

Abstract
Several studies have linked impaired glucose uptake and insulin resistance (IR) to functional impairment of the heart. Recently, endocannabinoids have been implicated in cardiovascular disease. However, the mechanisms involving endocannabinoid signaling, glucose uptake, and IR in cardiomyocytes are understudied. Here we report that the endocannabinoid 2-arachidonoylglycerol (2-AG), via stimulation of cannabinoid type 1 (CB1) receptor and Ca2+/calmodulin-dependent protein kinase β, activates AMP-activated kinase (AMPK), leading to increased glucose uptake. Interestingly, we have observed that the mRNA expression of CB1 and CB2 receptors was decreased in diabetic mice, indicating reduced endocannabinoid signaling in the diabetic heart. We further establish that TNFα induces IR in cardiomyocytes. Treatment with 2-AG suppresses TNFα-induced proinflammatory markers and improves IR and glucose uptake. Conversely, pharmacological inhibition or knockdown of AMPK attenuates the anti-inflammatory effect and reversal of IR elicited by 2-AG. Additionally, in human embryonic stem cell-derived cardiomyocytes challenged with TNFα or FFA, we demonstrate that 2-AG improves insulin sensitivity and glucose uptake. In conclusion, 2-AG abates inflammatory responses, increases glucose uptake, and overcomes IR in an AMPK-dependent manner in cardiomyocytes.
Keywords: AMP-activated kinase (AMPK), cell signaling, endocannabinoid, glucose transporter type 4 (GLUT4), inflammation, insulin resistance, glucose uptake
Introduction
Cardiac insulin resistance (IR),4 at its most fundamental level, inhibits the myocardial uptake of glucose in response to insulin. Insulin-resistant cardiomyocytes therefore preferentially metabolize FFA rather than glucose. IR contributes to the development of myocardial dysfunction, ultimately leading to diabetic cardiomyopathy (DCM), a major cause of morbidity and mortality in developed nations (1). Chronic lipid overload leads to increased cardiac lipid uptake and, via processes remaining to be fully elucidated, causes IR (2). Furthermore, local and systemic inflammatory processes participate in the development of cardiac IR (3). Previous studies demonstrated the development of IR by application of the proinflammatory cytokine TNFα (4, 5). TNFα, via induction and increased nuclear translocation of its downstream effector nuclear factor κ light chain enhancer of activated B cells p105 subunit (NFKB1) and IL-6 and IL-8 (CXCL8), along with chemokines like monocyte chemoattractant protein 1 (CCL2), participate in the activation and maintenance of intracellular pathways that promote the development of IR. However, irrespective of whether cardiac IR is induced by lipid overload or inflammatory stress, in both cases, cardiomyocytes down-regulate glucose utilization in long-term precipitating DCM. Vice versa, improvement of glucose metabolism and anti-inflammatory actions could improve IR and therefore prevent DCM.
Recent developments identify the endocannabinoid system (ECS) as a physiological signaling network and therapeutic target for the treatment of various pathological conditions, including cardiovascular disease (6). The endocannabinoids 2-arachidonoylglycerol (2-AG) and anandamide (AEA) are endogenous lipid mediators that predominantly act as a ligand for the G protein-coupled cannabinoid type 1 (CB1R) and type 2 (CB2R) receptors, respectively. Before its recent withdrawal from the market because of its association with increased incidence of psychiatric adverse events, the CB1R antagonist rimonabant was shown to improve body weight and metabolic and inflammatory abnormalities in several trials in obese subjects. Conversely, evidence suggests that endocannabinoids have important protective roles in pathophysiological conditions such as ischemic shock and myocardial infarction (7, 8). However, the underlying molecular and cellular mechanisms of endocannabinoid action remain elusive.
In this study, we have demonstrated that 2-AG, in a CaMKKβ-dependent manner, activates the AMPK signaling pathway. In an inflammatory stress-induced model of IR, we have demonstrated that 2-AG exerts anti-inflammatory effects, restores the dysfunctional insulin signaling pathway, and stimulates glucose uptake in cardiomyocytes. Additionally, we have created a novel model mimicking human cardiomyocyte insulin resistance by differentiating human embryonic stem cells into functional cardiomyocytes and exposing them to inflammatory stress or lipid overload. In this unique model, we have demonstrated that 2-AG exerts strong anti-inflammatory effects and restores insulin sensitivity in differentiated cardiomyocytes. Overall, our study provides a novel and detailed analysis of the molecular mechanism linking 2-AG with cardiomyocyte insulin resistance and indicates a potential beneficial role in the treatment of cardiometabolic diseases.
Results
TNFα induces IR in HL-1 cardiomyocytes
Initially, we investigated the effect of TNFα on insulin signaling and insulin-induced glucose uptake in HL-1 cardiomyocytes. TNFα exposure significantly induced gene expression of the key proinflammatory markers Nfkb1, Il6, Cxcl8, and Ccl2, suggesting that TNFα induces an inflammatory response in cardiomyocytes (Fig. 1A). Next, untreated and TNFα-treated cardiomyocytes were stimulated with insulin for a short time. In TNFα-challenged cardiomyocytes, insulin-induced phosphorylation of protein kinase B (AKT), AKT substrate 160 (AS160), and glycogen synthase kinase 3β (GSK-3β) was dramatically reduced, suggesting a deregulation of the insulin signaling pathway by TNFα (Fig. 1B). Furthermore, TNFα exposure led to a significant decrease in basal Glut4 mRNA level (Fig. 1C) and insulin-stimulated plasmalemmal GLUT4 translocation (Fig. 1D). Subsequently, as a functional consequence of perturbed insulin signaling, insulin-stimulated glucose uptake was significantly abrogated upon TNFα challenge compared with untreated cardiomyocytes (Fig. 1E). Additionally, in TNFα-challenged cardiomyocytes, periodic acid-Schiff staining indicated lowered glycogen content, visible as pink coloration (Fig. 1F), and quantitative analysis by glucose oxidase assay confirmed a decrease in glycogen (∼25%) compared with untreated cells (Fig. 1G). Overall, these results demonstrate that TNFα induces an inflammatory response and perturbs the insulin signaling pathway, coupled with decreased GLUT4 translocation, glucose uptake, and glycogen content, ultimately leading to IR in cardiomyocytes.
Figure 1.

Characterization of TNFα-induced IR. A, relative mRNA levels of proinflammatory markers. *, p < 0.01 by unpaired Student's t test. B, representative Western-blotting analysis of the insulin (Ins) signaling pathway following insulin stimulation. p, phospho; t, total. C, relative mRNA level of Glut4. *, p < 0.01 by unpaired Student's t test. D, insulin-stimulated GLUT4-myc translocation. *, p = 0.047 by two-way ANOVA with Bonferroni's multiple comparisons post hoc test. E, insulin-stimulated glucose uptake. *, p = 0.041 by two-way ANOVA with Bonferroni's multiple comparisons post hoc test. ACTB, β-actin. F, representative periodic acid-Schiff staining for intracellular glycogen content. Scale bar = 100 μm, ×40 magnification. G, glucose oxidase assay for quantitation of intracellular glycogen content. *, p < 0.01 by unpaired Student's t test. Data are expressed as mean ± S.D. n = 4–5 independent experiments in HL-1 cardiomyocytes.
2-AG-induced activation of the AMPK signaling pathway is mediated via the upstream kinase CaMKKβ
Previously, it was reported that cannabinoid derivative trans-Δ9-tetrahydrocannabinol activates the AMPK signaling pathway (9). Therefore, we initially investigated the time- and dose-dependent effect of the endocannabinoid 2-AG on AMPK activation in HL-1 cardiomyocytes (supplemental Fig. 1). Based on previous reports (10, 11), a working concentration of 10 μm 2-AG was chosen for the experiments. We observed that 2-AG treatment led to rapid phosphorylation of AMPK within 1 h (supplemental Fig. 1A). Upon further investigation, we observed that 2-AG in the range of 1–20 μm dose-dependently increased AMPK phosphorylation in HL-1 cardiomyocytes (supplemental Fig. 1B). Additionally, 2-AG treatment also led to rapid phosphorylation (within minutes) of both AMPK and ACC in HL-1 cardiomyocytes and isolated primary rat cardiomyocytes (Fig. 2A). Furthermore, cells were pretreated with specific antagonists of CB1R (AM251, 5 μm) or CB2R (AM630, 5 μm), followed by treatment with 2-AG (Fig. 2B). In the presence of AM251, 2-AG failed to increase the phosphorylation of AMPK and ACC, but no significant inhibition was observed with AM630 treatment (Fig. 2B). Next, we explored the cardiac expression pattern of cannabinoid receptors in a physiological model of IR, viz. leptin receptor-deficient db/db mice. Interestingly, both Cb1r and Cb2r gene expression is significantly lowered in diabetic, insulin-resistant db/db mice in comparison with their heterozygous, non-diabetic (db/+) counterpart (Fig. 2C). This suggests that, in the context of cardiac tissue, the ECS is dysfunctional in type 2 diabetes. As an additional control for the receptor specificity, the well known 2-AG-induced gene expression of Cb1r was significantly inhibited in the presence of AM251 but not by AM630. 2-AG had no significant effect on Cb2r gene expression (Fig. 2D). Next, to understand the downstream mechanism involved in 2-AG-induced AMPK activation, we pretreated LKB1-deficient HeLa cells with pharmacological inhibitors of known upstream AMPK kinases—KN93 (KN, Ca2+/calmodulin-dependent protein kinase II, 10 μm), STO-609 (STO, a CaMKKβ inhibitor, 20 μm), or OZ/(5Z)-7-oxozeanol (OZ, TGFβ-activated kinase 1, 5 μm)—followed by 2-AG treatment. 2-AG-induced phosphorylation of AMPK was completely abrogated in the presence of STO but not KN or OZ (Fig. 2E). Similar results were obtained in primary rat cardiomyocytes, where pretreatment with STO or an AMPK inhibitor (compound C (CC), 10 μm) suppressed 2-AG-induced phosphorylation of AMPK, whereas KN or OZ did not show any significant effect (Fig. 2F). Additionally, 2-AG significantly induced glucose uptake in adult rat cardiomyocytes, whereas both STO and CC pretreatment markedly reduced this effect (Fig. 2G). Overall, these results indicate that 2-AG-induced AMPK activation and increased glucose uptake require the CB1 receptor and CaMKKβ in cardiomyocytes.
Figure 2.

2-AG activates the AMPK signaling pathway in cardiomyocytes. A, phospho (p-) and total (t-) ACC and AMPK protein levels upon 2AG treatment in HL-1 and primary cardiomyocytes. veh, vehicle. B, phospho and total ACC and AMPK protein levels upon 2-AG treatment for 1 h, preceded by treatment with AM251 or AM630 for 1 h in HL-1 cardiomyocytes. C, relative mRNA levels of Cb1r and Cb2r in cardiac tissues obtained from db/+ (n = 4) and db/db (n = 6) mice. *, p < 0.01 by unpaired Student's t test. D, relative mRNA levels of Cb1r and Cb2r in HL-1 cardiomyocytes upon 2-AG treatment for 1 h, preceded by treatment with AM251 or AM630 for 1 h in HL-1 cardiomyocytes. *, p < 0.01 by one-way ANOVA with Tukey's multiple comparisons post hoc test. Ctl, control. E, phospho and total AMPK protein levels upon 2-AG treatment for 1 h, preceded by treatment with KN, STO, or OZ for 1 h in LKB1-deficient HeLa cells. F, phospho and total AMPK protein levels upon 2-AG treatment for 1 h, preceded by treatment with CC, KN, STO, or OZ for 1 h in primary rat cardiomyocytes. G, glucose uptake in primary rat cardiomyocytes upon 2-AG treatment for 1 h, preceded by treatment with CC, KN, STO, or OZ for 1 h. ACTB, β-actin. *, p < 0.01 by one-way ANOVA with Tukey's multiple comparisons post hoc test. Data are expressed as mean ± S.D. n = 4–5 independent experiments.
2-AG inhibits TNFα-induced IR
Next, to determine the effect of 2-AG on reversing IR, cardiomyocytes were challenged with TNFα in the presence or absence of 2-AG, followed by insulin stimulation (Fig. 3A). 2-AG co-treatment markedly reversed the inhibitory effect of TNFα on the insulin signaling pathway (i.e. it restored AKT, AS160, and GSK3β phosphorylation upon insulin stimulation) and increased both AMPK and ACC phosphorylation. Additionally, TNFα-induced activation of NF-κB (as represented by the phosphorylation of the p65 subunit of NF-κB) and JNK/stress-activated protein kinase signaling pathway was significantly inhibited upon co-treatment with 2-AG (supplemental Fig. 2A). Furthermore, the inhibitory effect of TNFα on plasmalemmal GLUT4-myc translocation (Fig. 3B) and insulin-stimulated glucose uptake (Fig. 3C) was restored upon 2-AG co-treatment. Interestingly, AEA also partially restored insulin signaling and increased glucose uptake along with AMPK activation in TNFα-challenged cardiomyocytes (supplemental Fig. 2, B and C), indicating that both endocannabinoids ameliorate inflammatory stress-induced IR. However, in comparison with AEA, 2-AG exhibited stronger beneficial effects.
Figure 3.

2-AG reverses inflammatory stress-induced IR. A, representative Western-blotting analysis of the insulin (Ins) signaling pathway following insulin stimulation. p, phospho; t, total. B, insulin-stimulated GLUT4-myc translocation. *, p < 0.01; #, p = 0.043; two-way ANOVA with Bonferroni's multiple comparisons post hoc test. Ctl, control. C, insulin-stimulated glucose uptake. *, p < 0.01 by two-way ANOVA with Bonferroni's multiple comparisons post hoc test. ACTB, β-actin. D, relative mRNA levels of the proinflammatory markers Glut4 and Ppargc1a. *, p < 0.01 by one-way ANOVA with Tukey's multiple comparisons post hoc test. E, glucose oxidase assay for quantitative determination of intracellular glycogen content in cardiomyocytes subjected to 1-h pretreatment with STO or CC, followed by 16 h of TNFα and/or 2-AG treatment as indicated. *, p < 0.01 by one-way ANOVA with Tukey's multiple comparisons post hoc test. Data are expressed as mean ± S.D. n = 4–5 independent experiments in primary rat cardiomyocytes.
Next, to determine the molecular mechanism involved in 2-AG-mediated reversal of IR, we pharmacologically inhibited the CaMKKβ and AMPK signaling pathways in primary cardiomyocytes. In the presence of either STO or CC, 2-AG failed to increase AMPK or ACC phosphorylation and failed to restore the TNFα-induced reduction in AKT, AS160, or GSK3β phosphorylation (supplemental Fig. 2D). Similarly, in the presence of STO or CC, 2-AG failed to enhance glucose uptake or restore insulin-stimulated glucose uptake in TNFα-challenged cardiomyocytes (supplemental Fig. 2E).
Furthermore, we determined the anti-inflammatory effect of 2-AG and the involvement of AMPK in the context of TNFα-induced IR. Gene expression analysis demonstrated that TNFα significantly induced the mRNA levels of the proinflammatory markers Nfkb1, Il6, Cxcl8, and Ccl2 (Fig. 3D). Conversely, TNFα exposure led to a decrease in gene expression of both Glut4 and Ppargc1a, the latter being a master cardiac transcriptional coactivator (12). 2-AG co-treatment decreased inflammatory stress, as evidenced by a decrease in the mRNA levels of Nfkb1, Il6, Cxcl8, and Ccl2, and increased cardiomyocyte energy efficiency by up-regulating Glut4 and Ppargc1a mRNA levels. However, pretreatment with either STO or CC blocked the beneficial effects of 2-AG in cardiomyocytes. Similarly, the 2-AG-mediated increase in intracellular glycogen content, either in the presence or absence of TNFα, was dramatically reduced in the presence of the inhibitors (Fig. 3E). Overall, these results strongly indicate that 2-AG, via CaMKKβ, activates AMPK, exerts anti-inflammatory effects, reverses IR, and restores insulin-stimulated glucose uptake in TNFα-challenged cardiomyocytes.
2-AG-induced inhibition of IR is attenuated upon knockdown of CaMKKβ and AMPK
Next, we applied lentiviral knockdown of the AMPKβ2 subunit (shAMPKβ2), the predominant β isoform of the AMPK αβγ heterotrimer in the heart. Western-blotting analyses comparing the effects of shAMPKβ2 with scrambled shRNA lentivirus (shScr) on AMPKβ2 and total AMPKα (t-AMPKα) protein levels demonstrate the efficiency of AMPKβ2 knockdown in cardiomyocytes (Fig. 4A). In shScr lentivirus-infected cells, TNFα-induced inhibition of insulin-stimulated AKT, AS160, and GSK3β phosphorylation was restored upon 2-AG co-treatment, but this beneficial effect on insulin signaling was absent in shAMPKβ2-infected cells (Fig. 4A). Both increased AMPK and ACC phosphorylation, normally elicited by 2-AG treatment, was abrogated upon AMPKβ2 knockdown. Furthermore, in shScr cells, 2-AG inhibited the TNFα-induced increases in the mRNA levels of the proinflammatory marker genes Nfkb1, Il6, and Ccl2, but this inhibitory effect of 2-AG was significantly ablated in the presence of shAMPKβ2 (Fig. 4B). Similarly, TNFα-mediated inhibition of Glut4 and Ppargc1a mRNA levels was reversed upon co-treatment with 2-AG, whereas shAMPKβ2-infected cells failed to respond to 2-AG stimulation. Furthermore, in line with the mRNA and protein levels, 2-AG failed to reverse the TNFα-induced inhibition of insulin-stimulated GLUT4-myc translocation (Fig. 4C) and glucose uptake (Fig. 4D) in shAMPKβ2-infected cardiomyocytes. These results were further validated upon lentiviral knockdown of CaMKKβ in HL-1 cardiomyocytes (supplemental Fig. 3). As expected, knockdown of CaMKKβ disrupts the 2-AG signaling cascade. This was evidenced by the failure of 2-AG to restore insulin signaling (via AKT phosphorylation) or to activate AMPK (supplemental Fig. 3A) in shCaMKKβ-infected cardiomyocytes. Additionally, 2-AG failed to reverse the TNFα-induced inhibition of insulin-stimulated GLUT4-myc translocation (supplemental Fig. 3B) and glucose uptake (supplemental Fig. 3C) in shCaMKKβ-infected cardiomyocytes. Taken together, these results demonstrate that AMPK is the key mediator of 2-AG-induced attenuation of TNFα-induced inflammatory stress and IR in cardiomyocytes.
Figure 4.

Knockdown of AMPK attenuates the ameliorative effect of 2-AG on IR. HL-1 cardiomyocytes were infected with shRNA scramble (shScr) or AMPKβ2 (shAMPKβ2) and selected for puromycin resistance, followed by treatments as indicated. A, representative Western-blotting analysis of the insulin (Ins) signaling pathway following insulin stimulation. p, phospho; t, total. B, relative mRNA levels of the proinflammatory markers Glut4 and Ppargc1a. *, p = 0.03, 0.05, 0.041, 0.022, and 0.026, respectively, by one-way ANOVA with Tukey's multiple comparisons post hoc test. C, insulin-stimulated GLUT4-myc translocation. *, p = 0.044; #, p < 0.01; two-way ANOVA with Bonferroni's multiple comparisons post hoc test. D, insulin-stimulated glucose uptake. ACTB, β-actin. *, p = 0.047; #, p < 0.01; two-way ANOVA with Bonferroni's multiple comparisons post hoc test. Data are expressed as mean ± S.D. n = 5 independent experiments.
2-AG inhibits IR in hESC-CMs
To address the role of 2-AG in in vitro human pathological models of IR, we challenged hESC-CMs with either TNFα or FFA (the latter representing an overarching, well established cell model of diet-induced IR in vivo). Differentiation of hESC to functional cardiomyocytes was confirmed by gene expression analysis showing a robust induction of the key cardiac structural and functional markers α-myosin heavy chain (MYH6) and cardiac troponin T type 2 (TNNT2), respectively (Fig. 5A). Both TNFα (Fig. 5B) and FFA (supplemental Fig. 4A) increased gene transcription of the proinflammatory markers NFKB1, IL6, and CXCL8 and inhibited PPARGC1A gene expression. Conversely, co-treatment of 2-AG led to a marked decrease in the mRNA levels of all proinflammatory markers along with an increase in PPARGC1A gene expression. Pretreatment with the inhibitors AM251, STO, or CC, representing key checkpoints in the 2-AG-AMPK signaling pathway, abolished the anti-inflammatory effect of 2-AG on TNFα- or FFA-induced inflammation. Similarly, insulin-stimulated phosphorylation of AKT, AS160, and GSK3β was strongly inhibited by both TNFα (Fig. 5C) and FFA (supplemental Fig. 4B), and 2-AG co-treatment restored the phosphorylation levels of these key proteins involved in the insulin signaling pathway along with induction of AMPK and ACC phosphorylation. Finally, both TNFα- and FFA-mediated inhibition of insulin stimulated-glucose uptake were significantly reversed in the presence of 2-AG, and, as expected, pretreatment with inhibitors completely blocked the ameliorative effect of 2-AG in IR (Fig. 5D and supplemental Fig. 4C). The results obtained in hESC-CMs suggest that 2-AG, via CB1R and CaMKKβ, activates the AMPK pathway to counteract FFA- and TNFα-induced inflammatory stress and IR and subsequently restores metabolic homeostasis of cardiomyocytes by potentiating insulin-stimulated glucose uptake (Fig. 6). Overall, these results clearly indicate that 2-AG ameliorates FFA- and TNFα-induced IR.
Figure 5.

The ameliorative effect of 2-AG on inflammatory stress-induced insulin resistance in hESC-derived cardiomyocytes. A, relative mRNA level of MYH6 and TNNT2. *, p < 0.01 by unpaired Student's t test. B, relative mRNA level of proinflammatory markers and PGC1α. *, p = 0.037, 0.041, 0.044, and 0.041, respectively, by one-way ANOVA with Tukey's multiple comparisons post hoc test. Ctl, control. C, representative Western-blotting analysis of the insulin (Ins) signaling pathway following insulin stimulation. p, phospho; t, total; A, AM251; S, STO-609; C, compound C. D, insulin-stimulated glucose uptake. ACTB, β-actin. *, p = 0.03 and 0.044, respectively, by one-way ANOVA with Tukey's multiple comparisons post hoc test. Data are expressed as mean ± S.D. n = 5 independent experiments.
Figure 6.
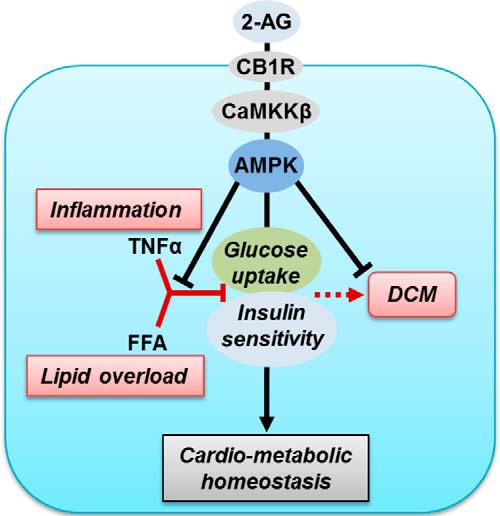
Proposed model of 2-AG action in stressed cardiomyocytes. Inflammation (induced by TNFα) and lipid oversupply (FFA) perturbs insulin sensitivity and restricts glucose uptake in cardiomyocytes in the long term, leading to DCM. The endocannabinoid 2-AG, via CB1R and CaMKKβ, activates the AMPK signaling pathway to inhibit inflammation, restore insulin sensitivity, and facilitate glucose uptake in cardiomyocytes, implying that 2-AG treatment can be a viable therapeutic approach to restore cardiometabolic homeostasis via energy balance in IR.
Discussion
Evidence points to inflammation-induced cytokine production and/or high plasma fatty acid levels as primary etiologic factors in the development of cardiac IR. Here we demonstrated that inflammation induces IR and perturbs glucose metabolism in cardiomyocytes. Similarly, exposure to FFA produced IR in cardiomyocytes, in accordance with earlier findings (2). We show that, irrespective of the cause, IR can be alleviated by 2-AG treatment. Mechanistically, 2-AG, via the CB1 receptor and CaMKKβ, activates AMPK. Treatment with 2-AG decreased the gene expression of proinflammatory cytokines, reversed IR, and enhanced glucose uptake, thus restoring metabolic homeostasis of cardiomyocytes. Pharmacological inhibition or genetic interference with the AMPK signaling pathway impaired these 2-AG actions, suggesting that AMPK is mediating the beneficial effects. These findings identify 2-AG as a key signaling molecule against IR and inflammation, prominent derangements observed during DCM (3), pointing to a possible therapeutic application.
Previous reports have demonstrated the detailed mechanism of TNFα-mediated inhibition of insulin signaling to induce IR in various metabolically relevant tissues (4, 5). Here we have delineated the effect of TNFα on insulin signaling and the functional consequences on cardiomyocytes, a topic that is remarkably understudied. Our data demonstrated that TNFα inhibits the insulin-stimulated phosphorylation of its downstream effectors AKT, AS160, and GSK3β to promote IR. The cardiac system normally responds to injury by altering substrate metabolism from the use of fatty acids toward glucose uptake and utilization. IR prevents this adaptive response and aggravates the injury by contributing to lipotoxicity and inflammation, among other stressors. These effects are in line with our observation of reduced insulin-stimulated GLUT4 translocation, glucose uptake, and intracellular glycogen content in TNFα-challenged cardiomyocytes, constituting a novel model to study metabolic derangements observed during cardiac inflammation.
A previous report (9) has indicated that the plant-derived cannabinoid trans-Δ9-tetrahydrocannabinol can activate AMPK, an important integrator of signals managing energy balance and acting as a protective response to energy stress during metabolic deregulations (13, 14). Being highly expressed in the cardiac system, AMPK has been demonstrated to have cardioprotective effects (15). We observed that the endocannabinoid 2-AG activates the AMPK signaling pathway in various cell types in a CB1R-CaMKKβ-dependent manner. This observation is in line with previous reports suggesting that 2-AG induces a rapid, transient increase in intracellular Ca2+ via CB1R stimulation (16); on the other hand, increased intracellular Ca2+ leads to AMPK activation (17). Additionally, we observed that 2-AG, via AMPK activation, enhances basal glucose uptake in cardiomyocytes, in accordance with the contraction-stimulated rise in AMPK activity and consequent increase of substrate uptake (15). However, conflicting results regarding the ECS under pathological conditions has emerged from recent studies, with experimental disease models demonstrating that pharmacological approaches that modulate the same specific targets of the ECS can have both positive and negative effects (18, 19). In this context, it should be noted that a substantial amount of 2-AG has been detected in various tissues, including the liver, kidney, spleen, several regions of the brain, lung, and plasma, as well as in human milk (20, 21). Furthermore, stimulus-induced generation of 2-AG in various tissues adds to the complexity in ascertaining the systemic effects of 2-AG. Therefore, the biosynthetic pathways for 2-AG appear to differ depending on the types of tissues and cells and the types of stimuli. Thus, on one hand, an elevated level of 2-AG upon chronic alcohol challenge has been implicated in the progression of alcoholic steatohepatitis (19), 2-AG-induced activation of JNK signaling pathway has been shown to aberrantly trigger hepatic gluconeogenesis (11), and rodent models with CB1R deficiency or antagonism have been demonstrated to be resistant to diet-induced obesity and insulin resistance, specifically in liver (22–24). On the other hand, our results as well as several previous studies have strongly indicated potential beneficial effects of 2-AG in the context of the cardiovascular system, neurotransmission, and immunomodulation (7, 8, 18, 20, 24). Nevertheless, the precise physiological function of 2-AG in acute and chronic inflammation and/or immune responses remains relatively obscure. Several questions still remain to be answered. What does the serum level of 2-AG indicate? How does it change in the context of metabolic disease, such as type 2 diabetes? In this context, our results demonstrating diminished cardiac Cb1r and Cb2r gene expression in db/db mice indicate a potential correlation between a dysfunctional ECS and cardiac IR. However, further studies are necessary for a better understanding of the metabolism and mode of action of 2-AG in a cell type- and tissue-specific manner to decipher the role of this highly complex signal transduction system in distinct pathological conditions.
Both in vitro and in vivo, the ECS has been shown to exhibit immunomodulatory properties (25). In our inflammation-induced IR model, 2-AG reverses IR by inhibiting the gene expression of proinflammatory cytokines, restoring the insulin signaling pathway, and enhancing glucose uptake. Furthermore, 2-AG treatment also led to an increase in Glut4 and Ppargc1a mRNA levels under these conditions. These findings are important in the context of DCM, as overexpression of human GLUT4 in db/db mice was demonstrated to normalize cardiac contractile dysfunction (26). Several reports indicate that PGC1α, in response to physiologic stressors, initiates biological responses that equip cardiomyocytes to meet energy demands by augmenting mitochondrial biogenesis, cellular respiration rates, and substrate utilization (12). Interestingly, both Ca2+ and AMPK have been positively implicated in the regulation of PGC1α. Indeed, we observed that pharmacological inhibition or genetic interference of CaMKKβ and AMPK resulted in attenuation of 2-AG-mediated induction of Ppargc1a as well as Glut4. Hence, we speculate that they are key contributors in mediating the beneficial effects of 2-AG in TNFα-induced IR and, possibly, for treatment of DCM.
Because the majority of ECS-related studies have been performed in mouse models, the relevance of 2-AG in humans, in the context of inflammation and IR, has remained largely elusive. In this study, we addressed this issue by utilizing hESC-CMs as a model to mimic adult human cardiomyocytes. The responsiveness of hESC-CMs to FFA- and TNFα-induced IR further indicates the possible value of this model as a novel tool for metabolic characterization of various human pathological conditions. Furthermore, in line with our previous results in rodent cardiomyocytes, the ameliorative effect of 2-AG on inflammatory stress, insulin signaling, and substrate utilization was observed to occur via the CB1R-CaMKKβ-AMPK signaling cascade, indicating that this novel signaling cascade is conserved in humans.
Overall, our study unravels an unprecedented role of 2-AG in the regulation of inflammation-induced IR. Endocannabinoids are crucial to bioregulation, and, because of their hydrophobic nature, their main actions are limited to autocrine or paracrine effects rather than systemic. With scientific evidence suggesting their conflictive roles in inflammation, IR, and energy metabolism, e.g. in the heart as opposed to the liver, tissue-specific application of endocannabinoids may be a valuable tool to counter cardiac IR. Further investigation of this exciting field will provide insights into the mechanisms of health and disease and provide novel therapeutic avenues.
Experimental procedures
Materials and methods
Noladin ether (2-AG ether, referred to as 2-AG), AM251, AM630, and (5Z)-7-oxozeaenol were purchased from Tocris Bioscience. Recombinant human TNFα was purchased from R&D Systems. BSA (fraction V, essentially fatty acid-free), DMSO, FFA (palmitic acid), STO-609, and KN93 were purchased from Sigma-Aldrich. CC was purchased from Calbiochem. [3H]2-deoxyglucose was obtained from GE Healthcare. LipofectamineTM 2000 was from Invitrogen. Ortho-phenylenediamine H2O2 fizzing tablets were from AbD Serotec (Kidlington, UK). The plasmid encoding GLUT4-myc was kindly provided by Dr. J. Eckel (German Diabetes Center, Düsseldorf, Germany). HL-1 cardiomyocytes (a gift from Dr. W. Claycomb), isolated primary rat cardiomyocytes, and hESC-CMs were incubated for 16 h with TNFα (10 ng/ml) or FFA (250 μm), in the presence or absence of 2-AG (10 μm) as indicated. For experiments with inhibitors, cardiomyocytes were pretreated with the indicated inhibitors for 1–2 h, followed by TNFα treatment in the absence or presence of 2-AG. For insulin stimulation experiments, cardiomyocytes were stimulated with insulin (100 nm, 30 min) following the indicated treatments.
Cell culture and transient transfection of HL-1 atrial cardiomyocytes
HL-1 cells were kindly provided by Dr. W. Claycomb (Louisiana State University, New Orleans, LA) and cultured in Claycomb medium (supplemented with 10% FBS, 0.1 mm norepinephrine, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin) at 37 °C and 5% CO2. Cells were transfected as described previously (27, 28).
Isolation and culturing of primary rat cardiomyocytes
Cardiac myocytes were isolated from male Lewis rats (200–250 g) using a Langendorff perfusion system and a Krebs-Henseleit bicarbonate medium equilibrated with a 95% O2/5% CO2 gas phase at 37 °C as described previously (27, 28).
Animals
Male heterozygous non-diabetic (db/+) and diabetic (db/db) C57BL/Ks mice (10 weeks of age) were purchased from Charles River Laboratories. The animals were housed in the specific pathogen-free animal facility of Eindhoven University of Technology in individually ventilated cages and in the conventional facility of Muenster University under controlled temperature (23 °C) and humidity (50%) with a 12:12-h dark-light cycle. The mice had ad libitum access to water and food (5K52 LabDiet, 22 kcal% protein, 16 kcal% fat, 62 kcal% carbohydrates). The animals were sacrificed by cervical dislocation under isoflurane anesthesia, and the heart was excised and frozen in liquid nitrogen for quantitative PCR analysis. All procedures conformed to Directive 2010/63/EU of the European Parliament and were approved by the Animal Experimental Committees of Maastricht University (The Netherlands).
Antibodies and Western-blotting analysis
Antibodies against the MYC epitope, AKT (Ser(P)-473 and total), ACC (Ser(P)-79 and total), AMPKα (Thr(P)-172 and total), total AMPKβ2, α-tubulin, GSK-3β (Ser(P)-9 and total), AS-160 (Thr(P)-642 and total), JNK/stress-activated protein kinase (Thr(P)-183/Tyr(P)-185 and total), NF-κB p65 (Ser(P)-536 and total), Caveolin 3, and β-actin were from Cell Signaling Technology. Cells were lysed in radioimmune precipitation assay buffer, and protein concentrations were determined by BCA (Thermo Scientific). Equal amounts of protein were separated by SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween, incubated with primary antibodies overnight, and washed prior to incubation with HRP-conjugated secondary antibodies. Proteins were detected by enhanced chemiluminescence (PerkinElmer Life Sciences).
RNA expression analysis
For quantitative PCR analysis, RNA was isolated using TRIzol (Amresco), and 1 μg was used to synthesize cDNA (high-capacity cDNA reverse transcription kit, Life Technologies). Amplification was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) and the ABI 7900HT fast real-time PCR system (Life Technologies). The genes Cyclophilin A (for HL-1 and primary rat cardiomyocytes) and TBP (for hESC-CMs) were used for normalization, and the average of the control cell values was used for calculating relative expression using the ΔΔCT method.
Glucose uptake
Following the indicated treatments, the medium was replaced with Krebs-Ringer bicarbonate buffer with 0.5% BSA, and cells were treated with PBS and 100 nm insulin for 30 min and then incubated with [3H]2-deoxyglucose (1:20 in cold 10 mm 2-deoxyglucose) for 10 min. Cells were lysed in 1% Triton X-100, and radioactivity was determined by β scintillation counting. Values were normalized to protein content (BCA).
Biochemical intracellular glycogen measurement
Extraction of intracellular glycogen was performed as described previously (29). Final hydrolysates were used for glucose determination using a glucose (GO) assay kit (Sigma) according to the instructions of the manufacturer and expressed as microgram of glucose per milligram of protein.
GLUT4-myc translocation assay (GLUT4-myc cell surface staining)
Plasmalemmal GLUT4 was detected using a GLUT4 variant carrying a myc tag on its first extracellular epitope, as described previously (30).
Lentiviral infection
pLKO.1 puro was a gift from Bob Weinberg (Addgene plasmid 8453). HEK-293T cells were co-transfected with psPAX2 and pMD2.G lentivirus packaging vectors with pLKO.1 scramble (shScr), pLKO.1 AMPKβ2 (shAMPKβ2), or pLKO.1 CaMKKβ (shCaMKKβ) using LipofectamineTM 2000 according to the protocol of the manufacturer. After a 48-h infection, the lentivirus particles were collected from the HEK-293T cells and transfected into HL-1 cells. Infected cells were selected for puromycin resistance (4 μg/ml) for 5 days, and Western-blotting analysis was performed to determine knockdown efficiency.
Cardiomyocyte differentiation from hES cells
All experiments were performed using the H7 (NIHhESC-10−0061) hESC lines, kindly provided by the WiCell Research Institute (Madison, WI). hESCs were maintained and differentiated according to the protocol of the manufacturer (Gibco) and as described previously (31). Briefly, undifferentiated hESCs were detached from the culture plate with 0.5 mm EDTA (UltraPure 0.5 m EDTA (pH 8.0), Invitrogen) and seeded onto Geltrex-coated plates (Geltrex LDEV-free hESC-qualified reduced growth factor basement membrane matrix, Gibco). Cells were refed daily with Essential 8 medium (Essential 8 basal medium containing 15 mm HEPES, l-glutamine, and sodium bicarbonate at 1.743 g/liter, supplemented with Essential 8 supplement (Gibco)) to expand the culture. When the cells reached a 30–50% confluent state (± 4 days of culture), cardiac differentiation was induced by replacement of Essential 8 medium with cardiomyocyte differentiation medium (PSC cardiomyocyte differentiation kit (Gibco) containing differentiation medium A, differentiation medium B, and maintenance medium). Cells were kept in differentiation medium A for 2 days, which was replaced with differentiation medium B for the following 2 days, after which cells were further maintained on maintenance medium. Cultures were refed every other day and monitored daily for signs of contractile capacity. After 30 days of in vitro differentiation, cells were ready for experimental purposes. All treatments were performed as indicated for HL-1 and primary rat cardiomyocytes.
Statistical analysis
Student's unpaired t tests or ANOVA were performed in GraphPad Prism v.6.01. Differences were considered statistically significant at p < 0.05.
Author contributions
D. C. and D. N. conceived and coordinated the study and wrote the paper. D. C., Y. O., I. G., Y. L., and J. J. F. P. L. designed, performed, and analyzed the experiments shown in Figs. 1–3. D. C. and X. Z. designed, performed, and analyzed the experiments shown in Fig. 4. D. C., I. G., and M. N. designed, performed, and analyzed the experiments shown in Fig. 5. W. C., J. J. F. P. L., and J. F. C. G. provided technical assistance and contributed to the preparation of the figures. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Florence Tienen and Dr. Arthur van den Wijngaard (Maastricht University Medical Center) for expertise and help with hESC-CM experiments.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. 1–4.
- IR
- insulin resistance
- DCM
- diabetic cardiomyopathy
- ECS
- endocannabinoid system
- 2-AG
- 2-arachidonoyl glycerol
- AEA
- anandamide
- CaMKKβ
- Ca2+/calmodulin-dependent protein kinase β
- AMPK
- AMP-activated protein kinase
- KN
- KN93
- STO
- STO-609
- OZ
- OZ/(5Z)-7-oxozeanol
- CC
- compound C
- hESC-CM
- human embryonic stem cell-derived cardiomyocyte
- ACC
- acetyl-CoA carboxylase
- ANOVA
- analysis of variance.
References
- 1. Jia G., DeMarco V. G., and Sowers J. R. (2016) Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 12, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dirkx E., Schwenk R. W., Glatz J. F., Luiken J. J., and van Eys G. J. (2011) High fat diet induced diabetic cardiomyopathy. Prostaglandins Leukot. Essent. Fatty Acids 85, 219–225 [DOI] [PubMed] [Google Scholar]
- 3. Palomer X., Salvadó L., Barroso E., and Vázquez-Carrera M. (2013) An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 168, 3160–3172 [DOI] [PubMed] [Google Scholar]
- 4. Hotamisligil G. S., Shargill N. S., and Spiegelman B. M. (1993) Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259, 87–91 [DOI] [PubMed] [Google Scholar]
- 5. Shoelson S. E., Lee J., and Goldfine A. B. (2006) Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szmitko P. E., and Verma S. (2008) The endocannabinoid system and cardiometabolic risk. Atherosclerosis 199, 248–256 [DOI] [PubMed] [Google Scholar]
- 7. Hiley C. R. (2009) Endocannabinoids and the heart. J. Cardiovasc. Pharmacol. 53, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montecucco F., and Di Marzo V. (2012) At the heart of the matter: the endocannabinoid system in cardiovascular function and dysfunction. Trends Pharmacol. Sci. 33, 331–340 [DOI] [PubMed] [Google Scholar]
- 9. Kola B., Hubina E., Tucci S. A., Kirkham T. C., Garcia E. A., Mitchell S. E., Williams L. M., Hawley S. A., Hardie D. G., Grossman A. B., and Korbonits M. (2005) Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J. Biol. Chem. 280, 25196–25201 [DOI] [PubMed] [Google Scholar]
- 10. Mukhopadhyay B., Liu J., Osei-Hyiaman D., Godlewski G., Mukhopadhyay P., Wang L., Jeong W. I., Gao B., Duester G., Mackie K., Kojima S., and Kunos G. (2010) Transcriptional regulation of cannabinoid receptor-1 expression in the liver by retinoic acid acting via retinoic acid receptor-γ. J. Biol. Chem. 285, 19002–19011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chanda D., Kim D. K., Li T., Kim Y. H., Koo S. H., Lee C. H., Chiang J. Y., and Choi H. S. (2011) Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J. Biol. Chem. 286, 27971–27979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Finck B. N., and Kelly D. P. (2006) PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 116, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arad M., Seidman C. E., and Seidman J. G. (2007) AMP-activated protein kinase in the heart: role during health and disease. Circ. Res. 100, 474–488 [DOI] [PubMed] [Google Scholar]
- 14. O'Neill L. A., and Hardie D. G. (2013) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355 [DOI] [PubMed] [Google Scholar]
- 15. Zaha V. G., and Young L. H. (2012) AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 111, 800–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sugiura T., Kodaka T., Nakane S., Miyashita T., Kondo S., Suhara Y., Takayama H., Waku K., Seki C., Baba N., and Ishima Y. (1999) Evidence that the cannabinoid CB1 receptor is a 2-arachidonoylglycerol receptor: structure-activity relationship of 2-arachidonoylglycerol, ether-linked analogues, and related compounds. J. Biol. Chem. 274, 2794–2801 [DOI] [PubMed] [Google Scholar]
- 17. Woods A., Dickerson K., Heath R., Hong S. P., Momcilovic M., Johnstone S. R., Carlson M., and Carling D. (2005) Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 [DOI] [PubMed] [Google Scholar]
- 18. Di Marzo V., and Petrosino S. (2007) Endocannabinoids and the regulation of their levels in health and disease. Curr. Opin. Lipidol. 18, 129–140 [DOI] [PubMed] [Google Scholar]
- 19. Jeong W. I., Osei-Hyiaman D., Park O., Liu J., Bátkai S., Mukhopadhyay P., Horiguchi N., Harvey-White J., Marsicano G., Lutz B., Gao B., and Kunos G. (2008), Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 7, 227–235 [DOI] [PubMed] [Google Scholar]
- 20. Sugiura T., Kobayashi Y., Oka S., and Waku K. (2002) Biosynthesis and degradation of anandamide and 2-arachidonoylglycerol and their possible physiological significance. Prostaglandins Leukot. Essent. Fatty Acids 66, 173–192 [DOI] [PubMed] [Google Scholar]
- 21. Di Marzo V., Sepe N., De Petrocellis L., Berger A., Crozier G., Fride E., and Mechoulam R. (1998) Trick or treat from food endocannabinoids? Nature 396, 636–637 [DOI] [PubMed] [Google Scholar]
- 22. Nogueiras R., Diaz-Arteaga A., Lockie S. H., Velásquez D. A., Tschop J., López M., Cadwell C. C., Diéguez C., and Tschöp M. H. (2009) The endocannabinoid system: role in glucose and energy metabolism. Pharmacol. Res. 60, 93–98 [DOI] [PubMed] [Google Scholar]
- 23. Irwin N., Hunter K., Frizzell N., and Flatt P. R. (2008) Antidiabetic effects of sub-chronic administration of the cannabinoid receptor (CB1) antagonist, AM251, in obese diabetic (ob/ob) mice. Eur. J. Pharmacol. 581, 226–233 [DOI] [PubMed] [Google Scholar]
- 24. Maccarrone M., Bab I., Bíró T., Cabral G. A., Dey S. K., Di Marzo V., Konje J. C., Kunos G., Mechoulam R., Pacher P., Sharkey K. A., and Zimmer A. (2015) Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 36, 277–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nagarkatti P., Pandey R., Rieder S. A., Hegde V. L., and Nagarkatti M. (2009) Cannabinoids as novel anti-inflammatory drugs. Future Med. Chem. 1, 1333–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Semeniuk L. M., Kryski A. J., and Severson D. L. (2002) Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am. J. Physiol. Heart Circ. Physiol. 283, H976-H982 [DOI] [PubMed] [Google Scholar]
- 27. Luiken J. J., van Nieuwenhoven F. A., America G., van der Vusse G. J., and Glatz J. F. (1997) Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: involvement of sarcolemmal proteins. J. Lipid Res. 38, 745–758 [PubMed] [Google Scholar]
- 28. Dirkx E., van Eys G. J., Schwenk R. W., Steinbusch L. K., Hoebers N., Coumans W. A., Peters T., Janssen B. J., Brans B., Vogg A. T., Neumann D., Glatz J. F., and Luiken J. J. (2014) Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J. Mol. Cell Cardiol. 76, 208–217 [DOI] [PubMed] [Google Scholar]
- 29. Oligschlaeger Y., Miglianico M., Chanda D., Scholz R., Thali R. F., Tuerk R., Stapleton D. I., Gooley P. R., and Neumann D. (2015) The recruitment of AMP-activated protein kinase to glycogen is regulated by autophosphorylation. J. Biol. Chem. 290, 11715–11728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwenk R. W., Dirkx E., Coumans W. A., Bonen A., Klip A., Glatz J. F., and Luiken J. J. (2010) Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia 53, 2209–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lundy S. D., Zhu W. Z., Regnier M., and Laflamme M. A. (2013) Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 22, 1991–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.