

Abstract
The protein optic atrophy 1 (OPA1) is a dynamin-related protein associated with the inner mitochondrial membrane and functions in mitochondrial inner membrane fusion and cristae maintenance. Inner membrane-anchored long OPA1 (L-OPA1) undergoes proteolytic cleavage resulting in short OPA1 (S-OPA1). It is often thought that S-OPA1 is a functionally insignificant proteolytic product of L-OPA1 because the accumulation of S-OPA1 due to L-OPA1 cleavage is observed in mitochondrial fragmentation and dysfunction. However, cells contain a mixture of both L- and S-OPA1 in normal conditions, suggesting the functional significance of maintaining both OPA1 forms, but the differential roles of L- and S-OPA1 in mitochondrial fusion and energetics are ill-defined. Here, we examined mitochondrial fusion and energetic activities in cells possessing L-OPA1 alone, S-OPA1 alone, or both L- and S-OPA1. Using a mitochondrial fusion assay, we established that L-OPA1 confers fusion competence, whereas S-OPA1 does not. Remarkably, we found that S-OPA1 alone without L-OPA1 can maintain oxidative phosphorylation function as judged by growth in oxidative phosphorylation-requiring media, respiration measurements, and levels of the respiratory complexes. Most strikingly, S-OPA1 alone maintained normal mitochondrial cristae structure, which has been commonly assumed to be the function of OPA1 oligomers containing both L- and S-OPA1. Furthermore, we found that the GTPase activity of OPA1 is critical for maintaining cristae tightness and thus energetic competency. Our results demonstrate that, contrary to conventional notion, S-OPA1 is fully competent for maintaining mitochondrial energetics and cristae structure.
Keywords: bioenergetics, mitochondria, mitochondrial disease, mitochondrial metabolism, mitochondrial respiratory chain complex, OPA1, cristae, mitochondrial fusion
Introduction
Changes in mitochondrial shape, number, and location in cells are now well appreciated collectively as mitochondrial dynamics. Among many types of mitochondrial shape changes, fission and fusion are the most characterized processes. The main molecular factors controlling mitochondrial fission and fusion are dynamin-related membrane remodeling proteins. Dynamin-like/-related protein 1 (DLP1/Drp1)2 is a cytosolic protein that becomes bound to the mitochondrial surface and constricts mitochondria for fission (1, 2). Two isoforms of mitofusin (Mfn1 and Mfn2) are anchored at the outer membrane and mediate tethering and fusion of outer membranes of apposing mitochondria through homotypic and heterotypic interactions between the two isoforms (3, 4). Optic atrophy 1 (OPA1) mediates fusion of the inner membrane following Mfn-mediated outer membrane fusion (5–7).
In a steady state, a balance between fission and fusion maintains mitochondrial size and number. However, metabolic changes, stimuli, or stress from inside and outside of the cells shifts the fission-fusion balance to induce temporary or permanent changes in mitochondrial morphology (8–13). Not surprisingly, growing evidence indicates that mitochondrial fission and fusion are intimately associated with energetic maintenance of mitochondria. However, molecular components regulating this connection are poorly understood. OPA1, one of the mitochondrial dynamics proteins, has been implicated in mitochondrial energetic maintenance. Mitochondria of OPA1-deficient cells are fragmented and show greatly diminished cristae, which is functionally reflected as a defect in oxidative phosphorylation (OXPHOS) (14–16). As such, although mutations in OPA1 in human are best known to cause optic atrophy, they are associated with extraocular systemic manifestations such as deafness, myopathy, ataxia, peripheral neuropathy, and more rarely spastic paraplegia and multiple sclerosis-like illness known as autosomal dominant optic atrophy plus (17–20). These observations suggest that OPA1 is a critical factor connecting mitochondrial morphology and energetics.
Inside mitochondria, OPA1 exists in both inner membrane-anchored and soluble forms facing the intermembrane space. Human OPA1 mRNA undergoes alternative splicing, generating eight different splice variants (21, 22). Upon expression, OPA1 is imported to the mitochondria by the N-terminal mitochondrial targeting sequence (MTS). The transmembrane (TM) domain downstream of MTS acts as a stop transfer signal, anchoring the OPA1 protein at the inner membrane, leaving most of the protein in the intermembrane space (23). The membrane-anchored OPA1 (“L-OPA1” for long OPA1) is further cleaved by proteases downstream of the TM region, generating TM-free OPA1 (“S-OPA1” for short OPA1). At least two cleavage sites, each cleaved by OMA1 and Yme1L, have been identified (6, 24–27), and additional sites are likely present (28). Although the fusion activity of L- and S-OPA1 has been extensively investigated (6, 24, 25), their roles in energetic maintenance are poorly understood.
It has been shown that the presence of both L- and S-OPA1 is necessary for mitochondrial fusion, whereas L- or S-OPA1 alone is insufficient for fusion in normal conditions (6). However, other studies showed that L-OPA1 alone can elongate mitochondria, which was more prominent under cellular stress, indicating that L-OPA1 is fusion-competent (24, 29, 30). Upon mitochondrial dysfunction associated with depolarization, apoptosis, or permeability transition, L-OPA1 is cleaved by OMA1 to form S-OPA1, which is suggested to prevent fusion of dysfunctional mitochondria (26, 27, 31, 32). Regarding the role of OPA1 in OXPHOS, knockdown or knock-out of OPA1 disrupts cristae structure and respiration and increases apoptotic sensitivity (15, 16, 33, 34), whereas OPA1 overexpression enhances cristae tightness, apoptotic resistance, respiratory supercomplex formation, and OXPHOS (35–38).
Currently available information concerning L- and S-OPA1-specific roles in mitochondrial energetics is from studies with the manipulations of OMA1, Yme1L, or prohibitin 2 (Phb2) that perturb OPA1 processing. OMA1/Yme1L double knock-out cells that have only L-OPA1 are fusion-competent and maintain normal cristae structure, indicating that L-OPA1 is sufficient for mitochondrial fusion, cristae maintenance, and conferring apoptotic resistance (29). In addition, deletion of Phb2 induces OPA1 cleavage, resulting in mitochondrial fragmentation, cristae disruption, and increased apoptotic sensitivity, and reintroduction of L-OPA1, not S-OPA1, to Phb2-null cells restored these defects, suggesting that S-OPA1 is incompetent for mitochondrial fusion and cristae maintenance (39). However, Phb2 has broad functions in other cellular processes (40–43), and OPA1 is not the sole substrate of OMA1 and Yme1L (44–50). Thus, it is possible that the mitochondrial phenotypes observed by knocking out Phb2, OMA1, or Yme1L may include effects unrelated to OPA1 function. Furthermore, contrary to the notion that OPA1 cleavage is causal for mitochondrial fragmentation and OXPHOS defect, an increase in OXPHOS enhances OPA1 cleavage and induces mitochondrial fusion (51). Therefore, the role of OPA1 cleavage in mitochondrial fusion is controversial, and specific roles of L- and S-OPA1 in energetic maintenance are ill-defined.
In the present study, we used cells exclusively expressing L-OPA1, S-OPA1, or both to evaluate the differential functions of L- and S-OPA1. Our data elaborate and confirm that L-OPA1 has an intrinsic activity for mitochondrial fusion, whereas the fusion activity of S-OPA1 is insignificant. On the other hand, we found that either S- or L-OPA1 alone is sufficient to support mitochondrial energetic activity. Most surprisingly, cells having S-OPA1 alone maintain normal cristae structure. These experimental data show that S-OPA1 has full competency for mitochondrial energetic maintenance despite lacking fusion activity. We propose that S-OPA1 may expedite cristae reorganization, providing energetic flexibility.
Results
Cell lines expressing L- and S-OPA1
OPA1 is expressed in eight different splice variants (v1–v8) (22). These splice variants undergo partial or full proteolytic cleavage at S1 and S2 (Fig. 1), presenting the mixture of L- and S-OPA1 in cells (6, 24–27). As a representative OPA1 that produces both L- and S-OPA1, we used variant 1 (OPA1-v1). When expressed in cells, OPA1-v1 is cleaved incompletely at S1, generating cleaved S-OPA1 along with uncleaved L-OPA1. Overexpression of the OPA1-v1 variant in cells and mice has been shown to increase mitochondrial length, cristae tightness, supercomplex assembly, and OXPHOS (36–38). OPA1-v1 carrying a deletion of the S1 cleavage site (OPA1-v1ΔS1) was used as non-cleavable L-OPA1. Variant 5 (OPA1-v5) was used as S-OPA1. OPA1-v5 contains exon 4b that allows complete cleavage at the S1 site, generating only S-OPA1 (6). These constructs were expressed in OPA1 knock-out (KO) mouse embryonic fibroblasts (MEFs), and stable clones were isolated. As shown in Fig. 1, immunoblots of cell lysates from these cells show the presence of L/S-OPA1, L-OPA1, and S-OPA1 in MEFs expressing OPA1-v1, OPA1-v1ΔS1, and OPA1-v5, respectively.
Figure 1.
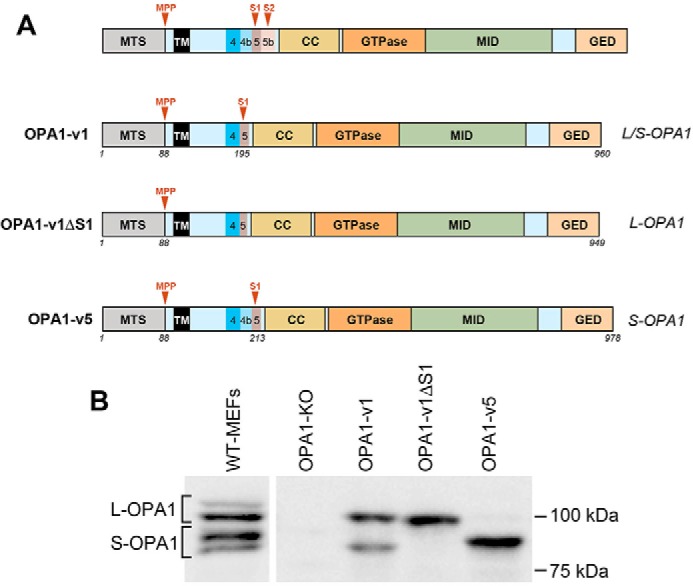
OPA1 variants and MEFs differentially expressing these variants. A, OPA1 proteolytic cleavages occur at S1 and S2 in exons 5 and 5b. CC, coiled-coil domain; MID, middle domain; GED, GTPase effector domain; MPP, mitochondrial processing peptidase. The variant 1 (OPA1-v1) contains the S1 site where partial cleavage occurs, producing both L- and S-OPA1 (L/S-OPA1). OPA1-v1ΔS1 only produces L-OPA1 due to the deletion of the S1 site in OPA1-v1. OPA1-v5 generates S-OPA1 only because of full cleavage at the S1 site. Amino acid numbers are indicated for each construct. B, three OPA1 variants were expressed in OPA1-KO MEFs, and stable clones were isolated. Immunoblots of cell lysates from these cells show exclusive expression of L-OPA1 and S-OPA1 in MEFs expressing OPA1-v1ΔS1 and OPA1-v5, respectively, and the presence of both L- and S-OPA1 in cells expressing OPA1-v1.
L-OPA1 has the capacity to elongate mitochondria in pro-fusion conditions
We observed that when cells were grown in normal culture conditions, mitochondria in cells expressing OPA1-v1 (both L- and S-OPA1) were mostly short, whereas long and intermediate mitochondria were observed in ∼30% of these cells (Fig. 2A). In contrast, mitochondria in cells expressing OPA1-v1ΔS1 (L-OPA1) or OPA1-v5 (S-OPA1) are fragmented, suggesting that the presence of both L- and S-OPA1 is necessary for mitochondrial fusion (Fig. 2A) as reported previously (6). However, unlike completely fragmented mitochondria in OPA1-KO cells, mitochondria in some of these cells (∼5%) form short tubules, suggesting that these OPA1 variants may be fusion-competent. Because mitochondrial morphology is determined by fission-fusion balance, we inhibited fission by Drp1 silencing and examined the mitochondrial elongation capacity in these cells. Upon Drp1 siRNA treatment, we found large increases in long and intermediate tubules in cells expressing OPA1-v1 or OPA1-v1ΔS1 but a minimal increase with OPA1-v5 expression (Fig. 2B). We observed similar results in conditions evoking stress-induced mitochondrial hyperfusion (SIMH) (Fig. 2, C and D). SIMH occurs in cellular stresses such as nutrient depletion and treatments with UV light, actinomycin D, and cycloheximide (30, 52, 53). Under nutrient starvation or cycloheximide treatment, similar to Drp1 siRNA-treated cells, more elongated mitochondria were observed in OPA1-v1 cells and OPA1-v1ΔS1 cells, whereas OPA1-v5 cells exhibited only a small increase in cells containing tubules (Fig. 2, C and D). Of note, mitochondrial elongation by OPA1-v1ΔS1 in SIMH conditions was shown previously (30). These observations indicate that L-OPA1 alone is fusion-competent without S-OPA1 or proteolytic cleavage. SIMH has been shown to occur through suppression of mitochondrial fission (52, 53). It is likely that the fusion activity of L-OPA1 is low, thus unable to overcome ongoing fission, displaying short fragmented mitochondria in normal conditions.
Figure 2.
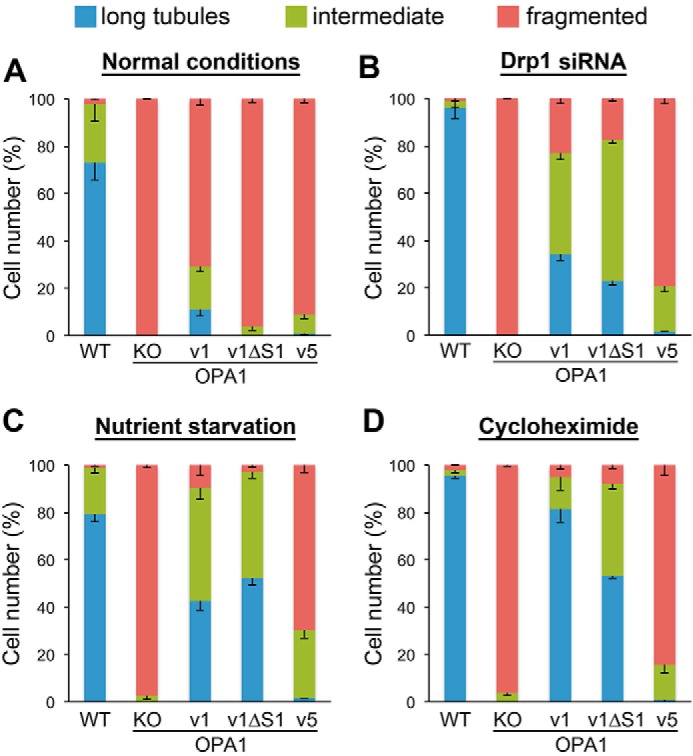
L-OPA1 alone has the capacity to elongate mitochondria in pro-fusion conditions. Mitochondrial tubule formation was assessed in normal conditions (A) and conditions of fission inhibition (B) and SIMH (C and D) in cells expressing different OPA1 variants. Mitochondrial lengths were categorized into long tubules, intermediate, and fragmented. In both fission inhibition and SIMH conditions, OPA1-v1 and v1ΔS1 significantly increased the formation of tubular mitochondria. Mitochondrial tubule formation by OPA1-v5 was minimal. n = 4. Error bars are S.E.
To further elaborate fusion activity of L- and S-OPA1, we examined mitochondrial fusion in hybrid cells formed by polyethylene glycol (PEG) treatment. Unlike the morphological evaluation described above, the PEG assay tests bona fide mitochondrial fusion by assessing mixing of matrices regardless of mitochondrial elongation/size. It has been shown that inner membrane fusion requires the presence of OPA1 only in one of the fusion partners (7). Therefore, we analyzed mitochondrial fusion activity between OPA1-KO cells expressing matrix-targeted DsRed and OPA1 variant cells expressing matrix-targeted GFP. The presence of cycloheximide to prevent expression of DsRed and GFP during the assay inherently also renders SIMH conditions. Fig. 3A shows examples of mitochondrial images from the fusion assay. We tested mitochondrial fusion with 4 and 8 h of fusion reaction. We observed that ∼60% of OPA1-v1 or OPA1-v1ΔS1 cells showed mitochondrial fusion by 4 h after PEG treatment, which was similar to wild-type (WT) cells (Fig. 3B). By 8 h, most cells showed mitochondrial fusion in these cells. Consistent with the mitochondrial morphology analyses in pro-fusion conditions, cells expressing OPA1-v5 showed no mitochondrial fusion after 4 h but only a small increase after 8-h incubation (Fig. 3). These results indicate that L-OPA1 without undergoing proteolytic cleavage is intrinsically fusion-competent, whereas the fusion activity of S-OPA1 is minimal.
Figure 3.

Assessments of fusion competency by PEG cell hybrid assays. Mixed cultures of OPA1-KO cells with DsRed-labeled mitochondria and OPA1 variant cells with GFP-labeled mitochondria were treated with PEG, and mixing of red and green fluorescence was evaluated in hybrid cells. A, examples of mitochondrial images from the fusion assay representing no fusion, partial fusion, and complete fusion. Scale bar, 20 μm. B, OPA1-v1 and OPA1-v1ΔS1 are fusion-competent, whereas OPA1-v5 has very little fusion capacity. n = 3. Error bars are S.E.
L- or S-OPA1 alone is sufficient to support mitochondrial respiratory function
Lack of OPA1 function has been shown to cause loss of mitochondrial DNA (mtDNA) and OXPHOS activity and disruption of cristae structure (14–16, 33, 54, 55). To test the contributions of L- and S-OPA1 to OXPHOS activity, we examined cell growth in galactose media in which cells are forced to use OXPHOS for energy production (56). All cell lines tested including OPA1-KO cells grew well in the glycolytic media containing glucose at a similar rate of ∼18 h of doubling time (Fig. 4A). In OXPHOS culture conditions containing galactose, OPA1-KO cells were unable to grow, confirming lack of OXPHOS activity. However, we found that growth rates of OPA1-v1ΔS1 and OPA1-v5 cells were indistinguishable from those of OPA1-v1 and WT cells (Fig. 4A). Doubling time in OXPHOS conditions was ∼24 h, indicating slower growth compared with glycolytic conditions. These results demonstrate that either L- or S-OPA1 alone is sufficient to restore the OXPHOS function lost by OPA1 deficiency. Consistent with cell growth in OXPHOS conditions, we found that all three variants showed oxygen consumption rates similar to that of WT cells, whereas OPA1-KO cells were respiration-impaired (Fig. 4, B and C). The three variants and WT cells have no distinguishable differences in basal, leak, and maximum respirations (Fig. 4B) and respiratory control index (Fig. 4C).
Figure 4.

L- or S-OPA1 alone is sufficient to support mitochondrial respiratory function. A, OXPHOS assessment by cell growth in galactose media. All cell lines including OPA1-KO cells grew well in the glycolytic media. In galactose media, OPA1-KO did not grow, whereas WT and OPA1 variant cells grew with no discernible difference in their growth rates. Experiments were done in quadruplicate and repeated three times. Representative data are presented. B and C, oxygen consumption rates (OCR) of OPA1 variant cells show no difference from that of WT, whereas OPA1-KO cells are respiration-deficient. The respiration control ratios of uncoupled maximum respiration (state 3u) to leak respiration under oligomycin (state 4o) were indistinguishable in OPA1 variants and WT. n = 6. Error bars are S.E. ***, p < 0.0001; #, p = 0.0002; **, p = 0.0089 (one-way ANOVA with Tukey's post hoc test). D and E, immunoblotting of WT and OPA1 variant cells in galactose and SIMH conditions. Cells were incubated in 10 mm glucose (Glc) or galactose (Gal) for 24 h (D). For SIMH, cells were incubated in HBSS for 3 h (Strv) or in 10 μm cycloheximide (Chx) for 6 h (E). There are no changes in L- and S-OPA1 in galactose and HBSS incubations. Cycloheximide incubation appears to increase L-OPA1 cleavage in WT and OPA1-v1 cells. Olm, oligomycin; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; Con, control.
It is remarkable that S-OPA1 alone without L-OPA1 is sufficient to support OXPHOS. It is possible that OXPHOS conditions (galactose media) may generate L-OPA1 in OPA1-v5 cells to provide OXPHOS activity. However, we found that growth in galactose media does not produce L-OPA1 in OPA1-v5 cells (Fig. 4D). Furthermore, galactose had no effect on the L- and S-OPA1 levels in other variant and wild-type cells (Fig. 4D). Additional tests of L- and S-OPA1 patterns in SIMH conditions showed no change in OPA1-v1ΔS1 and -v5 cells, whereas cycloheximide appeared to increase the cleavage of L-OPA1 in wild-type and OPA1-v1 cells possibly due to the induction of apoptosis (Fig. 4E).
L- and S-OPA1 are indistinguishable in maintaining mitochondrial respiratory complexes
To examine the mechanisms behind the cell growth and respiration restorations by L- or S-OPA1, we examined respiratory complexes and supercomplexes using blue native gel electrophoresis (BNGE) followed by immunoblotting with complex-specific antibodies. OPA1-KO cells showed lower levels of respiratory complexes with the exception of complex I (Fig. 5). Complex I was detected in the form of complexes I + III2 in BNGE that was not affected by the loss of OPA1 (Fig. 5). In complex III blots, despite the decreased complex III level, the pattern of supercomplex assembly in OPA1-KO cells was similar to that of WT, exhibiting residual III2 + IV, I + III2, and I + III2 + IV. This observation suggests that the OPA1 function is not required for the formation of supercomplexes. OPA1-KO cells did not show complex IV, but a low level of complex IV was detected as supercomplex (I + III2 + IV). In addition, OPA1-KO cells showed an impairment in complex V assembly, displaying the accumulation of F1 domain, a soluble functional subcomplex situated in the mitochondrial matrix, consistent with a previous report (54) (Fig. 5A).
Figure 5.

L- and S-OPA1 are indistinguishable in restoring mitochondrial respiratory complexes. BNGE was used to examine respiratory complexes and supercomplexes. A, OPA1-KO cells have significantly decreased levels of respiratory complexes except complex I. Note that supercomplex assembly was not affected by OPA1-KO. OPA1-KO cells accumulate F1 domain of complex V (shown in the long exposure). OPA1-v1, -v1ΔS1, and -v5 restored the levels of complexes III, IV, and V with a minimal recovery of complex II. There is no distinction between OPA1-v1ΔS1 and OPA1-v5 in restoring respiratory complexes. Numbers in parentheses indicate the size of the complexes in kDa. B, quantification of total band density for individual complexes in each cell line. n = 5. Error bars are S.E. (one-way ANOVA with Tukey's post hoc test). IB, immunoblotting.
We found that expression of OPA1-v1, -v1ΔS1, or -v5 in OPA1-KO cells restores respiratory complexes. Importantly, all three variants restored respiratory complex levels indistinguishably (Fig. 5), supporting the cell growth and respiration data. The recoveries of the levels of complexes III, IV, and V were seemingly significant, whereas complex II showed minimal recovery (Fig. 5A). Although the extent of recoveries of individual complexes appears different and statistically varying (Fig. 5B), no distinction between L- and S-OPA1 was found in the abilities of restoring respiratory complexes and supercomplexes.
We detected a difference in fusion activity of L- and S-OPA1 in nutrient depletion (Figs. 2 and 3). Therefore, we tested whether applying SIMH-inducing conditions would reveal differences in respiratory complexes in OPA1 variant cells. We found that starvation increased complex I levels in all samples including OPA1-KO MEFs, but no significant changes were found in other complexes (Fig. 6A). The reason for the complex I increase in nutrient starvation is currently unclear (see “Discussion”). Importantly, however, the lack of distinction between OPA1 variants in restoring respiratory complexes was still maintained in starvation (Fig. 6A). These results suggest that OPA1 fusion activity is not linked to the activity controlling energetics as reported before (54).
Figure 6.

No functional distinction between L- and S-OPA1 in maintaining respiratory complexes and mtDNA. A, respiratory complexes after starvation assessed by BNGE. Starvation does not significantly affect the complex levels except for complex I. Increased complex I levels were seen after starvation across the cell lines. * in the complex IV blot indicates a potential dimer. B, expression of OPA1 variants in OPA1-KO cells restores mtDNA levels. mtDNA and nuclear DNA were quantified using PCR with primer sets for ATP6 and GAPDH, respectively. OPA1-KO cells contain ∼30% of mtDNA copy number compared with WT cells. All three OPA1 variant cells showed ∼60% of the WT level, indicating partial recovery of mtDNA copy numbers. n = 4. Error bars are S.E. (Student's t test (two-tailed)).
The restoration of OXPHOS and respiratory complexes by L- and/or S-OPA1 expression was also reflected in recovery of mtDNA (Fig. 6B). OPA1-KO cells have reduced mtDNA copy number (54, 55). We found that OPA1-KO cells contain ∼30% of mtDNA copy number compared with WT cells. All three OPA1 variant cells showed increased mtDNA copy numbers, which were ∼60% of the WT level (Fig. 6B). Altogether our observations support the notion that L- or S-OPA1 alone has the intrinsic ability to maintain respiratory complexes and thus OXPHOS activity.
S-OPA1 is able to maintain cristae structure
Disrupted cristae are the hallmark of OPA1 deficiency for energetic incompetence. Therefore, we examined mitochondrial and cristae structures in OPA1 variant cells using electron microscopy (EM). EM images of WT MEFs show tubular mitochondria with ordered cristae (Fig. 7). In contrast, OPA1-KO MEFs have fragmented mitochondria that contain no or very few cristae (Fig. 7). We also noticed greatly decreased matrix electron density in OPA1-KO cells, reflecting the loss of energetic activity (Figs. 7 and 8B). In addition, mitochondria in OPA1-KO cells occasionally have septa that may have resulted from outer membrane fusion without subsequent inner membrane fusion (Fig. 8C, arrowheads) consistent with a previous report (7).
Figure 7.

EM images of mitochondria in WT, OPA1-KO, and OPA1 variant cells. No or a few cristae are in OPA1-KO mitochondria. OPA1 variant cells restored cristae structure, showing increased numbers of cristae and tight lamellae similar to those in WT cells. Scale bar, 1 μm.
Figure 8.

Analyses of cristae structure in OPA1 variant cells. A, quantification of cristae density. The number of cristae was counted for each mitochondrion and divided by the mitochondrial area in megapixels. Six to 16 mitochondria were analyzed in each cell for 11 cells per cell line. n = 111, 82, 83, 91, and 79 for WT, OPA1-KO, -v1, -v1ΔS1, and -v5, respectively. Error bars are S.E. (one-way ANOVA with Tukey's post hoc test). B, matrix electron density is presented by matrix gray value (GV[mat]) after background subtraction (GV[cyt]) in individual mitochondria. Distribution of matrix electron density was plotted with the average gray value (horizontal line) for each cell line along with low magnification EM images. n = 139, 130, 135, 150, and 139 for WT, OPA1-KO, -v1, -v1ΔS1, and -v5, respectively. #, p < 0.0001 (one-way ANOVA with Tukey's post hoc test). Scale bar, 0.5 μm. C, septa (arrowheads) in mitochondria of OPA1-KO cells. Scale bars, 0.5 μm. D, STJ (arrows) in OPA1-KO and OPA1-v5 cells. Scale bars, 0.5 μm. E, quantification of CCJ and STJ. The numbers of CCJ and STJ were separately counted and divided by the mitochondrial area in megapixels. 11 cells were analyzed for each cell line. n = 101, 79, 68, 83, and 69 for WT, OPA1-KO, -v1, -v1ΔS1, and -v5, respectively. Error bars are S.E. (one-way ANOVA with Tukey's post hoc test). F, quantification of cristae width for assessing cristae tightness. Distribution of cristae width in nm was plotted with the median width (horizontal line) for each cell line. n = 146, 171, 177, 125, and 159 for WT, OPA1-KO, -v1, -v1ΔS1, and -v5, respectively. #, p < 0.001 (one-way ANOVA with Tukey's post hoc test).
We found that cells expressing different OPA1 variants contained significantly increased cristae numbers in their mitochondria (Figs. 7 and 8A). Even the cells containing only S-OPA1 (OPA1-v5) showed normal cristae structure (Fig. 7). This result is surprising because it has been shown that S-OPA1 is not able to maintain normal cristae (39). Quantitative analyses revealed that cristae densities of OPA1-v1ΔS1 and -v5 cells were significantly higher than that of OPA1-KO cells, whereas they were lower than that of OPA1-v1 cells (Fig. 8A). Cristae density of OPA1-v1 cells is similar to that of WT cells, indicating maximal recovery by the presence of both L- and S-OPA1 (Fig. 8A). Cristae densities of OPA1-v1ΔS1 and -v5 cells were similar to each other. These data suggest that either L- or S-OPA1 alone is sufficient for cristae formation. Based on full recovery of OXPHOS function by L- or S-OPA1 (Fig. 4) despite partially reduced cristae densities, these EM observations indicate that submaximal cristae formation by L- or S-OPA1 alone is sufficient to support the OXPHOS activity. Supporting this notion, the normal recovery of matrix electron density was observed in cells expressing OPA1-v1, -v1ΔS1, and -v5 (Fig. 8B).
Although disputed, OPA1 was suggested to regulate cristae junction (CJ) (35, 57, 58). Therefore, we analyzed cristae junction density by quantifying the number of cristae with CJ (CCJ). In addition, short tubules attached to the inner boundary membrane were found in OPA1-KO cells (Fig. 8D, arrows). Because these structures have not been described before and may not be true cristae, we analyzed these short tubules (<100 nm in length) with junctions (STJ) separately from CCJ (>100 nm). Similar to a recent report (58), we observed the presence of CJ in OPA1-KO cells, but CJ density was significantly decreased. CCJ in OPA1-KO cells were reduced by ∼50% compared with CCJ in wild-type cells (Fig. 8E). Interestingly, OPA1-v5 appeared to have only a small effect on restoring CCJ, whereas OPA1-v1ΔS1 increased CCJ to the wild-type level (Fig. 8E). Because overall cristae density is similar in OPA1-v5 and OPA1-v1ΔS1 cells (Fig. 8A), the decreased CCJ density in OPA1-v5 cells suggests that S-OPA1 is less effective in maintaining CJ. The nature of STJ is currently unknown. Possibly STJ are nascent cristae that become mature cristae. Assuming that, the abundance of STJ in OPA1-KO cells may suggest that the initial formation of cristae occurs independently of OPA1, whereas cristae maturation requires OPA1 function.
It has been suggested that both L- and S-OPA1 are required for maintaining cristae tightness by functioning as molecular staples (35, 59). Therefore, we analyzed cristae width in OPA1 variant cells. We found that cristae widths of OPA1-v1ΔS1 and OPA1-v5 cells were similar to those in WT and OPA1-v1 cells (Fig. 8F), demonstrating that L- or S-OPA1 alone can maintain cristae tightness. In contrast, OPA1-KO cells have much wider cristae, showing ∼26-nm average width in comparison with 13–15 nm in WT and OPA1 variant cells. Overall, these EM analyses indicate that, contrary to conventional notion, S-OPA1 has the capacity in the absence of L-OPA1 to maintain the number and tightness of cristae, which is sufficient for supporting normal OXPHOS activity.
Requirement of GTPase activity in OPA1-mediated cristae maintenance
S-OPA1 support of cristae structure and energetic activity, despite the lack of mitochondrial fusion activity, indicates that OPA1 activities controlling mitochondrial fusion and energetics are independent of each other. Because mitochondrial fusion by OPA1 requires its GTPase activity, we asked whether the GTPase activity is necessary for S-OPA1 to support mitochondrial energetics. We generated the lysine-to-alanine mutations in the GTPase domain of OPA1 variants (OPA1-v1-K301A, OPA1-v1ΔS1-K291A, and OPA1-v5-K319A). This specific Lys-to-Ala mutation has been shown to cause defects in GTPase activity and mitochondrial fusion (5, 14, 29, 60). We expressed the OPA1 variants carrying the Lys-to-Ala mutations in OPA1-KO MEFs and first tested their fusion capacity by SIMH. We found that, unlike OPA1-v1 and -v1ΔS1 cells showing SIMH (Fig. 2), mitochondria in cells expressing the mutant OPA1 variants remained fragmented after incubation in cycloheximide (Fig. 9A), confirming the lack of fusion activity.
Figure 9.

OPA1 GTPase activity is required for mitochondrial fusion and OXPHOS. A, mitochondrial tubule formation was assessed by SIMH using cycloheximide in cells expressing Lys-to-Ala mutant OPA1 variants. In both normal and SIMH conditions, mitochondria in mutant OPA1-expressing cells remained fragmented. n = 4. Error bars are S.E. B, OXPHOS assessment of Lys-to-Ala mutant OPA1 variant cells by cell growth in galactose media. Although all cells grew well in the glycolytic media, Lys-to-Ala mutant-expressing cells grew for 3 days and abruptly died on day 4. Experiments were done in quadruplicate and repeated three times. Representative data are presented.
Next, we evaluated these mutant OPA1 variant cells for their growth in galactose media. We found that all OPA1 variant Lys-to-Ala mutant cells were able to grow initially, whereas OPA1-KO cells were not (Fig. 9B), similar to the non-mutant OPA1 variant cells, raising the possibility that the GTPase activity may not be necessary for the OXPHOS maintenance function of OPA1. Interestingly, however, cells expressing the Lys-to-Ala mutant versions of OPA1-v1, -v1ΔS1, and -v5 died abruptly at day 4 in galactose media (Fig. 9B). Although the reason is unclear, the sudden cell death after initial growth in galactose media suggests that the mutant proteins may have residual amounts of GTPase activities and thus OXPHOS function by which mutant cells are able to grow until stress overwhelms them. Nevertheless, because non-mutant OPA1 variant cells can grow in galactose, whereas mutant cells die, these results indicate that the OXPHOS function of OPA1 still requires the GTPase activity.
Finally we examined cristae structure of these OPA1 mutant variant cells by EM. Remarkably, although the presence of cristae structure is obvious in all three Lys-to-Ala mutant OPA1 variant cells, we found that, unlike non-mutant variants that restore normal cristae morphology (Fig. 7), cristae in mutant cells mostly appeared swollen and round, indicating the loss of cristae tightness (Fig. 10, A–C). Lys-to-Ala mutations in all three variants had the same effect on cristae structure. Quantification of cristae width shows mean widths of 46, 42, and 44 nm in OPA1-v1-K301A, -v1ΔS1-K291A, and -v5-K319A, respectively, widths even larger than that in OPA1-KO cells (Figs. 10D and 8F). These results indicate that OPA1 GTPase activity is required for maintaining cristae tightness. The presence of numerous cristae in mutant cells indicates that cristae formation is unaffected by the mutation. Although ballooned and varying morphologies of cristae made the quantification less reliable, the average cristae density appeared similar in three mutant variant cells (Fig. 10E). The restoration of cristae number by GTPase-defective OPA1 versus the lack of cristae by OPA1-KO suggests that the presence of OPA1 molecules, regardless of their GTPase activities, is sufficient for membrane expansion necessary for cristae formation, whereas the GTPase activity of OPA1 is critical for maintaining cristae tightness. Interestingly, matrix electron density appeared restored (Fig. 10, A–C), supporting the observed initial growth of the mutant cells in OXPHOS conditions. The fact that these mutant cells eventually succumb to death in strictly OXPHOS conditions indicates that maintaining cristae tightness is critical for sustaining energetic competency. Because complex V plays a role in regulating cristae curvature, we examined the state of complex V by BNGE. We found that the mutant OPA1 variants are unable to complete complex V assembly, a phenotype identical to that in OPA1-KO cells (Fig. 10F). Although it is unclear that the lack of complex V assembly is the cause or consequence of cristae ballooning, it is possible that OPA1 is involved in complex V biogenesis through its GTPase activity. Our data indicate that the GTPase activity of OPA1 is necessary for energetic competency through proper maintenance of cristae tightness.
Figure 10.

OPA1 GTPase activity is critical for maintaining cristae tightness. A–C, EM images of OPA1 GTPase-defective mutant cells. Cristae are swollen and round. Scale bar, 0.5 μm. D, quantification of cristae width. Distribution of cristae width in nm was plotted with the mean width (horizontal line) for each cell line. n = 103, 94, and 124 for OPA1-v1-K301A, -v1ΔS1-K291A, and -v5-K319A, respectively. E, quantification of cristae density. The number of cristae was counted for each mitochondrion and divided by the mitochondrial area in μm2. Three to 10 mitochondria were analyzed in each cell for 10 cells per cell line. n = 44, 54, and 56 for OPA1-v1-K301A, -v1ΔS1-K291A, and -v5-K319A, respectively. Error bars are S.E. F, immunoblotting of BNGE for complex V. Lys-to-Ala mutant OPA1 variant cells show disrupted complex V assembly, accumulating F1 domain of complex V, identical to OPA1-KO cells. Numbers in parentheses indicate the size in kDa.
Discussion
In this study, we analyzed cells differentially expressing L- and S-OPA1 to elaborate the roles of L- and S-OPA1 in mitochondrial fusion and energetic maintenance. OPA1 deficiency results in mitochondrial fragmentation and causes an OXPHOS defect and the loss of mtDNA, suggesting that OPA1-mediated mitochondrial fusion plays a role in maintaining energetic activity. In contrast, in the mechanistic aspect, it has been shown that the OPA1 function in fusion is independent of cristae maintenance and thus energetics (35, 54). Our finding that S-OPA1 without L-OPA1 has a sufficient capacity for maintaining mitochondrial energetic function despite lacking fusion activity is consistent with separate OPA1 mechanisms for fusion and cristae maintenance.
The information available for differential roles of L- and S-OPA1 in mitochondrial energetics was obtained via manipulating OMA1 and/or Yme1L, the mitochondrial proteases that generate S-OPA1 species, as well as Phb2 whose deficiency causes the loss of L-OPA1 by increased cleavage (29, 39). These studies concluded that L-OPA1 is competent for cristae maintenance, whereas S-OPA1 is not. In contrast, our functional and structural analyses of mitochondrial energetics demonstrated that S-OPA1 is fully competent for OXPHOS and maintenance of cristae structure. This discrepancy could be attributed to the potential pleiotropic effects from knocking out the proteins regulating OPA1 processing. For example, Phb2 complexed with Phb1 functions as a mitochondrial chaperone that stabilizes mitochondrially encoded translation products such as complex IV subunits for proper assembly of the complex (61, 62). In addition, OMA1 degrades misfolded complex IV subunits and a factor necessary for complex III assembly, and its loss impedes supercomplex stability (45–47). Yme1L degrades Tim17A, a subunit of Tim23, and regulates levels of respiratory complex subunits (49, 50). Therefore, it is predicted that altering Phb2, OMA1, or Yme1L directly will affect respiratory complexes and thus OXPHOS activity in addition to OPA1 processing. Although our approach circumvents manipulation of OPA1 processing, the limitation is that the expression level of S-OPA1 exceeds that of wild-type cells, which potentially contributes to the discrepancy. On the other hand, the aforementioned studies used AIF-230 as an S-OPA1 alternative. AIF-230 is the fusion protein between AIF and OPA1 containing MTS and TM of AIF (aa 1–95) and aa 230–997 of OPA1-v7 (24). S1 and S2 sites in OPA1-v7 are at aa 195 and aa 217–223, respectively (24), and the apoptotic cleavage site of AIF is at aa 101 (63). Therefore, AIF-230 does not contain the known cleavage sites of OPA1 and AIF except for the mitochondrial processing peptidase site to remove MTS and thus is predicted to be a constitutive TM protein. Because cellular S-OPA1 does not contain a TM domain, it is possible that the surrogate S-OPA1, AIF-230, exerts different functions from the TM-free soluble S-OPA1.
Our data demonstrate that S-OPA1, when expressed in OPA1-KO cells, restores the OXPHOS function to the normal level. Although we observed full functional restoration by S- or L-OPA1 as judged by growth in galactose media and respiration, we found, to some extent, incomplete restorations in structural components such as cristae, respiratory complexes, and mtDNA when compared with WT cells. These observations indicate that the maintenance of cellular energetics uses only parts of the structural components of OXPHOS. Indeed, it has been shown that respiratory complex activities can be considerably inhibited without affecting the rate of mitochondrial respiration or ATP synthesis, namely the mitochondrial threshold effect (64–66).
Our analysis shows that the absence of OPA1 function causes significant loss of respiratory complexes except for complex I. In contrast, a previous report of pancreatic β cell-specific OPA1-KO indicated that the assemblies of all respiratory complexes were normal, although subunit levels of complex IV were decreased (67). This discrepancy could be due to the presence of other cell types containing intact OPA1 in isolated islets, cell type differences, and different methods of respiratory complex analyses. Our results are consistent with another report showing defective assembly of complex V in OPA1-KO cells (54). Regardless, OPA1-KO causing an overall decrease of respiratory complexes (except complex I) appears to be congruous with a significant decrease of mtDNA. However, our results show that OPA1-KO cells have the normal level of complex I that contains seven mtDNA-encoded subunits. Moreover, OPA1-KO cells also showed a decrease of complex II, which is not encoded by mtDNA. This suggests that the decreases of respiratory complexes by OPA1-KO are more related to the absence of cristae structure and less related to mtDNA loss. Without cristae membranes, the assembly platform, respiratory complexes likely become unstable and degraded. In this regard, it has been noted that the loss of mtDNA by OPA1-KO is the process controlled by the fusion function of OPA1, which is suggested to be independent of the OPA1 function for cristae maintenance (54). However, our data indicate that fusion-deficient S-OPA1 still supports recovery of mtDNA and OXPHOS. Further studies will be necessary to understand the mechanisms by which OPA1 controls complex assembly and cristae structure.
It is unclear how the complex I level is maintained even with the decreased cristae in OPA1-KO cells. It has been suggested that complex I acts as a scaffold for respirasome assembly (a supercomplex of complexes I + III + IV) by incorporating subunits of complexes III and IV (68). Hence, it is possible that the maintenance of complex I is a prerequisite for proper OXPHOS function. Complex I may be able to assemble onto specific regions of the inner membrane or the nascent cristae in the absence of mature cristae structure to support respiratory complex assembly upon cristae biogenesis and maturation. The importance of complex I maintenance even in the absence of mature cristae can be further considered in the context of our observation that nutrient starvation increases the level of complex I. Nutrient depletion has been shown to increase complex V dimerization and cristae density to maintain ATP levels (52). It is possible that the complex I increase facilitates respiratory complex assembly necessary for efficient energetic maintenance under limited substrate supply during starvation. As for the mechanism of the starvation-induced complex I increase, considering that cells depend on β-oxidation of lipid droplets in nutrient starvation (69), we speculate that cells respond to the energy demand by an increase of complex I to utilize NADH generated by β-oxidation.
A proposed model of OPA1 maintenance of cristae tightness is that S-OPA1 binds to cristae membrane-anchored L-OPA1, functioning as a molecular staple to keep the cristae tight (35, 59). However, our data demonstrate that cells containing S-OPA1 alone maintain cristae density and tightness. It is currently unclear how S-OPA1 maintains cristae structure without the membrane-anchoring TM domain. A previous report showed that deletion of Phb2 results in the loss of L-OPA1 and aberrant cristae structures (39). It was found that expression of non-cleavable L-OPA1 partially restored normal cristae structures in Phb2-null cells, whereas no restoration was observed with S-OPA1 expression in which the aforementioned AIF-230 was used as S-OPA1 (39). Nonetheless, these observations suggest that Phb2 inhibits OPA1 cleavage, and thus L-OPA1 maintains cristae. In contrast, we observed that S-OPA1 restores cristae structures to a similar extent as L-OPA1, suggesting that the cause of cristae disruption observed in Phb2-null cells is not the conversion of L-OPA1 to S-OPA1 but likely the loss of prohibitin function. Because normal prohibitins are present in our experimental system, our results indicate that S-OPA1 may work with prohibitins for cristae maintenance. S-OPA1 binds to lipids such as cardiolipin (70). Prohibitins form ring complexes in the inner membrane to define local membrane domains (71). Unlike membrane-anchored L-OPA1, S-OPA1 may require prohibitins, which provide a favorable lipid environment, for membrane binding and cristae maintenance.
Our data using GTPase-defective Lys-to-Ala mutants show that OPA1 GTPase activity is dispensable for cristae formation but necessary for maintaining cristae tightness as GTPase-defective mutant cells have round and swollen cristae. The cells that have these abnormal cristae are OXPHOS-defective, indicating the energetic importance of maintaining cristae tightness. BNGE data indicate that expressing OPA1 variants in OPA1-KO cells restores respiratory complexes III, IV, and V (Fig. 5). However, Lys-to-Ala mutant OPA1 variants were unable to restore them (Fig. 10F and results not shown), suggesting that the round and swollen cristae are not suitable for respiratory complex biogenesis and assembly. However, considering the role of complex V in cristae morphogenesis, the lack of complex V assembly in OPA1 mutant cells could be the cause of the round swollen cristae. If that is the case, it is possible that OPA1 plays a role in complex V assembly through its GTPase activity. In contrast to our findings using the Lys-to-Ala mutation, a previous study reported that the GTPase-defective Q297V mutant was able to restore normal cristae morphology and support growth in galactose (54). The Q297V mutant is considered to be locked in the GTP-bound form and to remain oligomerized (57, 60). It is possible that oligomerization of OPA1, not the GTPase activity per se, is necessary for maintaining cristae tightness. As OPA1 oligomerization is a dynamic process regulated by GTP binding and hydrolysis, detailed analyses of enzymatic and biochemical properties of these two types of OPA1 mutants will elaborate the role of GTPase activity in maintaining cristae structure and energetics.
The OPA1 function for mitochondrial fusion has been suggested to be independent of the role of OPA1 for cristae maintenance based on a fusion-defective OPA1 mutant maintaining mitochondrial energetics (54). We made a similar observation in which S-OPA1 alone is fusion-incompetent but is capable of energetic maintenance. However, increasing the OPA1 level has been shown to enhance both fusion and energetic activity (36, 37, 72). Furthermore, OXPHOS stimulates inner membrane fusion through OPA1 cleavage (51), suggesting a close connection between mitochondrial fusion and cristae maintenance. It is likely that, following fusion of two structurally and functionally distinct mitochondria, inner membrane reorganization is necessary for functional optimization of newly fused mitochondria. The membrane-anchored L-OPA1 would be restricted to two-dimensional movement, whereas soluble S-OPA1 would be trans diffusible in the intermembrane space to expedite cristae reorganization. It is possible that mitochondrial fusion and inner membrane remodeling are coupled events in which generation of diffusible S-OPA1 promotes the postfusion inner membrane remodeling process.
Experimental procedures
DNA constructs, cell culture, and cell growth assay
Plasmids carrying cDNAs for human OPA1 variants (OPA1-v1, OPA1-v1ΔS1, and OPA1-v5) were kind gifts from David Chan (6). OPA1- KO MEFs were from American Type Culture Collection (ATCC CRL2995) (6). The lysine-to-alanine mutations were made using a standard PCR-based method. The OPA1 variants were transfected to OPA1-KO MEFs, and stable MEF clones were isolated by puromycin selection (2 μg/ml). Cells were maintained in complete media (DMEM high-glucose medium with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2. For cell growth assays, cells were plated in DMEM basal medium containing 1 mm pyruvate, 4 mm glutamine, and 10% fetal bovine serum with either 10 mm glucose for the glycolytic growth condition or 10 mm galactose as the sole sugar source to force cells to rely on mitochondrial OXPHOS (56). Cells were counted each day for 4 days. Medium was changed once on day 2.
Mitochondrial morphology analysis
Cells subjected to morphology analyses were grown on coverslips. For pro-fusion conditions, WT, OPA1-KO, and OPA1 variant MEFs expressing matrix-targeted GFP or DsRed were incubated in Hanks' balanced salt solution (HBSS) for 3 h for starvation (53) or treated with 10 μm cycloheximide for 6 h (30) before they were fixed in 4% paraformaldehyde in PBS. For Drp1 siRNA treatment, cells were transfected with double-strand RNA (sense, 5′-UCCGUGAUGAGUAUGCUUU-3′; antisense, 5′-AAAGCAUACUCAUCACGGA-3′) for 48 h (73, 74). Coverslips were mounted with ProLong antifade reagent (Molecular Probes) on glass slides, and cells were viewed with an epifluorescence microscope (Olympus IX71). Fluorescence images were acquired with a 60× objective for mitochondrial morphology quantification. Mitochondrial morphologies were divided into three classifications: “long tubules” when more than half of mitochondria in a cell displayed a tubular shape longer than 30 pixels, “intermediate” when more than half of mitochondria in a cell were 20–30 pixels, and “fragmented” when the majority of mitochondria in a cell displayed a short, fragmented shape shorter than 20 pixels.
Mitochondrial fusion assessment by PEG-induced cell hybrid assay
An equal number of OPA1-KO cells expressing matrix-targeted DsRed and OPA1 variants or WT expressing matrix-targeted GFP were mixed and cultured overnight on coverslips. The next day, cells were pretreated with cycloheximide (50 μm) for 30 min and washed with PBS. Cells were treated with 50% PEG 3350 for 60 s in serum-free DMEM. The cells were washed and incubated for 4 and 8 h in medium containing cycloheximide (50 μm) before fixation in 4% paraformaldehyde in PBS. Hybrid cells containing both red and green mitochondria were scored for complete, partial, and no fusion based on overlap of red and green fluorescence.
Whole-cell respiration
Cells were plated in complete media and cultured for 24 h before respiration analysis. Oxygen consumption was measured using a Clark-type oxygen electrode in a sealed chamber (Mitocell 200 system, Strathkelvin Instruments). Decreases of the oxygen concentration in the chamber were measured as whole-cell oxygen consumption. Respiration was measured in glucose-free DMEM containing 5 mm pyruvate, 2 mm glutamine, and 5 mm HEPES (pH 7.2). Oligomycin (1 μm) was added to measure oxygen consumption in the absence of ATP synthase activity. Oxygen consumption rate in the presence of oligomycin represents the leak rate. Maximal respiration was obtained by adding carbonyl cyanide p-trifluoromethoxyphenylhydrazone (2 μm). Oxygen consumption rate in the presence of antimycin A (1 μm) was subtracted to obtain mitochondrial oxygen consumption.
Transmission electron microscopy
Cells cultured in complete media were initially fixed in a combination of 0.1 m sodium cacodylate, 4% paraformaldehyde, and 2% glutaraldehyde. Resin infiltration and thin sectioning were performed by the Electron Microscopy Core facility of Medical College of Georgia. Quantifications of cristae density (cristae number/mitochondria area) and cristae width were performed with mitochondria from 10 or more cells. For quantification of matrix electron density, the cytosolic gray values were subtracted from matrix gray values.
Blue native gel electrophoresis
Sample preparation and blue native gel electrophoresis were carried out as described with modifications (75, 76). Briefly, cells in culture were washed twice in ice-cold PBS, collected using a cell lifter, and pelleted at 100 × g for 3 min. Pellets were frozen at −80 °C to increase cell breakage and resuspended at 10 mg/ml in sample buffer (1 m 6-aminohexanoic acid, 50 mm Bis-Tris-HCl (pH 7.0)). Digitonin was added to 100 μg of protein at a ratio of 2 g/g of protein for complex V dimer detection and 4 g/g for other complex assembly and incubated for 30 min in ice. The mixture was spun at 13,000 × g for 30 min, and the supernatant was collected. One-third volume of the sample buffer (5% Coomassie Blue G-250 in 0.5 m 6-aminohexanoic acid) was added, and samples were loaded on blue native 3–13% gradient gels. After electrophoresis, proteins were transferred to PVDF membrane and probed with specific antibodies.
Mitochondrial DNA quantitation
To quantify the amount of mtDNA present per nuclear genome, we used a primer set that detects the ATP6 gene: mtDNA forward primer, CGCCTAATCAACAACCGTCTC; mtDNA reverse primer, TACGGCTCCAGCTCATAGTG. To quantify nuclear DNA, we used a primer set that detects the GAPDH gene: nuclear DNA forward primer, GTGCCGTTGAATTTGCCGT; nuclear DNA reverse primer, ACTACAGACCCATGAGGAGTTCT. Quantification of relative copy number differences was carried out using analysis of the difference in threshold amplification between mtDNA and nuclear DNA.
Western blotting analysis
Western blotting analyses were performed using the following primary antibodies: mouse anti-OPA1 (BD Biosciences, 612606; 0.25 μg/ml); rabbit anti-NDUFA5 (GeneTex, GTX111016; 1:1000) for complex I, rabbit anti-succinate dehydrogenase subunit B (GeneTex, GTX113833; 1:1000) for complex II, rabbit anti-UQCRC2 (GeneTex, GTX114873; 1:5000) for complex III, rabbit anti-COX4 (GeneTex, GTX114330; 1:1000) for complex IV, and mouse anti-ATP synthase subunit β for complex V (ThermoFisher, A-21351; 0.5 μg/ml). Densitometry of immunoblots was performed using Image Lab (Bio-Rad).
Author contributions
H. L. and Y. Y. designed the study; performed experiments; analyzed the data; and wrote, reviewed, and edited the manuscript. S. B. S. contributed to data analyses and reviewed the manuscript.
Acknowledgment
We thank David Chan of California Institute of Technology for providing OPA1 variant constructs.
This work was supported by American Heart Association Grant 16GRNT31170032 (to Y. Y.) and National Institutes of Health Grant R01 EY012830 (to S. B. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DLP1/Drp1
- dynamin-like/-related protein 1
- OPA1
- optic atrophy 1
- L-OPA1
- long OPA1
- S-OPA1
- short OPA1
- OXPHOS
- oxidative phosphorylation
- Mfn
- mitofusin
- MTS
- mitochondrial targeting sequence
- TM
- transmembrane
- Phb
- prohibitin
- MEF
- mouse embryonic fibroblast
- v1–v8
- variants 1–8
- SIMH
- stress-induced mitochondrial hyperfusion
- BNGE
- blue native gel electrophoresis
- CJ
- cristae junctions
- CCJ
- cristae with CJ
- STJ
- short tubules with junctions
- AIF
- apoptosis-inducing factor
- aa
- amino acid(s)
- HBSS
- Hanks' balanced salt solution
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance.
References
- 1. Smirnova E., Griparic L., Shurland D. L., and van der Bliek A. M. (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoon Y., Pitts K. R., and McNiven M. A. (2001) Mammalian dynamin-like protein DLP1 tubulates membranes. Mol. Biol. Cell 12, 2894–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen H., Detmer S. A., Ewald A. J., Griffin E. E., Fraser S. E., and Chan D. C. (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koshiba T., Detmer S. A., Kaiser J. T., Chen H., McCaffery J. M., and Chan D. C. (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862 [DOI] [PubMed] [Google Scholar]
- 5. Cipolat S., Martins de Brito O., Dal Zilio B., and Scorrano L. (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U.S.A. 101, 15927–15932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song Z., Chen H., Fiket M., Alexander C., and Chan D. C. (2007) OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 178, 749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Song Z., Ghochani M., McCaffery J. M., Frey T. G., and Chan D. C. (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu T., Robotham J. L., and Yoon Y. (2006) Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 103, 2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim H., Scimia M. C., Wilkinson D., Trelles R. D., Wood M. R., Bowtell D., Dillin A., Mercola M., and Ronai Z. A. (2011) Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 44, 532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang W., Wang Y., Long J., Wang J., Haudek S. B., Overbeek P., Chang B. H., Schumacker P. T., and Danesh F. R. (2012) Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15, 186–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoon Y., Galloway C. A., Jhun B. S., and Yu T. (2011) Mitochondrial dynamics in diabetes. Antioxid. Redox Signal. 14, 439–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoon Y. (2005) Regulation of mitochondrial dynamics: another process modulated by Ca2+ signal? Sci. STKE 2005, pe18. [DOI] [PubMed] [Google Scholar]
- 13. Galloway C. A., and Yoon Y. (2012) Perspectives on: SGP Symposium on Mitochondrial Physiology and Medicine: what comes first, misshape or dysfunction? The view from metabolic excess. J. Gen. Physiol. 139, 455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griparic L., van der Wel N. N., Orozco I. J., Peters P. J., and van der Bliek A. M. (2004) Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 279, 18792–18798 [DOI] [PubMed] [Google Scholar]
- 15. Chen H., Chomyn A., and Chan D. C. (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280, 26185–26192 [DOI] [PubMed] [Google Scholar]
- 16. Kushnareva Y. E., Gerencser A. A., Bossy B., Ju W. K., White A. D., Waggoner J., Ellisman M. H., Perkins G., and Bossy-Wetzel E. (2013) Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. 20, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexander C., Votruba M., Pesch U. E., Thiselton D. L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., Bhattacharya S. S., and Wissinger B. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215 [DOI] [PubMed] [Google Scholar]
- 18. Delettre C., Lenaers G., Griffoin J. M., Gigarel N., Lorenzo C., Belenguer P., Pelloquin L., Grosgeorge J., Turc-Carel C., Perret E., Astarie-Dequeker C., Lasquellec L., Arnaud B., Ducommun B., Kaplan J., et al. (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210 [DOI] [PubMed] [Google Scholar]
- 19. Amati-Bonneau P., Valentino M. L., Reynier P., Gallardo M. E., Bornstein B., Boissière A., Campos Y., Rivera H., de la Aleja J. G., Carroccia R., Iommarini L., Labauge P., Figarella-Branger D., Marcorelles P., Furby A., et al. (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes. Brain 131, 338–351 [DOI] [PubMed] [Google Scholar]
- 20. Yu-Wai-Man P., Griffiths P. G., Gorman G. S., Lourenco C. M., Wright A. F., Auer-Grumbach M., Toscano A., Musumeci O., Valentino M. L., Caporali L., Lamperti C., Tallaksen C. M., Duffey P., Miller J., Whittaker R. G., et al. (2010) Multi-system neurological disease is common in patients with OPA1 mutations. Brain 133, 771–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Olichon A., Elachouri G., Baricault L., Delettre C., Belenguer P., and Lenaers G. (2007) OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. 14, 682–692 [DOI] [PubMed] [Google Scholar]
- 22. Delettre C., Griffoin J. M., Kaplan J., Dollfus H., Lorenz B., Faivre L., Lenaers G., Belenguer P., and Hamel C. P. (2001) Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 109, 584–591 [DOI] [PubMed] [Google Scholar]
- 23. Olichon A., Emorine L. J., Descoins E., Pelloquin L., Brichese L., Gas N., Guillou E., Delettre C., Valette A., Hamel C. P., Ducommun B., Lenaers G., and Belenguer P. (2002) The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 523, 171–176 [DOI] [PubMed] [Google Scholar]
- 24. Ishihara N., Fujita Y., Oka T., and Mihara K. (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Griparic L., Kanazawa T., and van der Bliek A. M. (2007) Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 178, 757–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Head B., Griparic L., Amiri M., Gandre-Babbe S., and van der Bliek A. M. (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 187, 959–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ehses S., Raschke I., Mancuso G., Bernacchia A., Geimer S., Tondera D., Martinou J. C., Westermann B., Rugarli E. I., and Langer T. (2009) Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 187, 1023–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sood A., Jeyaraju D. V., Prudent J., Caron A., Lemieux P., McBride H. M., Laplante M., Tóth K., and Pellegrini L. (2014) A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. U.S.A. 111, 16017–16022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anand R., Wai T., Baker M. J., Kladt N., Schauss A. C., Rugarli E., and Langer T. (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tondera D., Grandemange S., Jourdain A., Karbowski M., Mattenberger Y., Herzig S., Da Cruz S., Clerc P., Raschke I., Merkwirth C., Ehses S., Krause F., Chan D. C., Alexander C., Bauer C., et al. (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baricault L., Ségui B., Guégand L., Olichon A., Valette A., Larminat F., and Lenaers G. (2007) OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp. Cell Res. 313, 3800–3808 [DOI] [PubMed] [Google Scholar]
- 32. MacVicar T. D., and Lane J. D. (2014) Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J. Cell Sci. 127, 2313–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olichon A., Baricault L., Gas N., Guillou E., Valette A., Belenguer P., and Lenaers G. (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278, 7743–7746 [DOI] [PubMed] [Google Scholar]
- 34. Meeusen S., DeVay R., Block J., Cassidy-Stone A., Wayson S., McCaffery J. M., and Nunnari J. (2006) Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 127, 383–395 [DOI] [PubMed] [Google Scholar]
- 35. Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G. V., Rudka T., Bartoli D., Polishuck R. S., Danial N. N., De Strooper B., and Scorrano L. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189 [DOI] [PubMed] [Google Scholar]
- 36. Cogliati S., Frezza C., Soriano M. E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L. C., Perales-Clemente E., Salviati L., Fernandez-Silva P., Enriquez J. A., and Scorrano L. (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Civiletto G., Varanita T., Cerutti R., Gorletta T., Barbaro S., Marchet S., Lamperti C., Viscomi C., Scorrano L., and Zeviani M. (2015) Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 21, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Varanita T., Soriano M. E., Romanello V., Zaglia T., Quintana-Cabrera R., Semenzato M., Menabò R., Costa V., Civiletto G., Pesce P., Viscomi C., Zeviani M., Di Lisa F., Mongillo M., Sandri M., et al. (2015) The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 21, 834–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merkwirth C., Dargazanli S., Tatsuta T., Geimer S., Löwer B., Wunderlich F. T., von Kleist-Retzow J. C., Waisman A., Westermann B., and Langer T. (2008) Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 22, 476–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mishra S., Murphy L. C., and Murphy L. J. (2006) The prohibitins: emerging roles in diverse functions. J. Cell Mol. Med. 10, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bavelloni A., Piazzi M., Raffini M., Faenza I., and Blalock W. L. (2015) Prohibitin 2: at a communications crossroads. IUBMB Life 67, 239–254 [DOI] [PubMed] [Google Scholar]
- 42. Peng Y. T., Chen P., Ouyang R. Y., and Song L. (2015) Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 20, 1135–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kasashima K., Ohta E., Kagawa Y., and Endo H. (2006) Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 281, 36401–36410 [DOI] [PubMed] [Google Scholar]
- 44. Langer T., Käser M., Klanner C., and Leonhard K. (2001) AAA proteases of mitochondria: quality control of membrane proteins and regulatory functions during mitochondrial biogenesis. Biochem. Soc. Trans. 29, 431–436 [DOI] [PubMed] [Google Scholar]
- 45. Khalimonchuk O., Jeong M. Y., Watts T., Ferris E., and Winge D. R. (2012) Selective Oma1 protease-mediated proteolysis of Cox1 subunit of cytochrome oxidase in assembly mutants. J. Biol. Chem. 287, 7289–7300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Desmurs M., Foti M., Raemy E., Vaz F. M., Martinou J. C., Bairoch A., and Lane L. (2015) C11orf83, a mitochondrial cardiolipin-binding protein involved in bc1 complex assembly and supercomplex stabilization. Mol. Cell. Biol. 35, 1139–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bohovych I., Fernandez M. R., Rahn J. J., Stackley K. D., Bestman J. E., Anandhan A., Franco R., Claypool S. M., Lewis R. E., Chan S. S., and Khalimonchuk O. (2015) Metalloprotease OMA1 fine-tunes mitochondrial bioenergetic function and respiratory supercomplex stability. Sci. Rep. 5, 13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kaser M., Kambacheld M., Kisters-Woike B., and Langer T. (2003) Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J. Biol. Chem. 278, 46414–46423 [DOI] [PubMed] [Google Scholar]
- 49. Stiburek L., Cesnekova J., Kostkova O., Fornuskova D., Vinsova K., Wenchich L., Houstek J., and Zeman J. (2012) YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 23, 1010–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rainbolt T. K., Atanassova N., Genereux J. C., and Wiseman R. L. (2013) Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 18, 908–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mishra P., Carelli V., Manfredi G., and Chan D. C. (2014) Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 19, 630–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gomes L. C., Di Benedetto G., and Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rambold A. S., Kostelecky B., Elia N., and Lippincott-Schwartz J. (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U.S.A. 108, 10190–10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Patten D. A., Wong J., Khacho M., Soubannier V., Mailloux R. J., Pilon-Larose K., MacLaurin J. G., Park D. S., McBride H. M., Trinkle-Mulcahy L., Harper M. E., Germain M., and Slack R. S. (2014) OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 33, 2676–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M., and Chan D. C. (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gohil V. M., Sheth S. A., Nilsson R., Wojtovich A. P., Lee J. H., Perocchi F., Chen W., Clish C. B., Ayata C., Brookes P. S., and Mootha V. K. (2010) Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 28, 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamaguchi R., Lartigue L., Perkins G., Scott R. T., Dixit A., Kushnareva Y., Kuwana T., Ellisman M. H., and Newmeyer D. D. (2008) Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol. Cell 31, 557–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barrera M., Koob S., Dikov D., Vogel F., and Reichert A. S. (2016) OPA1 functionally interacts with MIC60 but is dispensable for crista junction formation. FEBS Lett. 590, 3309–3322 [DOI] [PubMed] [Google Scholar]
- 59. Pernas L., and Scorrano L. (2016) Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 78, 505–531 [DOI] [PubMed] [Google Scholar]
- 60. Misaka T., Miyashita T., and Kubo Y. (2002) Primary structure of a dynamin-related mouse mitochondrial GTPase and its distribution in brain, subcellular localization, and effect on mitochondrial morphology. J. Biol. Chem. 277, 15834–15842 [DOI] [PubMed] [Google Scholar]
- 61. Nijtmans L. G., de Jong L., Artal Sanz M., Coates P. J., Berden J. A., Back J. W., Muijsers A. O., van der Spek H., and Grivell L. A. (2000) Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 19, 2444–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Steglich G., Neupert W., and Langer T. (1999) Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 19, 3435–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Otera H., Ohsakaya S., Nagaura Z., Ishihara N., and Mihara K. (2005) Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 24, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rossignol R., Malgat M., Mazat J. P., and Letellier T. (1999) Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J. Biol. Chem. 274, 33426–33432 [DOI] [PubMed] [Google Scholar]
- 65. Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J. P., and Letellier T. (2003) Mitochondrial threshold effects. Biochem. J. 370, 751–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Houstek J., Mrácek T., Vojtisková A., and Zeman J. (2004) Mitochondrial diseases and ATPase defects of nuclear origin. Biochim. Biophys. Acta 1658, 115–121 [DOI] [PubMed] [Google Scholar]
- 67. Zhang Z., Wakabayashi N., Wakabayashi J., Tamura Y., Song W. J., Sereda S., Clerc P., Polster B. M., Aja S. M., Pletnikov M. V., Kensler T. W., Shirihai O. S., Iijima M., Hussain M. A., and Sesaki H. (2011) The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol. Biol. Cell 22, 2235–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moreno-Lastres D., Fontanesi F., García-Consuegra I., Martín M. A., Arenas J., Barrientos A., and Ugalde C. (2012) Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 15, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cabodevilla A. G., Sánchez-Caballero L., Nintou E., Boiadjieva V. G., Picatoste F., Gubern A., and Claro E. (2013) Cell survival during complete nutrient deprivation depends on lipid droplet-fueled beta-oxidation of fatty acids. J. Biol. Chem. 288, 27777–27788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ban T., Heymann J. A., Song Z., Hinshaw J. E., and Chan D. C. (2010) OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 19, 2113–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Osman C., Merkwirth C., and Langer T. (2009) Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 122, 3823–3830 [DOI] [PubMed] [Google Scholar]
- 72. Liu X., Weaver D., Shirihai O., and Hajnóczky G. (2009) Mitochondrial 'kiss-and-run': interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 28, 3074–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Koch A., Thiemann M., Grabenbauer M., Yoon Y., McNiven M. A., and Schrader M. (2003) Dynamin-like protein 1 is involved in peroxisomal fission. J. Biol. Chem. 278, 8597–8605 [DOI] [PubMed] [Google Scholar]
- 74. Wang L., Yu T., Lee H., O'Brien D. K., Sesaki H., and Yoon Y. (2015) Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc. Res. 106, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Acín-Pérez R., Fernández-Silva P., Peleato M. L., Pérez-Martos A., and Enriquez J. A. (2008) Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539 [DOI] [PubMed] [Google Scholar]
- 76. Wittig I., Braun H. P., and Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]