

Abstract
Mechanistic target of rapamycin (mTOR) signaling is necessary to generate a mechanically induced increase in skeletal muscle mass, but the mechanism(s) through which mechanical stimuli regulate mTOR signaling remain poorly defined. Recent studies have suggested that Ras homologue enriched in brain (Rheb), a direct activator of mTOR, and its inhibitor, the GTPase-activating protein tuberin (TSC2), may play a role in this pathway. To address this possibility, we generated inducible and skeletal muscle-specific knock-out mice for Rheb (iRhebKO) and TSC2 (iTSC2KO) and mechanically stimulated muscles from these mice with eccentric contractions (EC). As expected, the knock-out of TSC2 led to an elevation in the basal level of mTOR signaling. Moreover, we found that the magnitude of the EC-induced activation of mTOR signaling was significantly blunted in muscles from both inducible and skeletal muscle-specific knock-out mice for Rheb and iTSC2KO mice. Using mass spectrometry, we identified six sites on TSC2 whose phosphorylation was significantly altered by the EC treatment. Employing a transient transfection-based approach to rescue TSC2 function in muscles of the iTSC2KO mice, we demonstrated that these phosphorylation sites are required for the role that TSC2 plays in the EC-induced activation of mTOR signaling. Importantly, however, these phosphorylation sites were not required for an insulin-induced activation of mTOR signaling. As such, our results not only establish a critical role for Rheb and TSC2 in the mechanical activation of mTOR signaling, but they also expose the existence of a previously unknown branch of signaling events that can regulate the TSC2/mTOR pathway.
Keywords: cell growth, exercise, mechanotransduction, protein synthesis, skeletal muscle
Introduction
As the largest organ in the body, skeletal muscles comprise ∼45% of our total body mass and play essential roles in voluntary movement, metabolic health, and maintaining quality of life (1–4). Indeed, both sedentary and active adults will lose 35–40% of their skeletal muscle mass by the age of 80, and this loss in muscle mass is associated with disability, loss of independence, an increased risk of morbidity and mortality, as well as an estimated $18.5 billion in annual healthcare costs in the United States alone (2, 5–7). Thus, the development of therapies that can maintain, restore, or even enhance muscle mass is a clinically and fiscally significant goal (8). However, to succeed in developing such therapies, we must first understand the molecular mechanisms that regulate skeletal muscle mass.
Skeletal muscle is a highly plastic tissue, and it can change its mass in response to a number of environmental factors. At the most basic level, changes in muscle mass are driven by an alteration in the balance between the rate of protein synthesis and the rate of protein degradation, with a net positive balance leading to muscle growth (i.e. hypertrophy) and a net negative balance leading to muscle loss (i.e. atrophy) (9, 10). Over the last two decades, it has become apparent that a protein kinase called the mammalian/mechanistic target of rapamycin (mTOR)2 plays an essential role in the control of this balance, and mechanical signals have emerged as one of the most potent environmental factors that can regulate mTOR signaling and muscle mass (11–13). However, the mechanism(s) by which mechanical stimuli regulate mTOR signaling and muscle mass remain vaguely defined.
One of the most widely appreciated regulators of mTOR signaling is the Ras homologue enriched in brain (Rheb). Specifically, Rheb is a GTP-binding protein, and it has been shown that GTP-bound Rheb, but not GDP-bound Rheb, can directly stimulate mTOR kinase activity (14, 15). Moreover, it has been shown that the GTP-loading state of Rheb is largely regulated by tuberin (TSC2), which functions as a GTPase-activating protein, and converts active GTP-Rheb into inactive GDP-Rheb (16–19). As such, TSC2 functions as an inhibitor of mTOR signaling and, with this point in mind, a large number of studies have been aimed at understanding the mechanisms that enable TSC2 to control mTOR. To date, the vast majority of these studies have used growth factors (e.g. insulin) as a means for stimulating the TSC2/Rheb/mTOR pathway. According to these studies, it has been concluded that growth factors utilize a PI3K-dependent pathway to promote an increase in the phosphorylation of specific sites on TSC2 and that these changes in phosphorylation, in turn, inhibit the ability of TSC2 to act as a GTPase-activating protein for Rheb (18, 20, 21). As a result, growth factors promote an accumulation of active GTP-Rheb and the subsequent activation of mTOR signaling (20, 22, 23). Intriguingly, we recently discovered that, like growth factors, mechanical stimuli can also promote an increase in the phosphorylation state of TSC2, and this effect is associated with a robust activation of mTOR signaling (25). However, unlike growth factors, mechanical stimuli promote the activation of mTOR signaling via a PI3K-independent pathway (26, 36). Based on this point, we envisioned that mechanical stimuli might utilize a distinct set of phosphorylation sites on TSC2 to control the activation of Rheb/mTOR. However, to date, no studies have directly addressed whether TSC2 and/or Rheb play a role in the mechanical activation of mTOR signaling. Therefore, the initial goal of this study was to determine whether TSC2 and Rheb are necessary for the mechanical activation of mTOR signaling, and, if so, to explore whether changes in TSC2 phosphorylation contribute to this event.
Results
Characterization of skeletal muscle-specific and inducible knock-out mice
To determine whether Rheb and/or TSC2 are necessary for the mechanical activation of mTOR signaling, we developed skeletal muscle-specific and tamoxifen-inducible TSC2 (iTSC2KO+) and skeletal muscle-specific and tamoxifen-inducible Rheb (iRhebKO+), knock-out mice. The use of an inducible knock-out approach was considered to be very important because previous reports have shown that the chronic loss of TSC2, its direct binding partner TSC1, and other components of mTOR signaling can result in the development of various pathological conditions (e.g. chronic muscle-specific TSC1 knock-out mice present with severe kyphosis, a loss of muscle mass, and several other myopathies) (27–29). Therefore, we first set out to create iTSC2KO+ mice, and we did this by crossing floxed TSC2 mice, with mice that possess a transgene for human skeletal actin promoter-driven expression of a mutated estrogen receptor flanked cre-recombinase (HSA-MCM) (30, 31). Litter mate mice that possessed floxed TSC2 but did not express HSA-MCM were used as controls (iTSC2KO−) (supplemental Fig. S1). With these mice, we first determined the minimal amount of time following tamoxifen treatment (TAM) that was needed for a maximal knock-out of TSC2. As shown in Fig. 1A, the knock-out of TSC2 in TAs reached a maximum by 14 days after TAM, whereas the protein level of TSC2 in other tissues such as the brain, heart, and liver were unaffected (Fig. 1, A and B). At 14 days after TAM, TAs from iTSC2KO+ mice also exhibited a significant decrease in TSC1 protein levels, but the major components of the mTOR complexes including mTOR, Raptor, and Rictor were not altered (Fig. 1C). Consistent with the role that TSC2 plays in inhibiting Rheb/mTOR signaling, we determined that TAs from iTSC2KO+ mice exhibit an elevation in the basal levels of mTOR signaling (as revealed by the phosphorylation of p70 and S6) and protein synthesis (Fig. 1, D and E) (28, 32). Moreover, numerous studies have shown that a chronic elevation in mTOR signaling can activate a negative feedback loop that results in down-regulation of signaling through PKB, and consistent with these studies, we found that TAs from iTSC2KO+ mice exhibit a reduction in PKB phosphorylation (Fig. 1D) (33). Interestingly, we did not observe a significant alteration in the mass of the TAs (Fig. 1F). The reason for the lack of a change in muscle mass is not known, but it might be explained by a concomitant elevation in both protein synthesis and protein degradation as suggested by the increased expression of muscle-specific E3 ubiquitin ligases including muscle ring finger protein-1 and atrogin-1 (Fig. 1D). Finally, as shown in Fig. 1G, the gross appearance of the iTSC2KO+ mice was indistinguishable from that of the iTSC2KO− control mice.
Figure 1.

Characterization of the skeletal muscle-specific and inducible TSC2 knock-out mice. Six-week-old skeletal muscle-specific and inducible TSC2 knock-out mice (iTSC2KO+) or control mice (iTSC2KO−) were treated with 2 mg/day of tamoxifen for 5 days (TAM). A, at 7, 14, or 21 days after TAM, the TAs were subjected to Western blotting analysis for TSC2. The values above the blots are expressed relative to the 21-day TAM iTSC2KO− samples. B–G, all remaining analyses were performed on 14-day post-TAM iTSC2KO−/+ mice and included Western blotting analysis for the indicated proteins in soleus, brain, heart, and liver samples (B) or TAs (C and D). The values above the blots represent the total protein amount or the phosphorylated (P) to total protein ratio (P:T) for each group. E, rates of protein synthesis in TAs were assessed by Western blotting for puromycin-labeled peptides. F, muscle weight to body weight ratio of TAs. G, photograph of the TAM iTSC2KO−/+ mice. All values are presented as means (±S.E. in graph, n = 5–8/group). Symbols indicate significant difference (p ≤ 0.05) from 21-day iTSC2KO− (•), 7-day iTSC2KO+ (#), and 14-day iTSC2KO− (*).
Next, we created iRhebKO+ mice by using the same approach as described for the iTSC2KO+ mice, and with these mice we first sought to determine the earliest time point at which a maximal knock-out of Rheb could be observed. However, similar to what others have shown, we found that the protein level of Rheb in skeletal muscle is extremely low when compared with that of other tissues (Fig. 2A) (34). Because of this limitation, we were unable to reliably quantify Rheb protein levels with Western blotting analysis. Hence, to circumvent this issue, we measured Rheb mRNA levels in TAs at 14 days after TAM, and the results indicated that the Rheb mRNA levels were reduced by 77% in muscles from iRhebKO+ mice (p < 1 × 10−5; data not shown). However, a reduction in mRNA levels does not confirm a reduction at the protein level. Therefore, we performed an additional experiment in which an indirect readout of Rheb activity was used as a means for establishing when the maximal knock-out of Rheb occurred. Specifically, previous studies have concluded that Rheb is necessary for the insulin-induced activation of mTOR signaling, and therefore, we reasoned that Rheb expression levels would be maximally depleted at the time point during which the magnitude of the insulin-induced activation of mTOR signaling was maximally reduced (35). Our results demonstrated that, in TAs, this occurred within 7 days after TAM, and the knock-out remained at a maximum for at least 21 days after TAM (Fig. 2B). Therefore, to remain consistent with our studies in the iTSC2KO+ mice, we performed additional analyses on TAs at 14 days after TAM. The results from these analyses revealed that, unlike the loss of TSC2, the loss of Rheb did not significantly alter the basal levels of signaling through mTOR or PKB (Fig. 2C). Moreover, the loss of Rheb did not significantly alter the basal levels of protein synthesis or the mass of the TAs (Fig. 2, D and E). Finally, as shown in Fig. 2F, the gross appearance of the iRhebKO+ mice was indistinguishable from that of the iRhebKO− control mice.
Figure 2.

Characterization of the skeletal muscle-specific and inducible Rheb knock-out mice. Six-week-old skeletal muscle-specific and inducible Rheb knock-out (iRhebKO+) or control (iRhebKO−) mice were treated with 2 mg/day of Tamoxifen for 5 days (TAM). A, at 14 days after TAM, the indicated tissues were collected and Western blotted for Rheb. B, at 7, 14, or 21 days after TAM, mice were injected with insulin (INS+) or PBS (INS−), and after 30 min the TAs were collected and then Western blotted for the phosphorylated (P) to total protein ratio (P:T) of p70. The numbers above the blots represent the values for each group, whereas the graph represents these values when expressed as a percentage of each genotype- and days post-TAM-matched control (INS−) group. C and D, TAs from 14-day post-TAM iRhebKO−/+ mice were Western blotted for the indicated proteins (C) or puromycin-labeled peptides to measure the rates of protein synthesis (D). E and F, muscle weight to body weight ratio for TAs from 14-day post-TAM iRhebKO−/+ mice (E) and photographs of these mice (F). The values are presented as means (±S.E. in graph, n = 5–8/group). Symbols indicate significant difference (p ≤ 0.05) from genotype and days post-TAM-matched control (INS−) (*) and 14-day iRhebKO− INS+ (•).
TSC2 and Rheb significantly contribute to the eccentric contraction-induced activation of mTOR signaling
Having characterized the iTSC2KO+ and iRhebKO+ mice, we next wanted to determine whether TSC2 and/or Rheb are necessary for the mechanical activation of mTOR signaling. To accomplish this, we subjected TAs from 14-day post-TAM iTSC2KO−/+ or iRhebKO−/+ mice to a bout of ECs as a source of mechanical stimulation. The mice were allowed to recover for 60 min after the bout of ECs, and then the muscles were collected and analyzed for changes in p70(389) phosphorylation as a marker of mTOR signaling. As shown in Fig. 3, the outcomes revealed that the loss of either TSC2, or Rheb, resulted in a ≈50% reduction in the magnitude of the EC-induced activation of mTOR signaling. Given the very similar outcomes in the iTSC2KO+ and iRhebKO+ mice, we wanted to confirm that the results were not merely due to a nonspecific effect of the HSA-MCM transgene. Therefore, we performed an additional control experiment in which non-floxed 14-day post-TAM HSA-MCM−/+ mice were subjected to a bout of ECs, and the results demonstrated that the presence of the HSA-MCM transgene did not alter the EC-induced activation of mTOR signaling (supplemental Fig. S2). Moreover, because basal levels of mTOR signaling were highly elevated in muscles from iTSC2KO+ mice, we had to acknowledge that the reduction in the EC-induced activation of mTOR signaling might be an artifact that simply resulted from mTOR reaching a maximal level of activity. Therefore, to address this, we examined muscles at 20 min after the bout of ECs, a time point during which the activation of mTOR signaling is submaximal (36). As shown in supplemental Fig. S3, mTOR signaling was only elevated by 2.5-fold in muscles from iTSC2KO− mice at 20 min after EC, but the impaired EC-induced activation of mTOR signaling in muscles from the iTSC2KO+ mice was still readily apparent at this time point. In other words, the results indicated that impaired EC-induced activation of mTOR signaling could be detected under conditions in which mTOR signaling was not maximally activated. Thus, when taken together, the aforementioned results firmly indicate that both TSC2 and Rheb significantly contribute to the pathway through which ECs activate mTOR signaling (37, 38).
Figure 3.
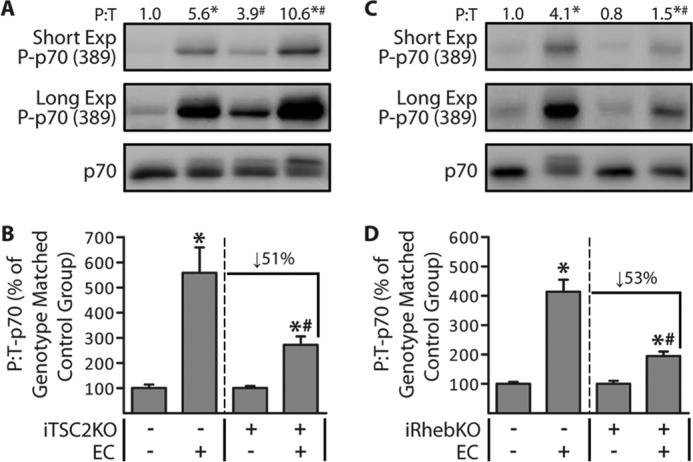
TSC2 and Rheb significantly contribute to the eccentric contraction-induced activation of mTOR signaling. Six-week-old iTSC2KO−/+ mice (A and B) and iRhebKO−/+ mice (C and D) were treated with 2 mg/day of tamoxifen for 5 days (TAM). At 14 days after TAM, TAs were stimulated with a bout of eccentric contractions (EC+) or the control condition (EC−). At 1 h after stimulation, the TAs were collected and subjected to Western blotting analysis for the indicated proteins. The values above the blots represent the phosphorylated (P) to total protein ratio (P:T) for each group. B, graphical representation of the data in A. D, graphical representation of the data in C, with the P:T ratio of p70 for each group expressed relative to its genotype-matched control (EC−) group. For example, in B, the values for the iTSC2KO+/EC+ group are expressed relative to the values obtained in the iTSC2KO+/EC− group. All values are presented as means (±S.E. in graph, n = 5–8/group). Symbols indicate significant difference (p ≤ 0.05) from the genotype-matched control (EC−) (*) or the stimulation-matched condition in iTSC2KO− (A and B) or iRhebKO− (C and D) (#).
Eccentric contractions induce TSC2 phosphorylation through a rapamycin-insensitive mechanism
As mentioned in the introduction, insulin-induced changes in the phosphorylation of TSC2 play a key role in its ability to control the activation of mTOR signaling (20, 39–41). As shown in Fig. 4, we determined that ECs also promote an increase in TSC2 phosphorylation. Furthermore, we determined that rapamycin could completely block the EC-induced activation of mTOR signaling, but it did not affect the EC-induced increase in TSC2 phosphorylation (Fig. 4). This was an important observation because it illustrated that the EC-induced increase in TSC2 phosphorylation is not an event that lies downstream of mTOR, but rather it might be a component of the upstream signaling events that ultimately lead to the activation of mTOR.
Figure 4.
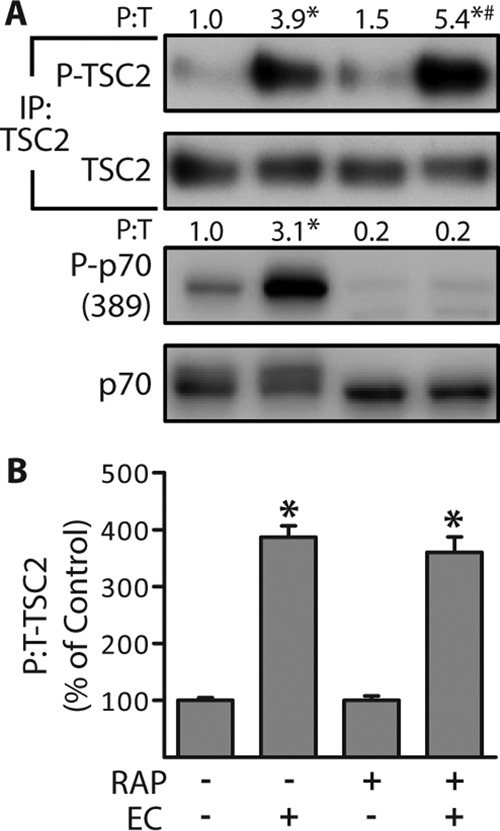
Eccentric contractions induce TSC2 phosphorylation through a rapamycin-insensitive mechanism. Wild-type male C57 mice were injected with 1.5 mg/kg of rapamycin (RAP+) or DMSO (RAP−) 100 min prior to being stimulated with a bout of eccentric contractions (EC+) or the control condition (EC−), and TAs were collected at 1 h after stimulation. A, whole homogenates and TSC2 immunoprecipitates (IP:TSC2) were subjected to Western blotting analysis for the indicated proteins. The values above the blots represent the phosphorylated (P) to total protein ratio (P:T) for each group. B, graphical representation of the data in A with the P:T ratio of TSC2 expressed relative to the drug-matched control (EC−). All values are presented as means (±S.E. in graph, n = 3–4/group). Symbols indicate significant difference (p ≤ 0.05) from drug-matched control (EC−) (*) and stimulation-matched control (RAP−) (#).
Identification of the eccentric contraction-regulated phosphorylation sites on TSC2
To determine whether the EC-induced alterations in TSC2 phosphorylation contribute to the activation of mTOR, we first needed to know which sites on TSC2 undergo a change in phosphorylation. Thus, to identify these sites, we used electroporation to transfect TAs with FLAG-tagged WT TSC2, allowed the muscles to recover for 7 days, and then subjected the muscles to a bout of ECs. Similar to endogenous TSC2, we confirmed that ECs promote an increase in the phosphorylation of WT TSC2 (Fig. 5A). The WT TSC2 was then immunopurified and subjected to LC/MS/MS to identify and quantify specific sites of phosphorylation. It total, we were able to perform quantitative analyses on 17 different phosphorylation sites, and it was concluded that six of these sites (Ser-664, Ser-1155, Ser-1254, Ser-1364, Ser-1449, and Ser-1452) experienced a significant EC-induced increase in phosphorylation (Fig. 5B). Intriguingly, the functional significance of many of these sites (Ser-1155, Ser-1364, Ser-1449, and Ser-1452) has never been reported. Moreover, we identified several of the known insulin-sensitive phosphorylation sites on TSC2 (e.g. Ser-939, Ser-981, and Ser-1132), but none of these sites were affected by ECs (20, 22, 39). Finally, to confirm the efficacy of our LC/MS/MS analyses, we created a FLAG-tagged phospho-defective mutant of TSC2 in which all of the EC-regulated phosphorylation sites were switched to non-phosphorylatable alanines (6A mutant). As shown in Fig. 5C, our results demonstrated that the EC-induced increase in TSC2 phosphorylation was effectively abolished by these mutations (Fig. 5C).
Figure 5.
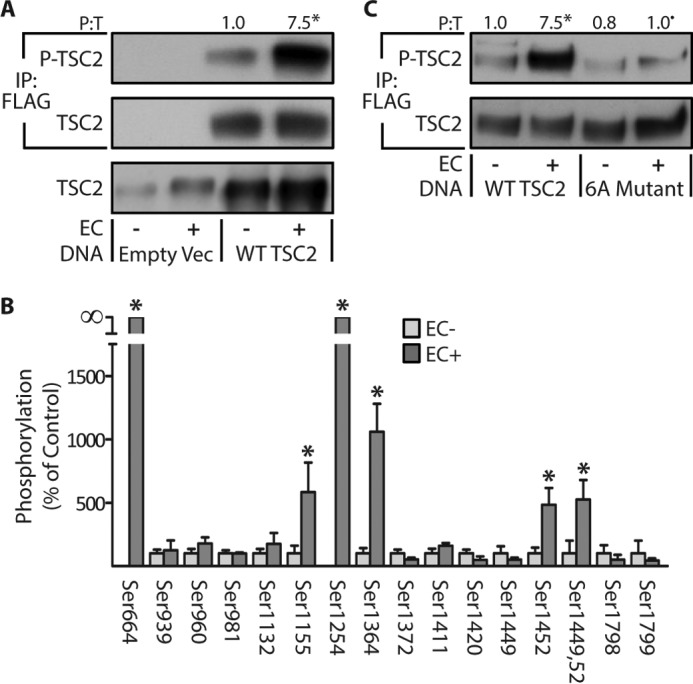
Identification of the eccentric contraction-regulated phosphorylation sites on TSC2. TAs from wild-type male C57 mice were transfected with 30 μg of either the control DNA (empty vector (Empty Vec)), FLAG-tagged WT TSC2, or a FLAG-tagged phosphodefective mutant of TSC2 in which the eccentric contraction-regulated phosphorylation sites were mutated to alanines (6A Mutant). Seven days later, the TAs were stimulated with a bout of eccentric contractions (EC+) or the control condition (EC−) and collected at 1 h after stimulation. A and C, whole homogenates and FLAG immunoprecipitations (IP:FLAG) were subjected to Western blotting analysis for the indicated proteins. B, FLAG immunoprecipitates of WT TSC2 were isolated and then subjected to in-gel trypsin digestion followed by LC/MS/MS to identify and quantify sites of phosphorylation. The values above the blots represent the phosphorylated (P) to total protein ratio (P:T) for each group. All values are presented as means (±S.E. in graph, n = 4–8/group). Note: the values presented for WT TSC2 in A and C are from the same data set. Symbols indicate significant difference (p ≤ 0.05) from DNA matched control (EC−) (*) and stimulation-matched WT TSC2 (•).
The role of TSC2 phosphorylation in the eccentric contraction- and insulin-induced activation of mTOR signaling
Having identified several EC-regulated sites of phosphorylation on TSC2, we next set out to determine whether these sites play a role in the EC-induced activation of mTOR signaling. To accomplish this, we employed a rescue-based model system in which endogenous TSC2 was inducibly knocked out of iTSC2KO+ mice, and then electroporation was used to re-express various forms of TSC2 or an empty vector as a control condition. To the best of our knowledge, we are the only group that has ever utilized this type of rescue approach, and thus, we first needed to establish whether the electroporated muscles could even respond with an EC-induced activation of mTOR signaling. To test this, we electroporated TAs of 14-day post-TAM control mice (iTSC2KO−) with Myc-tagged p70S6k (Myc-p70) and an empty vector, allowed the muscles to recover for 7 days, and then subjected the muscles to a bout of ECs. As shown in Fig. 6A, the results confirmed that mTOR signaling (i.e. Thr-389 phosphorylation on Myc-p70) could be activated by ECs in electroporated muscles. We then performed the same experiment in muscles from 14-day post-TAM iTSC2KO+ mice, and as expected, we determined that endogenous TSC2 was successfully knocked out, the basal levels of mTOR signaling were significantly elevated, and the ability of ECs to elicit and increase in mTOR signaling was dramatically reduced (Fig. 6, A and B). Next, muscles from 14-day post-TAM iTSC2KO+ mice were electroporated with Myc-p70 and either WT TSC2 or the 6A mutant and then subjected to the same experimental conditions described above. Our results confirmed that both of the TSC2 constructs were efficiently expressed, and both were able to abolish the elevation in basal mTOR signaling. As anticipated, we also found that the expression of WT TSC2 rescued the ability of ECs to elicit an increase in mTOR signaling; however, this effect was not observed with the 6A mutant (Fig. 6, A and B). This was a critically important observation, and therefore, we wanted to further ensure that our results were not due to a nonspecific artifact. Hence, we also examined the electroporated muscles for signaling through JNK, and the results demonstrated that EC-induced signaling through JNK was activated to the same degree in all of the aforementioned groups (Fig. 6A). This final result highlighted the specificity of the effect of TSC2 on mTOR signaling and enabled us to conclude that changes in the phosphorylation of TSC2 play a critical role in the EC-induced activation of mTOR signaling.
Figure 6.

The role of TSC2 phosphorylation in the eccentric contraction- and insulin-induced activation of mTOR signaling. Six-week-old iTSC2KO−/+ mice were treated with 2 mg/day of tamoxifen for 5 days (TAM). At 14 days after TAM, TAs were co-transfected with 2 μg of Myc-p70 and either 30 μg of the control plasmid DNA (empty vector), FLAG-tagged WT TSC2, or a FLAG-tagged phosphodefective mutant of TSC2 (6A Mutant). At 7 days after transfection, the TAs were stimulated with a bout of eccentric contractions (EC+) or the control condition (EC−) and collected at 1 h after stimulation (A and B), or the mice were injected with insulin (INS+) or PBS as a control condition (INS−), and TAs were collected at 30 min after injection (C and D). Whole homogenates and Myc immunoprecipitates (IP:Myc) were subjected to Western blotting analysis for the indicated proteins. The values above the blots represent the phosphorylated to total protein ratio (P:T) for each group when expressed relative to the mean value obtained in the iTSC2KO−/empty vector control condition (EC− in B, and INS− in D). B and D, graphical representation of the data in A (B) or C (D) with the P:T ratio of p70 for each EC+ or INS+ group expressed relative to its genotype and plasmid DNA matched control condition. For example, in B, the values for the iTSC2KO+/EC+/empty vector group are expressed relative to the values obtained in the iTSC2KO+/ EC−/empty vector group. In all cases, the values are presented as the group mean (±S.E. in graphs, n = 6–12/group). Symbols indicate significant difference (p ≤ 0.05) from the genotype and plasmid DNA matched control condition (*), the stimulation-matched condition within the empty vector groups (•), and the stimulation-matched empty vector group of iTSC2KO+ mice (#).
Next, we created two partial phospho-defective mutants of TSC2 with the hopes that they would enable us to further tease out which specific phosphorylation sites are necessary for the EC-induced activation of mTOR (supplemental Fig. S4A). Specifically, in one mutant, all of the EC-regulated phosphorylation sites on TSC2 that were immediately followed by a proline were mutated to non-phosphorylatable alanines (Ser-664, Ser-1155, Ser-1449, and Ser-1452, referred to as proline). In the other mutant, the EC-regulated phosphorylation sites on TSC2 that were preceded by a basic amino acid in the −3 position were mutated to non-phosphorylatable alanines (Ser-1254 and Ser-1364, referred to as basic). After creating these mutants, muscles from 14-day post-TAM iTSC2KO+ mice were electroporated with Myc-p70 and WT TSC2 or one of the partial phospho-defective mutants, allowed to recover for 7 days, and then subjected to a bout of ECs. As shown in supplemental Fig. S4, muscles electroporated with WT TSC2 revealed an EC-induced activation of mTOR signaling, but the magnitude of this effect was not altered by either of the partial phospho-defective mutations. Based on this observation, it would appear that the ability of TSC2 to regulate the EC-induced activation of mTOR signaling is not controlled by a single phosphorylation site, but instead, it is controlled by a combination of multiple phosphorylation sites.
In a final series of experiments, we set out to further define the role of the EC-regulated phosphorylation sites on TSC2. Specifically, we wanted to know whether the EC-regulated phosphorylation sites on TSC2 also control the ability of insulin to activate mTOR signaling. We were interested in this question because, as mentioned above, the results from our LC/MS/MS analyses suggested that insulin and ECs regulate a distinct set of phosphorylation sites on TSC2. Thus, to answer our question, we used the same rescue-based approach that was described for our EC studies, but in this case, the muscles were stimulated with insulin instead of ECs. Specifically, we first electroporated TAs of 14-day post-TAM control mice (iTSC2KO−) with Myc-p70 and an empty vector, allowed the muscles to recover for 7 days, and then subjected the muscles to insulin stimulation. As shown in Fig. 6C, the results confirmed that mTOR signaling could be robustly activated by insulin. We then performed the same experiment in muscles from 14-day post-TAM iTSC2KO+ mice, and as expected, we determined that endogenous TSC2 was successfully knocked out, the basal levels of mTOR signaling were significantly elevated, and the ability of insulin to elicit an increase in mTOR signaling was dramatically reduced (Fig. 6, C and D). Next, muscles from 14-day post-TAM iTSC2KO+ mice were electroporated with Myc-p70 and either WT TSC2 or the 6A mutant and then subjected to the same experimental conditions described above. Consistent with the results in Fig. 6A, we found that both of the TSC2 constructs were efficiently expressed, and both were able to abolish the elevated basal level of mTOR signaling. Furthermore, we confirmed that the expression of WT TSC2 rescued the ability of insulin to elicit an increase in mTOR signaling, but in stark contrast to what was observed with ECs, the insulin-induced activation of mTOR signaling was also efficiently rescued by the 6A mutant (Fig. 6, C and D). Again, these results did not appear to be due to any nonspecific artifacts, because insulin robustly activated signaling through PKB in all of the aforementioned groups. Thus, when combined, our results demonstrate that ECs and insulin utilize a distinct subset of phosphorylation sites on TSC2 to control the activation of mTOR signaling.
Discussion
The initial goal of this study was to determine whether TSC2 and/or Rheb are necessary for the mechanical activation of mTOR signaling. We addressed this question by developing the iTSC2KO and iRhebKO mice and then subjecting the muscles from these mice to a bout of ECs as a source of mechanical stimulation. Our results demonstrated that the knock-out of either TSC2, or Rheb, results in an ∼50% reduction in the magnitude of the EC-induced activation of mTOR signaling. The fact that the knock-out of these proteins did not completely abolish the EC-induced activation of mTOR signaling suggests that additional mechanisms contribute to the regulatory pathway. Indeed, recent studies using passive stretch as a source of mechanical stimulation have suggested that the synthesis of phosphatidic acid (another direct activator of mTOR) by diacylglycerol kinase ζ may also significantly contribute to the mechanical activation of mTOR signaling (36, 37). Thus, when combined with previous studies, it would appear that the mechanical activation of mTOR signaling requires inputs from multiple pathways including TSC2/Rheb and diacylglycerol kinase ζ/phosphatidic acid.
Our study also reveals that changes in the phosphorylation of TSC2 are necessary for the role that TSC2 plays in the EC-induced activation of mTOR signaling. Specifically, our work led to the identification of six EC-regulated phosphorylation sites on TSC2. The identification of these sites was particularly exciting because the functional significance of most of the sites had never been explored, and all of the sites were distinct from those that have been reported to be necessary for the growth factor-induced activation of mTOR signaling (Ser-939, Ser-981, Ser-1130, Ser-1132, and Thr-1462) (26, 39, 42). Based on this point, we reasoned that growth factors and ECs might utilize a distinct set of phosphorylation sites on TSC2 to control the activation of mTOR signaling. Indeed, we found that the EC-regulated phosphorylation sites on TSC2 are necessary for the EC-induced activation of mTOR signaling but not the insulin-induced activation of mTOR signaling (Fig. 6). Hence, our study has exposed a previously unknown branch of signaling events that can regulate the TSC2/mTOR pathway.
Given the functional significance of the EC-regulated phosphorylation sites on TSC2, we attempted to further define which specific phosphorylation sites play the most important role. To approach this, we utilized information from previous studies and motif prediction algorithms (e.g. NetworKIN) that indicate that all of the growth factor regulated phosphorylation sites on TSC2 can create a recognition motif for 14-3-3 proteins (43–45). Importantly, it is also known that phosphorylation-mediated binding to 14-3-3 proteins controls the intracellular localization of numerous proteins, including TSC2 (41, 46–48). Moreover, it has been shown that, under basal conditions, mTOR, Rheb, and TSC2 are all enriched on late endosomal/lysosomal (LEL) structures and that insulin-induced changes in TSC2 phosphorylation enable it to translocate away from the LEL (38, 41, 44, 49–53). Combined, these observations have led to a model in which it is thought that insulin-induced changes in TSC2 phosphorylation result in the binding of 14-3-3 proteins and, in turn, cause TSC2 to translocate away from the LEL. As a result, Rheb at the LEL is able to obtain its active GTP-bound state and subsequently promote the activation of the LEL-associated mTOR.
Using motif prediction algorithms, we found that two of the EC-regulated phosphorylation sites on TSC2 (Ser-1254 and Ser-1364) also form putative 14-3-3 binding sites (supplemental Fig. S5). Moreover, we have previously shown that, like growth factors, ECs cause TSC2 to translocate away from the LEL (25, 38). Therefore, we reasoned that the Ser-1254 and Ser-1364 residues might be the key sites that control the EC-induced activation of mTOR signaling. However, inserting phospho-defective mutations into these two sites did not significantly impact the role that TSC2 plays in controlling the EC-induced activation of mTOR signaling. Furthermore, inserting phospho-defective mutations into the four remaining EC-regulated sites also did not significantly impact the role that TSC2 plays in controlling the EC-induced activation of mTOR signaling. The lack of an effect of these partial phospho-defective mutants is very reminiscent of what has been observed in studies that have employed partial phospho-defective mutants of the insulin-regulated phosphorylation sites, and based on these studies, it has been concluded that all five insulin-regulated sites are necessary for the full insulin-induced activation of mTOR signaling (16, 39, 41, 54). Thus, just like insulin, it appears that the ability of TSC2 to regulate the EC-induced activation of mTOR signaling is dependent on a combination of multiple phosphorylation sites.
In summary, the data highlighted in this study establish Rheb and TSC2 as critical regulators of EC-induced activation of mTOR signaling. Moreover, we developed a novel methodology to demonstrate that alterations in TSC2 phosphorylation significantly contribute to this event. In the future, it will be important to further define which combination of phosphorylation sites on TSC2 play the most important role and which kinases/phosphatases control their phosphorylation. Such information will not only expand our knowledge about the upstream signaling cascades that can regulate mTOR, but it will also expand our understanding of the mechanisms through which mechanical stimuli ultimately regulate muscle mass. Furthermore, aberrant regulation of TSC2/mTOR has been implicated in a number of pathologies including tuberous sclerosis, breast cancer, and colorectal cancer (55–58). Thus, with the identification of a previously unknown branch of signaling events that can regulate this pathway, we expect that our results will exert a positive impact on a broad range of fields.
Experimental procedures
Animal care and use
All mice were housed in a room maintained with a 12-h light/12-h dark cycle (lights on from 6 a.m. to 6 p.m.) and received food and water ad libitum. All procedures involving mice were initiated between the hours of 4 and 6 p.m. Prior to surgical procedures, the mice were anesthetized with inhaled isoflurane (1–5%). After tissue extraction, the mice were sacrificed by cervical dislocation. The Institutional Animal Care and Use Committee at the University of Wisconsin–Madison approved all of the methods employed in this study.
Generation of skeletal muscle-specific and tamoxifen-inducible knock-out mice
Female mice homozygous for either TSC2 alleles floxed by loxP sites on exons 1 and 4 (described in Ref. 30; a generous gift from M. Gambello, University of Emory) or for Rheb alleles floxed by loxP sites on exon 3 (described in Ref. 35; a generous gift from P. Worley, Johns Hopkins University) were crossed with male mice that possessed hemizygotic expression of a transgene encoding an inducible Cre recombinase that is flanked by mutated estrogen receptor ligand-binding domains (MCM). In these mice, the expression of the MCM transgene is driven by the human skeletal actin promoter (HSA-MCM) (described in Ref. 59; a generous gift from K. Esser, University of Florida). Offspring were crossed until male mice homozygous for floxed TSC2 or Rheb and hemizygous for HSA-MCM were obtained (iTSC2KO+, iRhebKO+). Mice that were homozygous for floxed TSC2 or Rheb alleles but did not possess the HSA-MCM transgene were used as the control condition (iTSC2KO− and iRhebKO−). To control for the effects of the HSA-MCM transgene, a separate series of experiments were performed on HSA-MCM hemizygotic (HSA-MCM+) or null (HSA-MCM−) male mice that were obtained from crossing female C57BL6 mice (The Jackson Laboratory, Bar Harbor, ME) with male hemizygotic HSA-MCM mice (strain Tg(ACTA1-cre/Esr1*)2Kesr/J); The Jackson Laboratory). Genotypes were confirmed with tail snips followed by PCR using the primers outlined in supplemental Table S1.
Tamoxifen, rapamycin, and insulin injections
Tamoxifen (Sigma-Aldrich) was prepared for injections by first dissolving it in ethanol at a concentration of 85 mg/ml. The ethanol solution was further diluted with peanut oil, which resulted in a final solution that contained 12.75 mg/ml in a 15:85 ethanol:peanut oil mixture. Aliquots of the final tamoxifen solution were sonicated for 20 min at room temperature prior to i.p. injection at a dose of 1–2 mg/day. Rapamycin (LC Laboratories, Woburn, MA) was dissolved in DMSO to generate a 5 μg/μl stock solution. Rapamycin was administered via i.p. injection at a dose of 1.5 mg/kg in 200 μl of PBS, and an equal amount of DMSO dissolved in 200 μl of PBS was used for the vehicle condition. Novolog insulin (Novo Nordisk, Princeton, NJ) was administered via i.p. injection at a dose of 20 units/kg in 100 μl of PBS, and 100 μl of PBS alone was used for the vehicle condition.
Protein synthesis measurements
Protein synthesis was measured with the SUnSET technique as preciously described (60). Briefly, mice were given an i.p. injection of 0.04 μmol/g of puromycin dissolved in 100 μl of PBS. TAs were extracted 30 min after injection, frozen in liquid N2, and subjected to the Western blotting analysis for quantification of puromycin-labeled peptides as described below.
Eccentric contractions
The model previously described (36) was used to induce ECs in the TA. Specifically, electrodes were placed on the sciatic nerve of the right leg, and contractions were elicited by stimulating the sciatic nerve with an SD9E Grass stimulator (Grass Instruments, Quincy, MA) at 100 Hz, 4–8-V pulse, for 10 sets of 6 contractions. Each contraction lasted 3 s and was followed by a 10-s rest period, and a 1-min rest period was provided between each set. The stimulated right (EC+) and the contralateral control left (EC−) TAs were collected 1 h after the last set of contractions, frozen in liquid N2, and subjected to the various measurements below. Unless otherwise noted, the contralateral control left TA samples (EC−) were also used during the experiments aimed at characterizing the iTSC2KO and iRhebKO transgenic mouse lines (i.e. Figs. 1 and 2).
Plasmid constructs, mutagenesis, and purification
pRK7-FLAG-TSC2 (catalog no. 8996) was purchased from Addgene (Cambridge, MA). All TSC2 mutant plasmid constructs were generated from the pRK7-FLAG-TSC2 with the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Myc-p70S6k was also generated by using the QuikChange II site-directed mutagenesis kit by inserting a stop codon immediately before the region that encodes for the GST tag on the pRK5-myc-p70S6K-GST (described in Ref. 31; generous gift from K. Esser, University of Florida). All plasmid DNA was grown in DH5α Escherichia coli, purified with an Endofree plasmid kit (Qiagen), and resuspended in sterile PBS.
Skeletal muscle electroporation
Mice were anesthetized, and a small incision was made through the skin covering the TA. A 27-gauge needle was used to inject plasmid DNA solution (30 μg of pRK7-empty vector, pRK7-FLAG-WT TSC2, or TSC2 mutants (proline; basic; 6A) with or without 2 μg of Myc-p70S6k) into the proximal (6 μl) and distal (6 μl) ends of the muscle belly. Following the injections, two stainless steel pin electrodes (1-cm gap; Harvard Apparatus, Holliston, MA) connected to an ECM 830 electroporation unit (BTX/Harvard Apparatus, Holliston, MA) were laid on top of the proximal and distal myotendinous junctions and eight 20-ms square-wave electric pulses were delivered at a frequency of 1 Hz with a field strength of 160 V/cm as previously described (26). Following the electroporation procedure, the incision was closed with Vetbond surgical glue (Henry Schein, Melville, NY), and the mice were given an i.p. injection of buprenorphine (0.05 μg/g) dissolved in 100 μl of PBS.
Sample preparation
Upon collection, the muscles were immediately frozen in liquid nitrogen. The samples were homogenized with a Polytron for 20 s in either ice-cold buffer A (40 mm Tris, pH 7.5, 1 mm EDTA, 5 mm EGTA, 0.5% Triton X-100, 25 mm β-glycerophosphate, 25 mm NaF, 1 mm Na3VO4, 10 mg/ml leupeptin, and 1 mm PMSF) or buffer B for endogenous TSC2 immunoprecipitations (10 mm Tris-HCl, pH 7.5; 100 mm NaCl; 2 mm EDTA; 1% Nonidet P-40; 1 mm DTT; 1 mm PMSF; 20 μg/ml of leupeptin, pepstatin, aprotinin, and soybean trypsin inhibitor; 25 mm NaF; 25 mm β-glycerophosphate; and 1 mm Na3VO4). Either the whole homogenate was used for further analysis (puromycin Western blots only), or the homogenate was spun down at 2,500 × g for 5 min, and the supernatant was removed and used for further analysis. Sample protein concentration was determined with a DC protein assay kit (Bio-Rad).
Immunoprecipitations
For endogenous TSC2 immunoprecipitations, 400 μg of protein was diluted to a volume of 0.5 ml with fresh ice-cold buffer B. The samples were then incubated with anti-TSC2 (1:200) (Cell Signaling, Danvers, MA) at 4 °C for 2 h. During this incubation, 40 μl of protein A-agarose beads (Santa Cruz) were blocked in ice-cold 1% BSA-PBS for 1 h and then washed three times with PBS. The antibody-containing samples were incubated with the blocked beads at 4 °C for 2 h. The beads were then pelleted by centrifugation at 500 × g for 30 s and washed four times with buffer B and three times with ice-cold buffer C (40 mm Hepes, pH 7.4, 400 mm NaCl, 2 mm EDTA, 0.3% CHAPS). For immunoprecipitations of FLAG and Myc-tagged proteins, 800 μg (for FLAG IP) or 500 μg (for Myc IP) was diluted to a volume of 0.5 ml with fresh ice-cold buffer A. The samples were then incubated with 16 μl of EZview red anti-FLAG M2 agarose affinity gel beads (catalog no. F2426; Sigma-Aldrich) or 25 μl of EZview red anti-Myc agarose affinity gel beads (catalog no. E6654; Sigma-Aldrich) with gentle rocking at 4 °C for 2 h. Following the incubation, the beads were pelleted by centrifugation at 500 × g for 30 s and washed four times with fresh ice-cold buffer A. FLAG IP samples were then washed three times with ice-cold buffer C. After the washes, the pellets were dissolved in Laemmli buffer, heated to 100 °C for 5 min, and pelleted at 500 × g for 30 s, and then the supernatant was subjected to Western blotting analysis as described below.
Western blotting analysis
Western blot analyses were performed as previously described (61). Briefly, equivalent amounts of protein from each sample were dissolved in Laemmli buffer, heated to 100 °C for 5 min, and then subjected to electrophoretic separation by SDS-PAGE. Following electrophoretic separation, proteins were transferred to a PVDF membrane and blocked with 5% powdered milk in TBS containing 0.1% Tween 20 (TBST) for 1 h, followed by an overnight incubation at 4 °C with primary antibody dissolved in TBST containing 1% BSA. After an overnight incubation, the membranes were washed for 30 min in TBST and then probed with a peroxidase-conjugated secondary antibody for 1 h at room temperature. Following 30 min of washing in TBST, the blots were developed on film or with a Chemi410 camera mounted to a UVP Autochemi system (UVP, Upland, CA) using regular enhanced ECL reagent (Pierce) or ECL Prime reagent (Amersham Biosciences). Once the appropriate image was captured, the membranes were stained with Coomassie Blue to verify equal loading in all lanes. Images were quantified using ImageJ software (National Institutes of Health).
Antibodies
P-p70(389) (1A5) (catalog no. 9206, used to probe Myc-tagged P-p70), P-p70(389) (catalog no. 9234, used to probe endogenous P-p70), total p70 (49D7) (catalog no. 2708), anti-P-Akt substrate (RXRXX(S*/T*)) (23C8D2) for detection of P-TSC2 (catalog no. 10001), P-JNK(Thr-183/Tyr-185) (98F2) (catalog no. 4671), total JNK (catalog no. 9252), total mTOR (catalog no. 2972), total Rictor (D16H9) (catalog no. 9476), P-PKB (Ser-473) (catalog no. 4060), P-PKB (Thr-308) (catalog no. 9275S), total PKB (catalog no. 9272), P-S6 (Ser-240/244) (catalog no. 5364), P-S6 (Ser-235/236) (catalog no. 2211), total S6 (catalog no. 2217S), total TSC1 (D43E2) (catalog no. 6935), and total TSC2 (D93F12) (catalog no. 4308) were purchased from Cell Signaling Technology (Danvers, MA). Anti-puromycin (12D10) (catalog no. MABE343) was purchased from EMD Millipore (Billerica, MA). Anti-total muscle ring finger protein-1 and total atrogin-1 antibodies were obtained from Regeneron Pharmaceuticals (Tarrytown, NY). Anti-Rheb (2C11) (catalog no. M01) was purchased from Abnova (Taipei, Taiwan). Peroxidase-labeled anti-rabbit IgG (H+L) (catalog no. PI-1000) and anti-mouse IgG (H+L) (catalog no. NC9483837) secondary antibodies were purchased from Vector Laboratories Inc. (Burlingame, CA). Peroxidase-labeled anti-rabbit IgG (Light Chain Specific) (catalog no. 211-032-171, for Myc-tagged p70 blots) and anti-mouse IgG2a (catalog no. 115-035-206, for Myc-tagged p70 and puromycin blots) antibodies were purchased from Jackson ImmnunoResearch Laboratories Inc. (West Grove, PA).
Mass spectrometric analysis
Male C57BL6 mice (The Jackson Laboratory) at 8–12 weeks of age were randomly assigned to the experimental groups. FLAG-tagged TSC2 was immunoprecipitated and subjected to SDS-PAGE as described above. The gel was stained with Coomassie Blue, and the band corresponding to TSC2 was cut out of the gel and subjected to in-gel trypsin digestion followed by LC/MS/MS. In-gel digestion and mass spectrometric analysis were done at the Mass Spectrometry Facility (Biotechnology Center, University of Wisconsin–Madison) as previously described (24). A method of label-free quantification adapted from (40) was used to identify the individual phosphorylation sites that were significantly altered by eccentric contractions. Specifically, for each individual sample, the total ion current (TIC) for all of the phosphopeptides and non-phosphopeptides identified in that run which had ≥85% identification probability were determined with Scaffold version 4.0 software (Proteome Software, Portland, OR). For each identified phosphopeptide, the sum of the peak areas from the TIC values of the phosphopeptides was divided by the average TIC observed in a set of reference non-phosphopeptides (supplemental Table S2), and this value was used as a relative index of the phosphorylation state. These reference non-phosphopeptides were identified in every mass spectrometry run (n = 9) and never exhibited a phosphorylation event.
Statistical analysis
All values are expressed as the mean (±S.E. in graphs). Statistical significance was determined by using a two-tailed Student's t test for single comparisons and one or two-way analysis of variance followed by planned comparisons or Student Newman-Kuels pairwise comparisons, respectively, for multiple comparisons. Differences between groups were considered significant when p ≤ 0.05. All statistical analyses were performed using Excel or SigmaStat software (San Jose, CA).
Author contributions
B. L. J. and T. A. H. designed the experiments; B. L. J., R. M. M., K. J. K., R. B., R. E. P., J.-S. Y., and T. A. H. acquired and analyzed data; B. L. J. and T. A. H. wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
This work was supported by NIAMS, National Institutes of Health Grant AR057347 and Department of Defense Grant W81XWH-14-1-0105 (to T. A. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1 and S2 and Figs. S1–S5.
- mTOR
- mechanistic target of rapamycin
- EC
- eccentric contractions
- HSA
- human skeletal actin promoter
- iRhebKO
- inducible and skeletal muscle-specific knock-out mice for Rheb
- iTSC2KO
- inducible and skeletal muscle-specific knock-out mice for TSC2
- LEL
- late endosome/lysosome
- MCM
- mutated estrogen receptor flanked cre-recombinase
- PKB
- protein kinase B
- Rheb
- Ras homologue enriched in brain
- S6
- ribosomal S6 protein
- TA
- tibialis anterior muscle
- TAM
- tamoxifen
- TSC1
- hamartin
- TSC2
- tuberin
- TIC
- total ion current.
References
- 1. Pedersen B. K., and Febbraio M. A. (2012) Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 8, 457–465 [DOI] [PubMed] [Google Scholar]
- 2. Seguin R., and Nelson M. E. (2003) The benefits of strength training for older adults. Am. J. Prev. Med. 25, 141–149 [DOI] [PubMed] [Google Scholar]
- 3. Izumiya Y., Hopkins T., Morris C., Sato K., Zeng L., Viereck J., Hamilton J. A., Ouchi N., LeBrasseur N. K., and Walsh K. (2008) Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 7, 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Srikanthan P., and Karlamangla A. S. (2011) Relative muscle mass is inversely associated with insulin resistance and prediabetes: findings from the third National Health and Nutrition Examination Survey. J. Clin. Endocrinol. Metab. 96, 2898–2903 [DOI] [PubMed] [Google Scholar]
- 5. Proctor D. N., Balagopal P., and Nair K. S. (1998) Age-related sarcopenia in humans is associated with reduced synthetic rates of specific muscle proteins. J. Nutr. 128, 351S–355S [DOI] [PubMed] [Google Scholar]
- 6. Pahor M., and Kritchevsky S. (1998) Research hypotheses on muscle wasting, aging, loss of function and disability. J. Nutr. Health Aging 2, 97–100 [PubMed] [Google Scholar]
- 7. Srikanthan P., and Karlamangla A. S. (2014) Muscle mass index as a predictor of longevity in older adults. Am. J. Med. 127, 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hurley B. F., Hanson E. D., and Sheaff A. K. (2011) Strength training as a countermeasure to aging muscle and chronic disease. Sports Med. 41, 289–306 [DOI] [PubMed] [Google Scholar]
- 9. Goldberg A. L., Etlinger J. D., Goldspink D. F., and Jablecki C. (1975) Mechanism of work-induced hypertrophy of skeletal muscle. Med. Sci. Sports 7, 185–198 [PubMed] [Google Scholar]
- 10. Evans W. J. (2010) Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am. J. Clin. Nut.r 91, 1123S–1127S [DOI] [PubMed] [Google Scholar]
- 11. Schiaffino S., Dyar K. A., Ciciliot S., Blaauw B., and Sandri M. (2013) Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 280, 4294–4314 [DOI] [PubMed] [Google Scholar]
- 12. Hornberger T. A. (2011) Mechanotransduction and the regulation of mTORC1 signaling in skeletal muscle. Int. J. Biochem. Cell Biol. 43, 1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hall M. N. (2013) Talks about TORCs: recent advances in target of rapamycin signalling on mTOR nomenclature. Biochem. Soc. Trans. 41, 887–888 [DOI] [PubMed] [Google Scholar]
- 14. Sancak Y., Thoreen C. C., Peterson T. R., Lindquist R. A., Kang S. A., Spooner E., Carr S. A., and Sabatini D. M. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915 [DOI] [PubMed] [Google Scholar]
- 15. You J. S., Frey J. W., and Hornberger T. A. (2012) Mechanical stimulation induces mTOR signaling via an ERK-independent mechanism: implications for a direct activation of mTOR by phosphatidic acid. PLoS One 7, e47258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang J., and Manning B. D. (2008) The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y., Gao X., Saucedo L. J., Ru B., Edgar B. A., and Pan D. (2003) Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 5, 578–581 [DOI] [PubMed] [Google Scholar]
- 18. Inoki K., Li Y., Xu T., and Guan K. L. (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tee A. R., Fingar D. C., Manning B. D., Kwiatkowski D. J., Cantley L. C., and Blenis J. (2002) Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. U.S.A. 99, 13571–13576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manning B. D., Tee A. R., Logsdon M. N., Blenis J., and Cantley L. C. (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 10, 151–162 [DOI] [PubMed] [Google Scholar]
- 21. Potter C. J., Pedraza L. G., and Xu T. (2002) Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 4, 658–665 [DOI] [PubMed] [Google Scholar]
- 22. Inoki K., Li Y., Zhu T., Wu J., and Guan K.-L. (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657 [DOI] [PubMed] [Google Scholar]
- 23. Inoki K., Zhu T., and Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 24. Frey J. W., Jacobs B. L., Goodman C. A., and Hornberger T. A. (2014) A role for Raptor phosphorylation in the mechanical activation of mTOR signaling. Cell Signal. 26, 313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacobs B. L., You J. S., Frey J. W., Goodman C. A., Gundermann D. M., and Hornberger T. A. (2013) Eccentric contractions increase the phosphorylation of tuberous sclerosis complex-2 (TSC2) and alter the targeting of TSC2 and the mechanistic target of rapamycin to the lysosome. J. Physiol. 591, 4611–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goodman C. A., Miu M. H., Frey J. W., Mabrey D. M., Lincoln H. C., Ge Y., Chen J., and Hornberger T. A. (2010) A phosphatidylinositol 3-kinase/protein kinase B-independent activation of mammalian target of rapamycin signaling is sufficient to induce skeletal muscle hypertrophy. Mol. Biol. Cell 21, 3258–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bentzinger C. F., Romanino K., Cloëtta D., Lin S., Mascarenhas J. B., Oliveri F., Xia J., Casanova E., Costa C. F., Brink M., Zorzato F., Hall M. N., and Rüegg M. A. (2008) Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8, 411–424 [DOI] [PubMed] [Google Scholar]
- 28. Bentzinger C. F., Lin S., Romanino K., Castets P., Guridi M., Summermatter S., Handschin C., Tintignac L. A., Hall M. N., and Rüegg M. A. (2013) Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skeletal Muscle 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castets P., Lin S., Rion N., Di Fulvio S., Romanino K., Guridi M., Frank S., Tintignac L. A., Sinnreich M., and Rüegg M. A. (2013) Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 17, 731–744 [DOI] [PubMed] [Google Scholar]
- 30. Hernandez O., Way S., McKenna J. 3rd, Gambello M. J. (2007) Generation of a conditional disruption of the Tsc2 gene. Genesis 45, 101–106 [DOI] [PubMed] [Google Scholar]
- 31. Miyazaki M., and Esser K. A. (2009) REDD2 is enriched in skeletal muscle and inhibits mTOR signaling in response to leucine and stretch. Am. J. Physiol. Cell Physiol. 296, C583–C592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R., Gout I., Downes C. P., and Lamb R. F. (2004) The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Efeyan A., and Sabatini D. M. (2010) mTOR and cancer: many loops in one pathway. Curr. Opin. Cell Biol. 22, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Apró W., Moberg M., Hamilton D. L., Ekblom B., Rooyackers O., Holmberg H. C., and Blomstrand E. (2015) Leucine does not affect mechanistic target of rapamycin complex 1 assembly but is required for maximal ribosomal protein s6 kinase 1 activity in human skeletal muscle following resistance exercise. FASEB J. 29, 4358–4373 [DOI] [PubMed] [Google Scholar]
- 35. Zou J., Zhou L., Du X. X., Ji Y., Xu J., Tian J., Jiang W., Zou Y., Yu S., Gan L., Luo M., Yang Q., Cui Y., Yang W., Xia X., et al. (2011) Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 20, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Neil T. K., Duffy L. R., Frey J. W., and Hornberger T. A. (2009) The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J. Physiol. 587, 3691–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. You J. S., Lincoln H. C., Kim C. R., Frey J. W., Goodman C. A., Zhong X. P., and Hornberger T. A. (2014) The role of diacylglycerol kinase ζ and phosphatidic acid in the mechanical activation of mammalian target of rapamycin (mTOR) signaling and skeletal muscle hypertrophy. J. Biol. Chem. 289, 1551–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jacobs B. L., Goodman C. A., and Hornberger T. A. (2014) The mechanical activation of mTOR signaling: an emerging role for late endosome/lysosomal targeting. J. Muscle Res. Cell Motil. 35, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang H. H., Huang J., Düvel K., Boback B., Wu S., Squillace R. M., Wu C. L., and Manning B. D. (2009) Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One 4, e6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langlais P., Mandarino L. J., and Yi Z. (2010) Label-free relative quantification of co-eluting isobaric phosphopeptides of insulin receptor substrate-1 by HPLC-ESI-MS/MS. J. Am. Soc Mass. Spectrom. 21, 1490–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cai S. L., Tee A. R., Short J. D., Bergeron J. M., Kim J., Shen J., Guo R., Johnson C. L., Kiguchi K., and Walker C. L. (2006) Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 173, 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hornberger T. A., Stuppard R., Conley K. E., Fedele M. J., Fiorotto M. L., Chin E. R., and Esser K. A. (2004) Mechanical stimuli regulate rapamycin-sensitive signalling by a phosphoinositide 3-kinase-, protein kinase B- and growth factor-independent mechanism. Biochem. J. 380, 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Horn H., Schoof E. M., Kim J., Robin X., Miller M. L., Diella F., Palma A., Cesareni G., Jensen L. J., and Linding R. (2014) KinomeXplorer: an integrated platform for kinome biology studies. Nat. Methods 11, 603–604 [DOI] [PubMed] [Google Scholar]
- 44. Li Y., Inoki K., Vacratsis P., and Guan K. L. (2003) The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J. Biol. Chem. 278, 13663–13671 [DOI] [PubMed] [Google Scholar]
- 45. Li Y., Inoki K., Yeung R., and Guan K. L. (2002) Regulation of TSC2 by 14-3-3 binding. J. Biol. Chem. 277, 44593–44596 [DOI] [PubMed] [Google Scholar]
- 46. Miyazaki M., McCarthy J. J., and Esser K. A. (2010) Insulin like growth factor-1-induced phosphorylation and altered distribution of tuberous sclerosis complex (TSC)1/TSC2 in C2C12 myotubes. FEBS J. 277, 2180–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muslin A. J., and Xing H. (2000) 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal. 12, 703–709 [DOI] [PubMed] [Google Scholar]
- 48. van Heusden G. P. (2005) 14-3-3 proteins: regulators of numerous eukaryotic proteins. IUBMB Life 57, 623–629 [DOI] [PubMed] [Google Scholar]
- 49. Dibble C. C., Elis W., Menon S., Qin W., Klekota J., Asara J. M., Finan P. M., Kwiatkowski D. J., Murphy L. O., and Manning B. D. (2012) TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L., and Sabatini D. M. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., and Sabatini D. M. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flinn R. J., Yan Y., Goswami S., Parker P. J., and Backer J. M. (2010) The late endosome is essential for mTORC1 signaling. Mol. Biol. Cell 21, 833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saito K., Araki Y., Kontani K., Nishina H., and Katada T. (2005) Novel role of the small GTPase Rheb: its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 137, 423–430 [DOI] [PubMed] [Google Scholar]
- 54. Menon S., Dibble C. C., Talbott G., Hoxhaj G., Valvezan A. J., Takahashi H., Cantley L. C., and Manning B. D. (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ma L., Teruya-Feldstein J., Bonner P., Bernardi R., Franz D. N., Witte D., Cordon-Cardo C., and Pandolfi P. P. (2007) Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 67, 7106–7112 [DOI] [PubMed] [Google Scholar]
- 56. Ma L., Chen Z., Erdjument-Bromage H., Tempst P., and Pandolfi P. P. (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 [DOI] [PubMed] [Google Scholar]
- 57. Zhang Y., Wang X., Qin X., Liu F., White E., and Zheng X. F. (2015) PP2AC level determines differential programming of p38-TSC-mTOR signaling and therapeutic response to p38-targeted therapy in colorectal cancer. EBioMedicine 2, 1944–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Inoki K., and Guan K. L. (2009) Tuberous sclerosis complex, implication from a rare genetic disease to common cancer treatment. Hum. Mol. Genet. 18, R94–R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCarthy J. J., Srikuea R., Kirby T. J., Peterson C. A., and Esser K. A. (2012) Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skeletal Muscle 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goodman C. A., Mabrey D. M., Frey J. W., Miu M. H., Schmidt E. K., Pierre P., and Hornberger T. A. (2011) Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 25, 1028–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goodman C. A., Frey J. W., Mabrey D. M., Jacobs B. L., Lincoln H. C., You J. S., and Hornberger T. A. (2011) The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J. Physiol. 589, 5485–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.