D. L. (1997) How to make tubular crystals by reconstitution of detergent-solubilized Ca2+-ATPase. Biophys. J. 72, 2545–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baron S., Vangheluwe P., Sepúlveda M. R., Wuytack F., Raeymaekers L., and Vanoevelen J. (2010) The secretory pathway Ca2+-ATPase 1 is associated with cholesterol-rich microdomains of human colon adenocarcinoma cells. Biochim. Biophys. Acta 1798, 1512–1521 [DOI] [PubMed] [Google Scholar]
- 31. Kadowaki H., Grant M. A., and Seyfried T. N. (1994) Effect of Golgi membrane phospholipid composition on the molecular species of GM3 gangliosides synthesized by rat liver sialyltransferase. J. Lipid Res. 35, 1956–1964 [PubMed] [Google Scholar]
- 32. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lambert O., Levy D., Ranck J. L., Leblanc G., and Rigaud J. L. (1998) A new “gel-like” phase in dodecyl maltoside-lipid mixtures: implications in solubilization and reconstitution studies. Biophys. J. 74, 918–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamamoto S., Takehara M., Kabashima Y., Fukutomi T., and Ushimaru M. (2016) Identification of novel inhibitors of human SPCA2. Biochem. Biophys. Res. Commun. 477, 266–270 [DOI] [PubMed] [Google Scholar]
- 35. Lai P., and Michelangeli F. (2012) Bis(2-hydroxy-3-tert-butyl-5-methyl-phenyl)-methane (bis-phenol) is a potent and selective inhibitor of the secretory pathway Ca2+-ATPase (SPCA1). Biochem. Biophys. Res. Commun. 424, 616–619 [DOI] [PubMed] [Google Scholar]
- 36. Harper C., Wootton L., Michelangeli F., Lefièvre L., Barratt C., and Publicover S. (2005) Secretory pathway Ca2+-ATPase (SPCA1) Ca2+ pumps, not SERCAs, regulate complex [Ca2+], signals in human spermatozoa. J. Cell Sci. 118, 1673–1685 [DOI] [PubMed] [Google Scholar]
- 37. Plenge-Tellechea F., Soler F., and Fernandez-Belda F. (1997) On the inhibition mechanism of sarcoplasmic or endoplasmic reticulum Ca2+-ATPases by cyclopiazonic acid. J. Biol. Chem. 272, 2794–2800 [DOI] [PubMed] [Google Scholar]
- 38. Quynh Doan N. T., and Christensen S. B. (2015) Thapsigargin, origin, chemistry, structure-activity relationships and prodrug development. Curr. Pharm. Des. 21, 5501–5517 [DOI] [PubMed] [Google Scholar]
- 39. Winther A. M., Liu H., Sonntag Y., Olesen C., le Maire M., Soehoel H., Olsen C. E., Christensen S. B., Nissen P., and Møller J. V. (2010) Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J. Biol. Chem. 285, 28883–28892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Skytte D. M., Møller J. V., Liu H., Nielsen H. Ø., Svenningsen L. E., Jensen C. M., Olsen C. E., and Christensen S. B. (2010) Elucidation of the topography of the thapsigargin binding site in the sarco-endoplasmic calcium ATPase. Bioorg. Med. Chem. 18, 5634–5646 [DOI] [PubMed] [Google Scholar]
- 41. Toyoshima C., and Nomura H. (2002) Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 418, 605–611 [DOI] [PubMed] [Google Scholar]
- 42. Søhoel H., Liljefors T., Ley S. V., Oliver S. F., Antonello A., Smith M. D., Olsen C. E., Isaacs J. T., and Christensen S. B. (2005) Total synthesis of two novel subpicomolar sarco/endoplasmatic reticulum Ca2+-ATPase inhibitors designed by an analysis of the binding site of thapsigargin. J. Med. Chem. 48, 7005–7011 [DOI] [PubMed] [Google Scholar]
- 43. Christensen S. B., Andersen A., Poulsen J. C., and Treiman M. (1993) Derivatives of thapsigargin as probes of its binding site on endoplasmic reticulum Ca2+-ATPase. Stereoselectivity and important functional groups. FEBS Lett. 335, 345–348 [DOI] [PubMed] [Google Scholar]
- 44. Dubois C., Vanden Abeele F., Sehgal P., Olesen C., Junker S., Christensen S. B., Prevarskaya N., and Møller J. V. (2013) Differential effects of thapsigargin analogues on apoptosis of prostate cancer cells: complex regulation by intracellular calcium. FEBS J. 280, 5430–5440 [DOI] [PubMed] [Google Scholar]
- 45. Wootton L. L., and Michelangeli F. (2006) The effects of the phenylalanine 256 to valine mutation on the sensitivity of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) Ca2+ pump isoforms 1, 2, and 3 to thapsigargin and other inhibitors. J. Biol. Chem. 281, 6970–6976 [DOI] [PubMed] [Google Scholar]
- 46. Toyoshima C., Iwasawa S., Ogawa H., Hirata A., Tsueda J., and Inesi G. (2013) Crystal structures of the calcium pump and sarcolipin in the Mg2+-bound E1 state. Nature 495, 260–264 [DOI] [PubMed] [Google Scholar]
- 47. Strehler E. E., and Zacharias D. A. (2001) Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 81, 21–50 [DOI] [PubMed] [Google Scholar]
- 48. Benaim G., Pimentel A. A., Felibertt P., Mayora A., Colman L., Sojo F., Rojas H., and De Sanctis J. B. (2016) Sphingosine inhibits the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) activity. Biochem. Biophys. Res. Commun. 473, 572–577 [DOI] [PubMed] [Google Scholar]
- 49. Li Y., Ge M., Ciani L., Kuriakose G., Westover E. J., Dura M., Covey D. F., Freed J. H., Maxfield F. R., Lytton J., and Tabas I. (2004) Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 279, 37030–37039 [DOI] [PubMed] [Google Scholar]
- 50. Fairclough R. J., Dode L., Vanoevelen J., Andersen J. P., Missiaen L., Raeymaekers L., Wuytack F., and Hovnanian A. (2003) Effect of Hailey-Hailey disease mutations on the function of a new variant of human secretory pathway Ca2+/Mn2+-ATPase (hSPCA1). J. Biol. Chem. 278, 24721–24730 [DOI] [PubMed] [Google Scholar]
- 51. Rigaud J. L., and Lévy D. (2003) Reconstitution of membrane proteins into liposomes. Methods Enzymol. 372, 65–86 [DOI] [PubMed] [Google Scholar]
- 52. Selvy P. E., Lavieri R. R., Lindsley C. W., and Brown H. A. (2011) Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111, 6064–6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Holemans T., Sørensen D. M., van Veen S., Martin S., Hermans D., Kemmer G. C., Van den Haute C., Baekelandt V., Günther Pomorski T., Agostinis P., Wuytack F., Palmgren M., Eggermont J., and Vangheluwe P. (2015) A lipid switch unlocks Parkinson's disease-associated ATP13A2. Proc. Natl. Acad. Sci. U.S.A. 112, 9040–9045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee A. G. (2003) Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta 1612, 1–40 [DOI] [PubMed] [Google Scholar]
- 55. Takahashi M., Kondou Y., and Toyoshima C. (2007) Interdomain communication in calcium pump as revealed in the crystal structures with transmembrane inhibitors. Proc. Natl. Acad. Sci. U.S.A. 104, 5800–5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Simmonds A. C., East J. M., Jones O. T., Rooney E. K., McWhirter J., and Lee A. G. (1982) Annular and non-annular binding sites on the (Ca2+ + Mg2+)-ATPase. Biochim. Biophys. Acta 693, 398–406 [DOI] [PubMed] [Google Scholar]
- 57. Law B. Y., Wang M., Ma D. L., Al-Mousa F., Michelangeli F., Cheng S. H., Ng M. H., To K. F., Mok A. Y., Ko R. Y., Lam S. K., Chen F., Che C. M., Chiu P., and Ko B. C. (2010) Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol. Cancer Ther. 9, 718–730 [DOI] [PubMed] [Google Scholar]
- 58. Autzen H. E., Siuda I., Sonntag Y., Nissen P., Møller J. V., and Thøgersen L. (2015) Regulation of the Ca2+-ATPase by cholesterol: a specific or non-specific effect? Mol. Membr. Biol. 32, 75–87 [DOI] [PubMed] [Google Scholar]
- 59. Jensen A. M., Sørensen T. L., Olesen C., Møller J. V., and Nissen P. (2006) Modulatory and catalytic modes of ATP binding by the calcium pump. EMBO J. 25, 2305–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Obara K., Miyashita N., Xu C., Toyoshima I., Sugita Y., Inesi G., and Toyoshima C. (2005) Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+. Proc. Natl. Acad. Sci. U.S.A. 102, 14489–14496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Toyoshima C., Norimatsu Y., Iwasawa S., Tsuda T., and Ogawa H. (2007) How processing of aspartylphosphate is coupled to lumenal gating of the ion pathway in the calcium pump. Proc. Natl. Acad. Sci. U.S.A. 104, 19831–19836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Toyoshima C., Yonekura S., Tsueda J., and Iwasawa S. (2011) Trinitrophenyl derivatives bind differently from parent adenine nucleotides to Ca2+-ATPase in the absence of Ca2+. Proc. Natl. Acad. Sci. U.S.A. 108, 1833–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Andersen A., Cornett C., Lauridsen A., Olsen C. E., and Christensen S. B. (1994) Selective transformations of the Ca2+ pump inhibitor thapsigargin. Acta Chem. Scand. 48, 340–346 [Google Scholar]
- 64. Smitt U. W., and Christensen S. B. (1991) Nortrilobolide, a new potent guaianolide secretagogue from Thapsia garganica. Planta Med. 57, 196–197 [DOI] [PubMed] [Google Scholar]
- 65. Xu C., Ma H., Inesi G., Al-Shawi M. K., and Toyoshima C. (2004) Specific structural requirements for the inhibitory effect of thapsigargin on the Ca2+ ATPase SERCA. J. Biol. Chem. 279, 17973–17979 [DOI] [PubMed] [Google Scholar]
- 66. Yu M., Lin J., Khadeer M., Yeh Y., Inesi G., and Hussain A. (1999) Effects of various amino acid 256 mutations on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase function and their role in the cellular adaptive response to thapsigargin. Arch. Biochem. Biophys. 362, 225–232 [DOI] [PubMed] [Google Scholar]
- 67. Yu M., Zhong L., Rishi A. K., Khadeer M., Inesi G., Hussain A., and Zhang L. (1998) Specific substitutions at amino acid 256 of the sarcoplasmic/endoplasmic reticulum Ca2+ transport ATPase mediate resistance to thapsigargin in thapsigargin-resistant hamster cells. J. Biol. Chem. 273, 3542–3546 [DOI] [PubMed] [Google Scholar]
- 68. Büttner S., Faes L., Reichelt W. N., Broeskamp F., Habernig L., Benke S., Kourtis N., Ruli D., Carmona-Gutierrez D., Eisenberg T., D'hooge P., Ghillebert R., Franssens V., Harger A., Pieber T. R., et al. (2013) The Ca2+/Mn2+ ion-pump PMR1 links elevation of cytosolic Ca2+ levels to α-synuclein toxicity in Parkinson's disease models. Cell Death Differ. 20, 465–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kaneko M., Desai B. S., and Cook B. (2014) Ionic leakage underlies a gain-of-function effect of dominant disease mutations affecting diverse P-type ATPases. Nat. Genet. 46, 144–151 [DOI] [PubMed] [Google Scholar]
- 70. Okunade G. W., Miller M. L., Azhar M., Andringa A., Sanford L. P., Doetschman T., Prasad V., and Shull G. E. (2007) Loss of the Atp2c1 secretory pathway Ca2+-ATPase (SPCA1) in mice causes Golgi stress, apoptosis, and midgestational death in homozygous embryos and squamous cell tumors in adult heterozygotes. J. Biol. Chem. 282, 26517–26527 [DOI] [PubMed] [Google Scholar]
- 71. Vandecaetsbeek I., Christensen S. B., Liu H., Van Veldhoven P. P., Waelkens E., Eggermont J., Raeymaekers L., Møller J. V., Nissen P., Wuytack F., and Vangheluwe P. (2011) Thapsigargin affinity purification of intracellular P(2A)-type Ca2+ ATPases. Biochim. Biophys. Acta 1813, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 72. Jidenko M., Lenoir G., Fuentes J. M., le Maire M., and Jaxel C. (2006) Expression in yeast and purification of a membrane protein, SERCA1a, using a biotinylated acceptor domain. Protein Expr. Purif. 48, 32–42 [DOI] [PubMed] [Google Scholar]
- 73. Holemans T., Vandecaetsbeek I., Wuytack F., and Vangheluwe P. (2014) Measuring Ca2+ pump activity in overexpression systems and cardiac muscle preparations. Cold Spring Harb Protoc 2014, 876–886 [DOI] [PubMed] [Google Scholar]
- 74. Chemical Computing Group Inc. (2014) Molecular Operating Environment (MOE), version 2014.09, Chemical Computing Group Inc., Montreal, Quebec, Canada [Google Scholar]
- 75. Wang J. M., Cieplak P., and Kollman P. A. (2000) How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21, 1049–1074 [Google Scholar]
- 76. Jones G., Willett P., and Glen R. C. (1995) Molecular recognition of receptor-sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245, 43–53 [DOI] [PubMed] [Google Scholar]
- 77. Jones G., Willett P., Glen R. C., Leach A. R., and Taylor R. (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.