

Abstract
Toxoplasma gondii is among the most prevalent protozoan parasites, which infects a wide range of organisms, including one-third of the human population. Its rapid intracellular replication within a vacuole requires efficient synthesis of glycerophospholipids. Cytidine diphosphate-diacylglycerol (CDP-DAG) serves as a major precursor for phospholipid synthesis. Given the peculiarities of lipid biogenesis, understanding the mechanism and physiological importance of CDP-DAG synthesis is particularly relevant in T. gondii. Here, we report the occurrence of two phylogenetically divergent CDP-DAG synthase (CDS) enzymes in the parasite. The eukaryotic-type TgCDS1 and the prokaryotic-type TgCDS2 reside in the endoplasmic reticulum and apicoplast, respectively. Conditional knockdown of TgCDS1 severely attenuated the parasite growth and resulted in a nearly complete loss of virulence in a mouse model. Moreover, mice infected with the TgCDS1 mutant became fully resistant to challenge infection with a hyper-virulent strain of T. gondii. The residual growth of the TgCDS1 mutant was abolished by consecutive deletion of TgCDS2. Lipidomic analyses of the two mutants revealed significant and specific declines in phosphatidylinositol and phosphatidylglycerol levels upon repression of TgCDS1 and after deletion of TgCDS2, respectively. Our data suggest a “division of labor” model of lipid biogenesis in T. gondii in which two discrete CDP-DAG pools produced in the endoplasmic reticulum and apicoplast are subsequently used for the synthesis of phosphatidylinositol in the Golgi bodies and phosphatidylglycerol in the mitochondria. The essential and divergent nature of CDP-DAG synthesis in the parasite apicoplast offers a potential drug target to inhibit the asexual reproduction of T. gondii.
Keywords: glycerophospholipid, parasite metabolism, phosphatidylglycerol, phosphatidylinositol, Toxoplasma gondii
Introduction
Toxoplasma belongs to a group of unicellular parasites that comprises some of the most notorious pathogens of humans and animals, such as Plasmodium, Cryptosporidium, Eimeria, Trypanosoma, and Leishmania. One single species of Toxoplasma, i.e. Toxoplasma gondii, is able to infect and reproduce in most nucleated cells of virtually all warm-blood organisms. The widespread success of T. gondii depends on reversible switching of its two asexual stages known as tachyzoites and bradyzoites, which cause acute and chronic infection, respectively (1). Although infection is usually asymptomatic in healthy human adults, the acute infection of developing fetus and individuals with deteriorated immunity can be potentially fatal due to severe tissue necrosis caused by successive rounds of lytic cycles. The rapid intracellular replication of tachyzoites and concurrent expansion of the enclosing vacuole necessitate a significant membrane biogenesis.
The parasite membranes consist of polar and neutral lipids (2, 3). We have shown that glycerophospholipids account for a major fraction of total membrane lipids isolated from purified tachyzoites. Phosphatidylcholine (PtdCho)2 is the most abundant glycerophospholipid present in tachyzoites followed by phosphatidylethanolamine (PtdEtn), phosphatidylthreonine (PtdThr), phosphatidylinositol (PtdIns), phosphatidylserine (PtdSer), phosphatidylglycerol (PtdGro), and phosphatidic acid (PtdOH) (2, 4). Our previous work has also identified the pathways of PtdCho, PtdEtn, PtdThr, and PtdSer synthesis in the parasite (4–7). Synthesis of PtdCho, PtdThr, and PtdSer occurs in the endoplasmic reticulum (ER), whereas PtdEtn can be made in the ER, mitochondrion, and parasitophorous vacuole. Endogenous production of glycerophospholipids begins with the synthesis of glycerol 3-phosphate and fatty acids from glucose-derived carbon (8–10). The parasite produces short acyl chains through a prokaryotic-type (type II) fatty acid synthase pathway located in the apicoplast (8), a non-photosynthetic plastid-like organelle acquired by secondary endosymbiosis of red algae (11). These acyl chains can be exported to the ER, where they are modified to generate long-chain and unsaturated fatty acids (9).
Glycerol 3-phosphate and fatty acids are utilized to synthesize PtdOH. In mammalian cells, PtdOH is a central intermediate lipid that serves as the precursor to make other glycerophospholipids (12, 13). PtdOH is first converted to diacylglycerol (DAG) or CDP-DAG (Fig. 1). DAG enables the synthesis of PtdCho and PtdEtn, whereas CDP-DAG is utilized to make PtdIns and PtdGro. Prokaryotes lack the enzyme to produce DAG and deploy CDP-DAG as the progenitor for all glycerophospholipids (Fig. 1, shaded). The enzyme CDP-DAG synthase is, therefore, regarded as one of the most central enzymes of lipid synthesis in prokaryotes as well as in eukaryotes. Given the functional integration of apicoplast with other organelles harboring lipid synthesis, understanding the mechanism and relevance of CDP-DAG synthesis is particularly interesting in T. gondii but has not yet been studied. The parasite may also access host-derived CDP-DAG; it is unknown, however, whether tachyzoites can scavenge this lipid from the environment.
Figure 1.
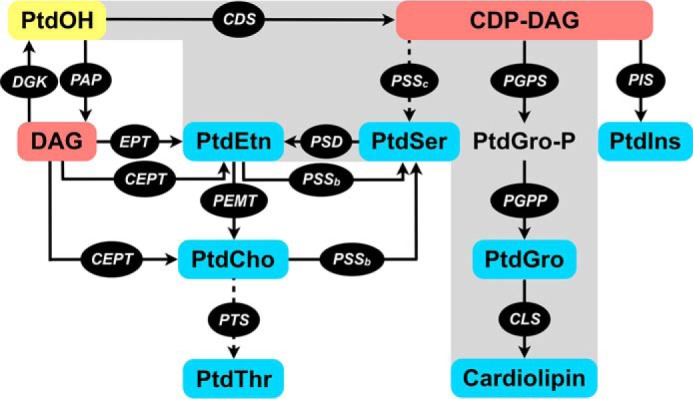
Synthesis of major phospholipids in eukaryotes and prokaryotes. Pathways shown in solid arrows correspond to mammalian cells (human/mouse) and/or eubacteria. All depicted pathways except for PSSc and PTS (shown with dotted arrows) are present in mammalian cells, whereas bacteria harbor only the gray-shaded part of the network including PSSc. A bona fide PTS has been recently identified in the apicomplexan parasite T. gondii. The end phospholipids are highlighted in blue; intermediates of synthesis are displayed in red, and underlying enzymes are in black. PtdOH (yellow) serves as the central precursor for the synthesis of all key phospholipids. CDS, CDP-DAG synthase; CEPT, choline/ethanolamine phosphotransferase; CLS, cardiolipin synthase; DGK, diacylglycerol kinase; EPT, ethanolamine phosphotransferase; PAP, phosphatidic acid phosphatase; PEMT, phosphatidylethanolamine N-methyltransferase; PGPP, phosphatidylglycerol phosphate phosphatase; PGPS, phosphatidylglycerol phosphate synthase; PIS, phosphatidylinositol synthase; PSD, phosphatidylserine decarboxylase; PSSb, phosphatidylserine synthase (base-exchange type); PSSc, phosphatidylserine synthase (CDP-DAG-dependent); PTS, phosphatidylthreonine synthase.
This work identified two phylogenetically distinct routes of CDP-DAG synthesis located in the ER and apicoplast of T. gondii, which have evolved to be primarily non-redundant and in fact functionally complementary to each other. We show that the two sources of CDP-DAG facilitate the synthesis of PtdIns and PtdGro, presumably in the Golgi complex and mitochondrion, respectively. The work suggests a substantial spatial segregation and inter-organelle trafficking of lipids. Metabolic autonomy of tachyzoites and evolutionary divergence of CDP-DAG pathway in the apicoplast also provide an opportunity to selectively inhibit the parasite development.
Results
T. gondii harbors two phylogenetically distinct CDS enzymes
Our bioinformatic searches using bona fide CDS protein sequences from yeast and human identified one CDS gene in the Toxoplasma database (ToxoDB), termed as TgCDS1 henceforth (ToxoDB, TGGT1_281980; GenBankTM, KU199242). Similar database mining using the prokaryotic CDS sequences indicated the unexpected existence of a second CDS in the parasite, which was designated as TgCDS2 (ToxoDB, TGGT1_263785; GenBankTM KU199243). Subsequently, we also found prokaryotic-type CDSs in selected protozoan parasites (Eimeria falciformis, Trypanosoma cruzi, and Leishmania major) albeit not in many others (Plasmodium falciparum, Cryptosporidium parvum, and Trypanosoma brucei). Phylogenetic clustering of CDS sequences revealed discrete eukaryotic and prokaryotic clades (Fig. 2A, supplemental Table S1). All eukaryotes possessed at least one CDS, which clustered with TgCDS1. In contrast, TgCDS2 segregated with prokaryotic-type CDS sequences from bacteria, plants, algae, and indicated parasites. Notably, TgCDS2 grouped with CDSs from cyanobacteria (PmCDS, SsCDS, and TeCDS) and red alga (CcCDS2 and GsCDS2), both of which are considered as ancestors of the apicoplast in apicomplexan parasites (11, 14).
Figure 2.

T. gondii expresses two distinct CDS enzymes located in the ER and apicoplast. A, phylogenetic analysis of TgCDS1, TgCDS2, and orthologs from various organisms representing major forms of life. Branch support was estimated by 100 bootstrap replicates. Other relevant information including the full names of organisms, accession numbers, identity, and similarity details for each CDS sequence are shown in supplemental Table S1. B, schematic drawing of the primary structures of TgCDS1 and TgCDS2. The numbers indicate positions of cytidylyltransferase domains, CDS signature motifs, and transmembrane regions as well as signal and transit peptides, as predicted by Simple Modular Architecture Research Tool (SMART), transmembrane hidden Markov model (TMHMM), SignalP 4.1, ChloroP 1.1, and PlasmoAP. C, multiple alignments of the signature motifs present in eukaryotic- and prokaryotic-type CDS sequences. The residues identical across all sequences are shaded in black, and amino acids that are conserved only in eukaryotes or prokaryotes are highlighted in red or blue, respectively. D and E, scheme for 3′-insertional tagging of TgCDS1 and TgCDS2 genes with a C-terminal HA tag. Plasmids harboring the COS of respective genes were linearized with the indicated enzymes and transfected into tachyzoites (RHΔku80-Δhxgprt) followed by drug selection. Immunofluorescence of stable transgenic tachyzoites expressing TgCDS1-HA (D) and TgCDS2-HA (E) under the control of endogenous promoters and TgSAG1–3′-UTR was performed using anti-HA and Alexa594 antibodies (24 h post-infection). TgCDS1-HA and TgCDS2-HA proteins were co-localized with TgDER1-GFP signal and anti-TgFd/Alexa488 antibodies, respectively. Scale bars: 2 μm. DIC, differential interference contrast.
The ORFs of TgCDS1 and TgCDS2 encode for 1068 and 1044 amino acids with several transmembrane regions (Fig. 2B). Both proteins contain an archetypal cytidylyltransferase domain encompassing a CDS signature motif (GX4SX2KRX4KDX5PGHGGX2DRXD; Fig. 2, B and C). These features are shared by homologs from other organisms irrespective of the phylogenetic origins. Sequence comparisons also revealed many signature residues that were differentially conserved in the eukaryotic- and prokaryotic-type CDS motifs (see red and blue-shaded residues, Fig. 2C). In particular, the prokaryotic-type proteins harbor acidic and basic residues (aspartate-lysine) instead of small and aromatic pair of amino acids (glycine-phenylalanine) occurring in the eukaryotic-type orthologs. Likewise, eukaryotic CDSs show a strictly conserved nucleophile-amide motif (cysteine-glutamine) absent in prokaryotic counterparts. The cytidylyltransferase domain of TgCDS1 exhibited a higher overall identity and similarity with the domains of other eukaryotic-type CDS sequences than with those of the prokaryotic types, whereas the reverse was true for TgCDS2 (supplemental Table S1).
TgCDS1 is expressed in the ER, whereas TgCDS2 is expressed in the apicoplast
The parasite database indicated a constitutive expression of TgCDS1 as well as TgCDS2 transcripts during the lytic cycle (ToxoDB). To test whether TgCDS1 and TgCDS2 proteins are indeed expressed during the asexual development of tachyzoites, we tagged them with a HA epitope by 3′-insertional tagging of the endogenous loci (Fig. 2, D and E). The same approach also enabled us to determine subcellular distributions in intracellular parasites by immunofluorescence assays. Consistent with transcript abundance, endogenous expression of TgCDS1 and TgCDS2 was readily detectable in stable transgenic strains. TgCDS1-HA was expressed primarily in the parasite ER, as confirmed by its co-localization with a known organelle marker TgDER1-GFP (15) (Fig. 2D). In contrast, TgCDS2-HA co-localized with TgFd, a bona fide marker of the apicoplast (16) (Fig. 2E). Distinct subcellular locations of the two CDSs in T. gondii agree with their phylogenetic origins.
The alignment of primary structures revealed prolonged N-terminal extensions in TgCDS1 and TgCDS2 (Fig. 3A). Moreover, a putative bipartite sequence composed of a signal peptide and a transit peptide starting from the second methionine was identified in the N-terminal extension of TgCDS2 (Fig. 3A and supplemental Fig. S1). To address the roles of extended N termini for subcellular targeting, the two mutant isoforms lacking the designated extensions and fused with a C-terminal Myc tag (TgCDS1398–1068-Myc and TgCDS2527–1044-Myc) were ectopically expressed and co-localized with the corresponding full-length proteins. TgCDS1398–1068-Myc was still targeted to the ER, as discerned by co-staining with wild-type TgCDS1 (Fig. 3B). In contrast, TgCDS2527–1044-Myc was not localized in the apicoplast anymore and appeared to be cytosolic instead (Fig. 3B), suggesting a crucial function of N-terminal peptide for correct location. We then generated additional mutants of TgCDS2 protein containing internal deletions of either signal peptide (TgCDS2ΔSP-Myc) or transit peptide (TgCDS2ΔTP-Myc) or both (TgCDS2ΔBS-Myc). Localization studies with these mutants revealed a clear role of bipartite sequence for targeting of TgCDS2 to the apicoplast (Fig. 3B).
Figure 3.

The N terminus of TgCDS2, but not of TgCDS1, is required for correct localization. A, comparison of the primary structures of CDS orthologs across different organisms. The numbers shown before cytidylyltransferase domains indicate the size of the N termini. The lengths of full-length proteins and the positions of the bipartite sequence in TgCDS2 are also mentioned. B, immunofluorescence of tachyzoites co-expressing full-length CDS (TgCDS1-HA or TgCDS2-HA) along with corresponding truncated isoforms lacking the extended N-terminal region (TgCDS1398–1068-Myc or TgCDS2527–1044-Myc), signal peptide (TgCDS2ΔSP-Myc), transit peptide (TgCDS2ΔTP-Myc), or the entire bipartite sequence (TgCDS2ΔBS-Myc). Transgenic parasites harboring epitope-tagged wild-type proteins (see Fig. 2, D and E, for details) were transfected with plasmids encoding the corresponding mutated isoforms (regulated by TgGRA1 elements). C, immunofluorescence of intracellular parasites expressing dual-tagged CDS proteins (Myc-TgCDS1-HA or Myc-TgCDS2-HA) under the control of the pTETO7SAG1 promoter. Immunostaining were performed 24 h post-infection using anti-HA/Alexa488 and anti-Myc/Alexa594 antibodies. Scale bars: 2 μm. DIC, differential interference contrast. Gs, Galdieria sulphuraria; Cr, Chlamydomonas reinhardtii; Ef, Eimeria falciformis; Tc, Trypanosoma cruzi; At, Arabidopsis thaliana; Sc, Saccharomyces cerevisiae; Dm, Drosophila melanogaster; Ec, Escherichia coli; Pm, Prochlorococcus marinus.
To examine whether the N termini were processed during maturation and targeting of the two proteins, we expressed dual-tagged isoforms containing a Myc tag at the N terminus and a HA epitope at the C terminus (Myc-TgCDS1-HA or Myc-TgCDS2-HA). In the case of TgCDS1, both Myc and HA epitopes were distributed in the ER (Fig. 3C). However, the N-terminal region of TgCDS2 (Myc-tagged) showed an evidently cytosolic signal, whereas the C-terminal peptide (HA-tagged) was still localized in the apicoplast (Fig. 3C), which suggested that the N terminus of the protein is cleaved off when the rest of the protein is imported and likely integrated into the organelle membrane.
CDS enzymes are indispensable for the lytic cycle of tachyzoites
To investigate the physiological importance of TgCDS1 and TgCDS2 in T. gondii tachyzoites, we first attempted to create mutants lacking either of the two genes. Our multiple endeavors to delete the TgCDS1 and TgCDS2 loci were futile, indicating essential nature of the two proteins in parasites. We, therefore, generated a conditional mutant of TgCDS1 using the tetracycline repressor-based system as described elsewhere (17). To achieve the mutant, TgCDS1-HA was first expressed under the control of a tetracycline-regulated promoter (pTETO7SAG1) targeted at the uracil phosphoribosyltransferase (UPRT) locus in the RHΔku80-TaTi strain (Fig. 4A, Step 1, making of a merodiploid strain). We then replaced the native TgCDS1 locus by the dihydrofolate reductase-thymidylate synthase (DHFR-TS) selection cassette (S.C.) via double homologous recombination (Fig. 4A, Step 2, making of a Δtgcds1r mutant). Genetic deletion of the TgCDS1 gene was confirmed by recombination-specific PCR screening, which showed the occurrence of 5′ and 3′ homologous recombination events in the mutant but not in the parental strain (Fig. 4B). RT-PCR validated a conditional repression of TgCDS1 transcript by anhydrotetracycline (ATc) in the Δtgcds1r strain (Fig. 4C). TgCDS1 mRNA was induced 2-fold under the control of pTETO7SAG1 promoter, which could be down-regulated by ∼14-fold in ATc-treated cultures. As shown by immunofluorescence and immunoblot analyses (Fig. 4, D and E), exposure to ATc also repressed the expression of TgCDS1-HA protein, which was undetectable after 4 days of cultures with the drug.
Figure 4.

Conditional mutagenesis of TgCDS1 and deletion of TgCDS2 in T. gondii. A, scheme for creating the Δtgcds1r and Δtgcds1r/Δtgcds2 mutants. Steps 1 and 2 depict the making of the Δtgcds1r mutant, which involved integrating a tetracycline-regulatable copy of TgCDS1 (TgCDS1-HA) at the TgUPRT locus in the RHΔku80-TaTi strain (Step 1) followed by deletion of the TgCDS1 gene by the TgDHFR-TS S.C. via double homologous recombination (Step 2). Subsequently, to generate the Δtgcds1r/Δtgcds2 strain (Step 3), the TgCDS2 gene was deleted in the Δtgcds1r mutant by the CAT selection marker. Primers used to screen for 5′ and 3′ recombination at the TgCDS1 and TgCDS2 loci are marked as arrows. B, genomic PCR of the Δtgcds1r and Δtgcds1r/Δtgcds2 strains confirming the events of 5′ and 3′ crossovers. Genomic DNA of the parental strain was included alongside (negative control). C, quantitative PCR of TgCDS1 and TgCDS2 transcripts in the Δtgcds1r and Δtgcds1r/Δtgcds2 mutants. The TgCDS1 transcript was repressed 14-fold in the off state (+ATc). TgCDS2 transcript was not detectable (N.D.) in the Δtgcds1r/Δtgcds2 strain. The average values with S.E. from three independent assays are shown. D and E, immunofluorescence and immunoblot analyses of the two mutants showing a conditional regulation of TgCDS1-HA by ATc. Staining was performed using anti-HA and anti-TgHSP90 antibodies (loading control). Scale bars: 2 μm.
Even though we were unsuccessful in generating a TgCDS2 knock-out mutant using the parental strain, we were able to delete the TgCDS2 locus in the Δtgcds1r mutant, which was probably due to overexpression of TgCDS1 in the latter strain (Fig. 4C). To make a double mutant, a plasmid with the 5′ and 3′-UTRs of TgCDS2 flanking the chloramphenicol acetyltransferase (CAT) S.C. was transfected in the Δtgcds1r strain (Fig. 4A, Step 3, making of a Δtgcds1r/Δtgcds2 mutant). The events of double homologous recombination and integration of the selection marker at the TgCDS2 locus in the double mutant were verified by crossover-specific PCR (Fig. 4B). RT-PCR corroborated the absence of TgCDS2 mRNA in the Δtgcds1r/Δtgcds2 strain (Fig. 4C). It was not feasible to test the protein level by immunofluorescence or immunoblot assays due to unavailability of antibody recognizing TgCDS2. With respect to the transcriptional and translational regulation of TgCDS1, the Δtgcds1r/Δtgcds2 mutant behaved similar to its parental Δtgcds1r strain (Fig. 4, C–E). For example, the Δtgcds1r/Δtgcds2 mutant also showed a 2-fold elevation of TgCDS1 transcript in the on state (−ATc), which could be repressed by 14-fold in the off state (+ATc). Likewise, TgCDS1 protein was expressed at comparable levels in both mutants (on state), and a chemical regulation was achievable within 96 h.
Conditional knockdown of TgCDS1 impairs biogenesis of PtdIns and PtdSer
To discern the functional importance of TgCDS1 and TgCDS2 for phospholipid synthesis, we performed lipidomic analysis. Lipids were isolated from tachyzoites of the parental, Δtgcds1r and Δtgcds1r/Δtgcds2 strains cultured in the absence or presence of ATc and subjected to high-performance liquid chromatography (HPLC) and mass spectrometry (MS). As anticipated, total phospholipids in the parental strain were not perturbed by treatment with ATc, whereas we found a modest increase in the Δtgcds1r mutant during on state, which was further increased in off state (Fig. 5A). In-depth analysis of individual lipids revealed a notable elevation of all major phospholipids, which were probably due to ectopic overexpression of TgCDS1 (Figs. 5, B–D, and 6, A and B). More importantly, a knockdown of TgCDS1 in the Δtgcds1r strain led to a significant and rather selective reduction in the amounts of PtdIns and PtdSer (Fig. 6, A and B), which was confirmed by thin-layer chromatography (not shown). Other phospholipids were either unaffected (PtdEtn, PtdThr, Fig. 5, C and D) or even elevated (PtdCho in Fig. 5B, PtdGro in Fig. 6C). Such a modest rise in PtdCho (accounting for >70% of total phospholipids; Refs. 2 and 3) was sufficient to induce the level of total lipids during off state despite a prominent decline in PtdIns and PtdSer (Fig. 5A), which together amounted to <10% of total phospholipids.
Figure 5.

Phospholipid profiles of the Δtgcds1r and Δtgcds1r/Δtgcds2 strains. Lipids isolated from tachyzoites were analyzed by HPLC/MS. Major phospholipids identified were PtdCho, PtdEtn, PtdThr, PtdIns, PtdSer, and PtdGro. A shows the cumulative sum of all lipids in the indicated strains cultured with or without ATc. Other panels illustrate PtdCho (B), PtdEtn (C), and PtdThr (D). PtdIns, PtdSer, and PtdGro are depicted in Fig. 6. Values are the means with S.E. from six independent assays (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Figure 6.

Conditional knockdown of TgCDS1 results in a reduction of PtdIns and PtdSer, whereas deletion of TgCDS2 impairs PtdGro biogenesis. Total lipids were extracted from tachyzoites of the indicated strains and subjected to HPLC and MS analysis. Amounts of PtdIns (A), PtdSer (B), PtdGro (C), and the corresponding major lipid species (D, E, and F) in the presence or absence of ATc are depicted in heat-maps. The color-coded gradient in each column is meant to compare the relative abundance of the given lipid species in the mutants with respect to the on-state parental strain. Actual amounts of lipid species (pmol/106 parasites) are also mentioned for comparing columns. The sums of individual lipid species in D–F are less than the total lipid amount in A–C because only major species are shown. Note that the reduction of PtdSer species in the off-state parental strain (+ATc) is relatively negligible even though they appear somewhat blue. All values show the mean values with S.E. from six independent assays (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Next, we examined the major species of PtdIns and PtdSer (Fig. 6, D and E). PtdIns species were composed primarily of short to medium saturated and unsaturated fatty acids (30–36 carbons). In particular, the amount of the most plentiful species 34:1 and 34:2 in the Δtgcds1r mutant changed dramatically in an ATc-regulated manner when compared with the parental strain. Other key species of PtdIns that were declined upon repression of TgCDS1 included 30:0, 32:1, and 36:2. One particular species 36:4 (C16:0/20:4) of PtdIns showed a surprisingly opposite trend in the mutant. Interestingly, the most abundant PtdIns species in human foreskin fibroblasts (HFFs) is PtdIns 38:4 (C18:0/20:4), which is negligible in the parasite extract (<3% of total PtdIns) leading to a suspicion that tachyzoites might salvage this species containing the C20:4 acyl chain from host cells and then remodel it to its own benefit. The most abundant PtdSer species contained mono- and polyunsaturated acyl chains with 34, 36, and 42 carbon atoms. All of them were induced noticeably during on state of the Δtgcds1r strain and reduced after a down-regulation of TgCDS1 (off state). These data together suggest a requirement of TgCDS1 to produce PtdIns and PtdSer via CDP-DAG-dependent enzymes.
Loss of TgCDS2 results in a selective impairment of PtdGro synthesis
Having evaluated the impact of TgCDS1 knockdown in the single mutant, we examined a role of TgCDS2 for phospholipid biogenesis and functional interrelationship of both enzymes using the Δtgcds1r/Δtgcds2 strain. In essence, biochemical phenotypes of the double mutant were similar to the parental and Δtgcds1r strains with few noteworthy exceptions. One of the most evident effects was >80% reduction in PtdGro biogenesis upon ablation of TgCDS2 (Fig. 6C). All species of PtdGro were significantly decreased irrespective of ATc treatment, which confirmed a need of TgCDS2 (but not TgCDS1) for PtdGro biogenesis (Fig. 6F). Consistently, the most common PtdGro species with short saturated fatty acids (30:0) was increased when TgCDS1 was repressed in the Δtgcds1r strain (Fig. 6F), suggesting utilization of subsequently surplus PtdOH species into PtdGro via TgCDS2. Cardiolipin was detectable only in the minor amounts even in the parental strain; we were, therefore, unable to reproducibly quantify and compare its levels across the parasite strains. The deletion of TgCDS2 in the Δtgcds1r strain also resulted in an apparent diminution of certain PtdIns (34:2, 30:0) and PtdSer (34:2) species in the Δtgcds1r/Δtgcds2 mutant during on state, which were further reduced by deficiency of TgCDS1 in off state (Fig. 6, D and E). Other major lipids were largely unaffected by loss of TgCDS2 when compared with the parental strain (Fig. 5).
The CDS mutants show a defective growth due to impaired replication
We next determined the phenotypic effects of TgCDS1 knockdown and TgCDS2 knock-out on the lytic cycle of tachyzoites in HFF cells. To measure the growth fitness, we first performed plaque assays, which recapitulate the consecutive lytic cycles of tachyzoites (Fig. 7A). The two mutants along with the parental strain were cultured in the absence or presence of ATc. The Δtgcds1r strain demonstrated normal growth during on state, whereas plaque size was reduced down to 25% in off state when compared with the parental strain (Fig. 7B). Plaque formation by the Δtgcds1r/Δtgcds2 strain declined by a modest 12% during the on state with respect to the parental strain and was severely reduced by ∼90% when the expression of TgCDS1 was turned off (Fig. 7B). Plaque numbers of both mutants were the same as the parental strain in on state, as expected, whereas they dropped down to ∼60 and 40% during off state, respectively (Fig. 7C). The addition of exogenous CDP-DAG, PtdIns, PtdGro, and cardiolipin to the plaque cultures did not restore the growth of either of the mutants (not shown), indicating that tachyzoites are unable to scavenge these lipids from the surrounding milieu.
Figure 7.

The Δtgcds1r and Δtgcds1r/Δtgcds2 mutants show an attenuated growth. A, plaque assays using the indicated parasite strains cultured in the presence or absence of ATc. HFF monolayers were infected with 250 tachyzoites (pre-cultured with or without ATc for 4 days) and incubated for 1 week without any perturbation to allow plaque formation. B, plaque sizes of the parasite strains from A. 90 plaques of each strain were measured for area using the ImageJ software. a.u., arbitrary units. C, plaque numbers of individual strains from A. D, yield assays in the presence or absence of ATc. Host cells were infected with 3 million tachyzoites pretreated with ATc for 4 days before the assay (m.o.i. 3). Parasites were syringe-released 40 h post-infection for counting. E, replication assays in HFFs (m.o.i. 1). Cells were fixed 40 h post-infection and stained with anti-TgGAP45 antibody to visualize intracellular tachyzoites. A total of 100–150 vacuoles were analyzed for each strain. The percentages of vacuoles containing different numbers of parasites are shown. Values are the means with S.E. for three (B, C, and E) or six (D) independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001). F, virulence assay in a mouse model. Animals (C57BL/6J) were infected intraperitoneally with tachyzoites (103) of the parental or Δtgcds1r strains (pre-cultured with or without ATc for 6 days). Drug treatment during infection (2 weeks) was performed via the drinking water. Infected mice were monitored for 28 days (2 assays, each with 4 mice for the parental strain and 6 mice for the Δtgcds1r strain). Animals surviving infection with the mutant strain (+ATc) were challenged with the parental strain (103, dotted arrow) and examined for additional 28 days.
A similar phenotype of the mutants was observed in yield assays when parasites were syringe-released and counted 40 h post-infection (Fig. 7D). The parasite yield dropped by 75%, 23%, and 96% after the loss of TgCDS1 (off-state Δtgcds1r), TgCDS2 (on-state Δtgcds1r/Δtgcds2) alone, or both (off-state Δtgcds1r/Δtgcds2), respectively. Based on the parasite yields, we also calculated the replication rate of all strains. The doubling time of the parental strain was ∼9.4 h, which was prolonged to 16.2 h when TgCDS1 expression was knocked down in the Δtgcds1r mutant. Knock-out of TgCDS2 alone caused only a modest delay in cell division (10.2 h). However, a loss of both enzymes severely attenuated the parasite proliferation (doubling time, 56.5 h). We also assessed the replication by numerating parasites in the parasitophorous vacuoles (Fig. 7E). We found an evident downshift in the vacuole sizes of both strains after treatment with ATc. In other words, mutants in off state contained a higher proportion of smaller vacuole, whereas the converse was true for on state.
A deficiency of CDP-DAG synthesis attenuates the parasite virulence
We then investigated the physiological relevance of CDP-DAG synthesis within the parasite for virulence in a mouse model (Fig. 7F). In this regard we employed the Δtgcds1r mutant along with the parental strain. Our rationale for using only the single mutant was based on a robust phenotypic impairment observed in vitro (off state, Fig. 7, A–E), which should ideally translate into a strong weakening of virulence in vivo. Hence, a double mutant (Δtgcds1r/Δtgcds2) with even more inhibited in vitro growth would be more attenuated in vivo. Indeed, most animals infected with the Δtgcds1r strain that were treated with ATc in drinking water survived, as opposed to the control parental strain, which was categorically lethal irrespective of ATc treatment. The mutant in its on state exhibited a lethal phenotype similar to the parental strain. Collectively, these results indicated an essential role of CDS for the lytic cycle and virulence in mice. The data also suggested that tachyzoites are unable to salvage sufficient amounts of CDP-DAG from the host to bypass an ablation of autonomous synthesis. We also explored the vaccination potential of the Δtgcds1r strain. Notably, all animals surviving the primary infection with the mutant developed sufficient immunity to resist the subsequent lethal challenge by a hypervirulent strain, which proves the prophylactic utility of a metabolically attenuated mutant to prevent acute toxoplasmosis.
PtdGro phosphate and PtdIns are made in the mitochondrion and Golgi bodies, respectively
The CDS-dependent synthesis of PtdIns and PtdGro resonates with the occurrence of a PtdIns synthase (PIS) and a PtdGro phosphate (PtdGro-P) synthase (PGPS) in tachyzoites. The second enzyme of PtdGro synthesis (PtdGro-P phosphatase) could not be identified in the parasite genome. We examined subcellular distribution of TgPGPS (ToxoDB, TGGT1_246530; GenBankTM, KX017550) by 3′-HA tagging of the endogenous locus in T. gondii. TgPGPS-HA was expressed in the parasite mitochondrion, as shown by its co-staining with a bona fide organelle marker TgF1B (7) (Fig. 8A). We also immuno-localized TgPIS-HA (ToxoDB, TGGT1_207710; GenBankTM, KX017549). It co-localized with a known marker of the Golgi body TgERD2 (Fig. 8B). Taken together, these results indicated synthesis of PtdGro and PtdIns in the mitochondrion and Golgi, respectively, requiring a transfer of CDP-DAG from the apicoplast and ER to the sites of phospholipid biogenesis.
Figure 8.

CDP-DAG-dependent biogenesis of PtdIns, PtdSer and PtdGro in the tachyzoite stage of T. gondii. A and B, immunofluorescent images of intracellular tachyzoites expressing either TgPGPS-HA or TgPIS-HA regulated by endogenous and pTETO7SAG1 elements, respectively. The indicated proteins were localized 24 h post-infection using anti-HA antibody and corresponding co-localization markers (TgF1B, TgERD2-Ty1). 3′-Insertional tagging was performed as described in Fig. 2, D and E. Scale bars: 2 μm. DIC, differential interference contrast. C, a model of lipid biogenesis embodying division of labor among the specified organelles in tachyzoites. The illustration depicts abridged interorganelle trafficking of selected lipids, especially those relevant to the context. Two distinct CDP-DAG sources are utilized to produce PtdIns, PtdSer, and PtdGro. The CDP-DAG pool in the ER is generated by TgCDS1 and used for biogenesis of PtdIns in the Golgi bodies. PtdIns should enable the synthesis of phosphatidylinositol phosphates (PIP) and glycosylphosphatidylinositol (GPI). TgCDS2 generates a second pool of CDP-DAG in the apicoplast that contributes to the making of PtdGro and cardiolipin in the mitochondrion. The confirmed pathways are marked with solid arrows, whereas those hypothesized are shown with dotted arrows. FASII, fatty acid synthase type II; FAE, fatty acid elongase.
Discussion
This study identified two phylogenetically distinct enzymes involved in the synthesis of one of the central lipid precursors CDP-DAG in T. gondii. TgCDS1 belongs to the eukaryotic-type CDSs that are conserved across the eukaryotic organisms. It is compartmentalized in the ER of tachyzoites, similar to what has been described for corresponding CDS proteins in yeast (18), plants (19), and mammals (20, 21). In protozoan parasites P. falciparum and T. brucei, only one eukaryotic-type CDS has been identified, which is required for the biosynthesis of PtdIns and its descendant lipid glycosylphosphatidylinositol (22–24). In mammals and plants there are two or more eukaryotic-type CDS enzymes with different expression patterns, all of which influence PtdIns and PtdOH levels (19, 25). Interestingly, a mitochondrial maintenance protein lacking the typical cytidylyltransferase domain and CDS signature motif (Tam41) was found to catalyze the formation of CDP-DAG from PtdOH in the yeast mitochondria (26); however, similar proteins have not been identified in any protozoan parasites. Instead, we discovered a prokaryotic-type TgCDS2 in the apicoplast of T. gondii and its orthologs in selected protozoan parasites, e.g. E. falciformis, T. cruzi, and L. major. No homologs were found in P. falciparum, C. parvum, and T. brucei, suggesting a loss of the prokaryotic-type CDS during the evolution of these parasites.
Apicoplast has evolved by two successive endosymbiotic events, first involving cyanobacteria and then red alga (11, 14, 27–29). Both events were ensued by horizontal gene transfer from the cyanobacterial genome to the algal genome and subsequently to the genome of the parasites. As a result, most of the apicoplast proteins, including TgCDS2, have prokaryotic or algal origins but are encoded by the parasite nucleus (14, 30, 31). Endosymbiotic gene transfer from the apicoplast to the nucleus enables parasites to control the foreign organelle; however, it requires post-translational targeting of proteins back to the apicoplast. In many apicoplast-resident proteins, this process is guided by an N-terminal bipartite sequence comprising a signal peptide and a transit peptide (32). These proteins are first imported into the ER and then transferred directly from the ER to the apicoplast either through the general secretory pathway or via vesicular trafficking (33–35). Recent studies also reveal involvement of the Golgi as a sorting point for soluble proteins destined to the apicoplast (36, 37). Both putative signal and transit peptides could be identified in the N terminus of the TgCDS2 sequence; however, they begin from the second methionine instead of the start codon. Consistently, our mutagenesis studies confirm the need of prolonged N-terminal extension and bipartite sequence for localization in the apicoplast. The data indicate that the targeting of TgCDS2 to the apicoplast is mediated by the bipartite signal mechanism.
TgCDS1 and TgCDS2 mutants showed severely reduced growth and attenuated virulence, which is associated with the loss of PtdIns and PtdGro in parasites. PtdIns serves not only as a structural component of membranes but also as a precursor for the biogenesis of central signaling molecules. Earlier studies on the PtdIns phosphates have revealed that PtdIns 3-monophosphate and PtdIns 3,5-bisphosphate are required for the apicoplast biogenesis in tachyzoites (38, 39). Another downstream product of PtdIns, glycosylphosphatidylinositol, has been implicated in host-cell attachment and modulation of immune response and is reported to be essential for the parasite survival (40, 41). By contrast, literally nothing is known about the structural and functional relevance of PtdGro in T. gondii. PtdGro typically serves as an intermediate for the synthesis of cardiolipin, which is indispensable for the mitochondrial homeostasis and viability of kinetoplastid parasites (42, 43). It is expected that changes in PtdIns, PtdGro, and cardiolipin would result in organelle dysfunction and eventual demise of T. gondii unless parasites could salvage CDP-DAG or its descendent lipids from the host cell to bypass the ablation of CDS enzymes, which seems not to be the case. Detailed biochemical and morphological analyses shall reveal the underlying basis of the growth impairment in the CDS mutants.
TgCDS1 and TgCDS2 mutants did not show a perturbation of other dominant phospholipids except for PtdCho, which can be explained by the endogenous pathways expressed in tachyzoites. We have previously reported the functional existence of PtdCho and PtdEtn syntheses in the ER through CDP-choline and CDP-ethanolamine pathways, respectively, both of which utilize DAG as the lipid backbone (2, 6, 7). Because synthesis of PtdCho occurs exclusively through the CDP-choline route, a significant increase in the lipid content during the off state of the two mutants is expected due to potential rerouting of PtdOH to DAG. On the other hand, PtdEtn can also be produced by decarboxylation of PtdSer in the parasite mitochondrion and parasitophorous vacuole (5, 7), which may have balanced its content in the single and double mutants. PtdSer and PtdThr are generated from PtdEtn and/or PtdCho in a base-exchange manner by two distinct routes located in the parasite ER (4). No change of PtdThr was observed in both mutants. Given its unique fatty acid chain composition (mostly 20:1, 20:4), it seems that a different source of glycerol backbone or lipid remodeling is required to make PtdThr. In the case of PtdSer, a sizeable increase during on state and a decline in off state of both mutants was surprising and suggests the presence of yet another PtdSer synthase (CDP-DAG-dependent) for making PtdSer (Fig. 8C). A perturbation in the amounts of PtdCho and PtdSer may also account for the phenotype observed in individual mutants. Although the detection of CDP-DAG and DAG in tachyzoite has been quite challenging, future studies using isotope labeling with lipid precursors (polar head groups, glycerol) should validate aforesaid postulations.
TgCDS1 and TgCDS2 are utilized for biogenesis of PtdIns, PtdSer, and PtdGro in a rather selective manner. Our data suggest a model in which ER-derived CDP-DAG fuels the synthesis of PtdIns in the Golgi bodies, whereas CDP-DAG originating in the apicoplast is utilized for making PtdGro in the mitochondrion (Fig. 8C). These data are consistent with the presence of two isoforms of each enzyme synthesizing lysophosphatidic acid and phosphatidic acid (ToxoDB). One such enzyme, glycerol-3-phosphate acyltransferase, has recently been reported to localize in the apicoplast (44), whereas others remain to be characterized. Such a spatial distribution of lipid syntheses necessitates a coordinated lipid transport among organelles in T. gondii. It will be interesting to examine the topological orientations of these enzymes in different organelles, which is imperative for a concerted lipid synthesis. Several lipid trafficking mechanisms have been reported, especially in yeast and mammalian cells, which involve lateral and transbilayer movements within the same organelle as well as membrane contact sites, vesicular trafficking, and protein-mediated transport between different organelles (45–47). In apicomplexan parasites, analogous membrane contact sites have been observed between the ER, Golgi body, apicoplast, and mitochondrion (48, 49); however, whether they serve as the privileged zones of inter-organelle exchange merits further investigation, in particular a “retrograde” transfer of PtdIns from Golgi to ER and of CDP-DAG from ER/apicoplast to Golgi/mitochondrion. Our prototype model of lipid synthesis and trafficking provides a framework for studying the paradigm in a well established model eukaryotic pathogen.
Experimental procedures
Biological reagents and resources
The RHΔku80-TaTi (30) and RHΔku80-Δhxgprt (50) strains of T. gondii were kindly provided by Boris Striepen (University of Georgia) and Vern Carruthers (University of Michigan). The pG152, pTKO-HXGPRT, and pTUB8-TgDER1-GFP plasmids were donated by Markus Meissner (University of Glasgow), John Boothroyd (Stanford University School of Medicine), and Boris Striepen (University of Georgia), respectively. The pTKO-CAT and pTKO-DHFR-TS plasmids were generated by modifying the pTKO-HXGPRT plasmid. Primary antibodies against TgFd, TgHSP90, TgGAP45, TgSAG1, and TgF1B proteins were provided from Frank Seeber (Robert-Koch Institute, Berlin), Sergio Angel (IIB-INTECH, Chascomús, Argentina), Dominique Soldati-Favre (University of Geneva), Jean-Francois Dubremetz (University of Montpellier), and Peter Bradley (University of California, Los Angeles, CA), respectively. Other primary antibodies (anti-HA, Myc or Ty1) were purchased from Sigma. Secondary antibodies (Alexa488 or Alexa594) as well as oligonucleotides were supplied from Life Technologies.
Parasite and host cell cultures
HFFs (Cell Lines Service, Eppelheim, Germany) were cultured in DMEM supplemented with 10% fetal bovine serum (PAN Biotech, Aidenbach, Germany), 2 mm glutamine, 1 mm sodium pyruvate, minimum Eagle's non-essential amino acids, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator (37 °C, 5% CO2). HFFs were harvested by trypsinization and grown to confluence in flasks, dishes, or plates as required. Tachyzoites were propagated by serial passage in HFF monolayers at a m.o.i. of 3. For all assays including lipidomics, parasites were mechanically released from late-stage cultures and used immediately. Briefly, parasitized cells (40–42 h infection) were scraped in fresh culture medium and squirted through 23- and 27-gauge syringes (2× each) to obtain extracellular tachyzoites for the experiments described herein.
Molecular cloning and real-time PCR
RNA was isolated from freshly syringe-released tachyzoites using TRIzol-based extraction method and subsequently reverse-transcribed into first-strand cDNA (Life Technologies). The gDNA was isolated using the genomic DNA preparation kit (Jena Bioscience, Jena, Germany). All amplicons were amplified by Pfu Ultra II Fusion polymerase (Agilent Technologies, Santa Clara, CA) and cloned into the corresponding vectors either by ligation-independent or by restriction enzyme-mediated cloning as indicated. Primers used for PCRs are listed in supplemental Table S2. Plasmids were transformed into Escherichia coli XL-1b strain for cloning and vector amplification. To perform the real-time PCR, total RNA was first reverse-transcribed using the oligo-dT primer and analyzed by SYBR Green-based assays (Mastercycler Pro Gradient S System, Eppendorf, Hamburg, Germany). The relative expression of transcripts (-fold induction) was calculated with respect to the parental strain using the ΔΔCT method. Transcripts of elongation factor A, tubulin A, and glucose transporter 1 were used as housekeeping genes to examine the normalized expression of TgCDS1 and TgCDS2 across samples.
Generation of transgenic parasites
For generating the transgenic strains, the respective plasmid constructs were transfected into fresh tachyzoites of specified strains suspended in filter-sterile Cytomix (120 mm KCl, 0.15 mm CaCl2, 10 mm K2HPO4/KH2PO4, 25 mm HEPES, 2 mm EGTA, 5 mm MgCl2, 5 mm glutathione, 5 mm ATP, pH 7.6) using a BTX electroporation instrument (50 μg of plasmid DNA, ∼107 parasites, 2 kV, 50 ohms, 25 microfarads, 250 μs). Transformed tachyzoites were selected for resistance to a drug corresponding to the selection marker encoded by transfected plasmid. The drug-resistant transgenic parasites were cloned by limiting dilution, and individual clones were screened by PCR and/or immunofluorescence assays.
For tagging of the TgCDS1, TgCDS2, and TgPGPS genes with a C-terminal HA tag, 1.0–1.3 kb of the 3′-end of these genes excluding stop codon (crossover sequence (COS)) were amplified using tachyzoite gDNA and gene-specific primers (supplemental Table S2). Amplicons were inserted into the pG152 vector by ligation-independent cloning. Constructs were linearized using an appropriate enzyme (NaeI, XhoI, or SacI as specified in figures) present in the COS and transfected into the RHΔku80-Δhxgprt strain. Parasites were selected for hypoxanthine-xanthine-guanine phosphoribosyltransferase (HXGPRT) expression using mycophenolic acid (25 μg/ml) and xanthine (50 μg/ml) (51). The resulting transgenic strains expressed TgCDS1-HA, TgCDS2-HA, or TgPGPS-HA under the control of conforming endogenous promoters and 3′-UTR of TgSAG1. Parasites expressing TgCDS1-HA were subsequently transfected with the construct encoding for TgDER1-GFP (regulated by pTUB8 promoter) for co-localization studies.
For ectopic expression of TgCDS1398–1068-Myc (lacking N-terminal extension and tagged with a C-terminal Myc epitope), the partial ORF was cloned into the pTKO-CAT plasmid at NsiI/PacI sites. The deletion variants of TgCDS2, including TgCDS2527–1044-Myc and those lacking the signal peptide (TgCDS2ΔSP-Myc), transit peptide (TgCDS2ΔTP-Myc), or the entire bipartite sequence (TgCDS2ΔBS-Myc) were engineered in the pTKO-DHFR-TS vector at NsiI/PacI sites (see primers in supplemental Table S2). The plasmid constructs were linearized with NotI and transfected into strains harboring the full-length TgCDS1-HA or TgCDS2-HA. To express TgCDS1 and TgCDS2 with dual epitopes (N-terminal Myc tag and C-terminal HA tag) as well as TgPIS with a C-terminal HA tag, their cDNAs were ligated into the pTETO7SAG1-UPKO plasmid at NcoI/PacI sites. Constructs were linearized by NotI and then transfected into the RHΔku80-TaTi strain. Parasites expressing TgPIS-HA were successively transfected with a construct encoding for TgERD2-Ty1 (regulated by pGRA1 promoter) to do co-localization studies.
The regulatable mutant of TgCDS1 (Δtgcds1r) was generated in two steps. First, TgCDS1-HA regulated by ATc-repressible promoter (pTETO7SAG1) was targeted at the TgUPRT locus. The ORF of TgCDS1 containing a C-terminal HA tag was inserted into the pTETO7SAG1-UPKO vector at NcoI/PacI restriction sites. The eventual construct was linearized by NotI and transfected into the RHΔku80-TaTi strain followed by negative selection for the disruption of the TgUPRT locus using 5-fluorodeoxyuridine (5 μm) (52). In the second step the TgCDS1 locus was deleted by double homologous recombination in the merodiploid strain expressing an ATc-regulatable copy of TgCDS1. To achieve this, the 5′- and 3′-UTRs of TgCDS1 were amplified from the tachyzoite gDNA and cloned into the pTKO-DHFR-TS vector using XcmI/SpeI and HindIII/ApaI enzyme pairs, respectively. The construct was linearized using ApaI and transfected into the merodiploid strain generated in the first step. The conditional mutant was selected for the expression of DHFR-TS using 1 μm pyrimethamine (53). To make a knock-out of the TgCDS2 gene, the 5′- and 3′-UTRs amplified from tachyzoite gDNA were cloned into the pTUB8-CAT plasmid at ApaI and XhoI/XbaI restriction sites, respectively. The plasmid was linearized (XbaI) and transfected into the Δtgcds1r strain followed by selection for the expression of CAT using 20 μm chloramphenicol (54). The resulting strain (Δtgcds1r/Δtgcds2) lacked the expression of TgCDS2 entirely and allowed conditional knockdown of TgCDS1 by ATc.
Indirect immunofluorescence and immunoblot assays
Parasitized HFFs cultured on glass coverslips were washed with phosphate-buffered saline (PBS) 24 h post-infection, fixed with 4% paraformaldehyde for 10 min, and neutralized with 0.1 m glycine in PBS for 5 min. Cells were permeabilized with 0.2% Triton X-100 in PBS for 20 min and treated with 2% bovine serum albumin in 0.2% Triton X-100/PBS for 30 min. Samples were stained with suitable combinations of the primary antibodies (anti-HA, 1:3000; anti-Myc, 1:3000; anti-Ty1, 1:50; anti-TgFd, 1:500; anti-TgF1B, 1:1000; anti-TgGAP45, 1:3000; anti-TgSAG1, 1:1500) for 1 h, as shown in the figures. Cells were washed 3 times with 0.2% Triton X-100 in PBS and then stained with Alexa488/594-conjugated antibodies for 45 min. After three additional washings with PBS, samples were mounted in Fluoromount G and DAPI mix (SouthernBiotech, Birmingham, AL) and stored at 4 °C. Imaging was done using a fluorescence microscope (ApoTome, Zeiss, Jena, Germany).
To perform immunoblot analysis, fresh extracellular parasites (1.5 × 107) were washed 2× with PBS, pelleted (400 × g, 10 min, 4 °C), resuspended in the protein loading buffer, and subjected to denaturing gel electrophoresis. Proteins were resolved by 10% SDS-PAGE and transferred to a nitrocellulose membrane (85 mA, 90 min). The membrane was treated with 5% skimmed dry milk suspended in Tris-buffered saline and 0.2% Tween 20 (overnight, 4 °C), incubated with anti-HA (1:1,500, mouse) and anti-TgHSP90 (1:1,000, rabbit) antibodies (2 h at room temperature), washed 3× for 5 min each, and then incubated with IR dyes-conjugated secondary antibodies (680RD, 800CW, each at 1:10,000 for 1 h; Li-COR Biosciences, Lincoln NE). Proteins were visualized using a Li-COR imaging system.
Lipid analysis
Parasite pellets were suspended in 0.8 ml of PBS, and lipids were extracted according to Bligh-Dyer method (55). Total lipids equivalent to 3 × 107 parasites were dried under a nitrogen gas stream and suspended in 1 ml of chloroform and methanol mixture (1:1). A 10-μl aliquot was introduced onto a Kinetex HILIC column with dimensions 50 × 4.6 mm and 2.6 μm (Phenomenex, Torrance, CA). Phospholipids were resolved at a flow rate of 1 ml/min as described before (56). The column effluent was introduced into a mass spectrometer instrument (LTQ-XL, Thermo Scientific, Waltham, MA) and analyzed by electrospray ionization in positive and negative ion modes. Calibration curves of authentic standards were used to quantify lipids. Fatty acid composition of individual lipids was determined by MS/MS. Data were processed using the package XCMS in R (R-project) (57). Only those lipid species that were reproducibly detectable in independent assays are shown in this work. Lipids that were not conclusively detectable or quantifiable, such as PtdOH, cardiolipin, DAG, and CDP-DAG, are excluded. The abbreviations reported here follow the nomenclature of the IUPAC-IUB convention.
Lytic cycle assays
Standard methods were used to determine the impact of genetic manipulation on the lytic cycle of the parasite. Tachyzoites of all strains were pretreated with or without ATc (1 μm) for 2 passages (4 days) in culture before setting up individual assays. Plaque assays were performed by infecting HFF monolayers in 6-well plates (250 parasites per well). Lipids, if supplemented, were dissolved in serum and added to plaque cultures (0.05–0.1 μm). Cultures were incubated unperturbed for 7 days in the presence or absence of ATc followed by fixation with ice-cold methanol and staining with crystal violet dye. Plaques were imaged and scored for sizes and numbers using ImageJ software (NIH, Bethesda, MD). For yield assays, 3 × 106 tachyzoites of each strain were used to infect confluent HFFs (m.o.i. 3) in the presence or absence of ATc as specified. Parasites were then syringe-released from host cells after 40 h and numerated. For replication assays, confluent HFFs cultured on coverslips in 24-well plates were infected with tachyzoites (m.o.i. 1; 40 h) and then subjected to immunostaining using anti-TgGAP45 antibody, as described above. The mean percentage of vacuoles containing variable numbers of parasites was determined to examine the replication phenotype.
Virulence assays
Tachyzoites of the parental (RHΔku80-TaTi) and Δtgcds1r strains were pretreated with or without ATc (1 μm) for 3 passages (6 days in cultures). ATc treatment of C57BL/6J mice (female, 6–8 weeks old) was initiated 2 days before inoculation by supplying the drug in drinking water (0.2 mg/ml) and continued for 2 weeks during infection. Animals were inoculated with fresh extracellular parasites (103) via an intraperitoneal route and monitored for mortality and morbidity over a period of 4 weeks. For re-infection, animals immunized with the Δtgcds1r strains (ATc-treated) were challenged with tachyzoites of the parental strain (103) and monitored for additional 28 days.
Statistics
All data are shown as the mean with S.E. from three to six independent assays using a representative parasite clone. Statistical analyses were performed by GraphPad Prism program (v5, Prism Software Inc., La Jolla, CA). Significance was tested by analysis of variance (between different strains) or unpaired two-tailed Student's t test with equal variances (between treated and untreated conditions). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Study approval
All animal experiments were performed in strict compliance with the Animal Protection Law of the People's Republic of China (Chapter 6; Legal Protection of Laboratory Animals) and the Institutional Animal Care and Use Committee of China Agricultural University (Beijing). Assays were approved by Beijing administration committee of laboratory animals (11401300021924).
Author contributions
N. G. and P. K. conceived and designed the study. P. K. performed the experiments. C.- M. U. and D. L. M. Z. helped with the phenotype and localization assays. Q. Y. and X. S. performed the animal assays. N. G., P. K., and J. F. B. analyzed the data. X. S., J. B. H, J. F. B., and N. G. contributed reagents or analysis tools. P. K. and N. G. wrote the paper. All authors approved the manuscript.
Supplementary Material
Acknowledgments
We thank Grit Meusel (Humboldt University Berlin) for technical assistance and Jeroen W. A. Jansen (Utrecht University) for aiding the lipidomic studies.
This work was supported by German Research Foundation (DFG) Grants GU1100/3-1 and GU1100/4-1 and a Heisenberg program award (to N. G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2 and Fig. S1.
- PtdCho
- phosphatidylcholine
- PtdEtn
- phosphatidylethanolamine
- PtdThr
- phosphatidylthreonine
- PtdIns
- phosphatidylinositol
- PtdSer
- phosphatidylserine
- PtdGro
- phosphatidylglycerol
- PtdOH
- phosphatidic acid
- ER
- endoplasmic reticulum
- DAG
- diacylglycerol
- CDP
- cytidine diphosphate
- CDS
- CDP-DAG synthase
- DHFR-TS
- dihydrofolate reductase-thymidylate synthase
- S.C.
- selection cassette
- ATc
- anhydrotetracycline
- CAT
- chloramphenicol acetyltransferase
- HFF
- human foreskin fibroblast
- PIS
- PtdIns synthase
- PtdGro-P
- PtdGro phosphate
- PGPS
- PtdGro-P synthase
- m.o.i.
- multiplicity of infection
- COS
- crossover sequence
- HXGPRT
- hypoxanthine-xanthine-guanine phosphoribosyltransferase
- Tg
- T. gondii
- gDNA
- genomic DNA
- UPRT
- uracil phosphoribosyltransferase.
References
- 1. Lyons R. E., McLeod R., and Roberts C. W. (2002) Toxoplasma gondii tachyzoite-bradyzoite interconversion. Trends Parasitol. 18, 198–201 [DOI] [PubMed] [Google Scholar]
- 2. Gupta N., Zahn M. M., Coppens I., Joiner K. A., and Voelker D. R. (2005) Selective disruption of phosphatidylcholine metabolism of the intracellular parasite Toxoplasma gondii arrests its growth. J. Biol. Chem. 280, 16345–16353 [DOI] [PubMed] [Google Scholar]
- 3. Welti R., Mui E., Sparks A., Wernimont S., Isaac G., Kirisits M., Roth M., Roberts C. W., Botté C., Maréchal E., and McLeod R. (2007) Lipidomic analysis of Toxoplasma gondii reveals unusual polar lipids. Biochemistry 46, 13882–13890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arroyo-Olarte R. D., Brouwers J. F., Kuchipudi A., Helms J. B., Biswas A., Dunay I. R., Lucius R., and Gupta N. (2015) Phosphatidylthreonine and lipid-mediated control of parasite virulence. PLos Biol. 13, e1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gupta N., Hartmann A., Lucius R., and Voelker D. R. (2012) The obligate intracellular parasite Toxoplasma gondii secretes a soluble phosphatidylserine decarboxylase. J. Biol. Chem. 287, 22938–22947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sampels V., Hartmann A., Dietrich I., Coppens I., Sheiner L., Striepen B., Herrmann A., Lucius R., and Gupta N. (2012) Conditional mutagenesis of a novel choline kinase demonstrates plasticity of phosphatidylcholine biogenesis and gene expression in Toxoplasma gondii. J. Biol. Chem. 287, 16289–16299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hartmann A., Hellmund M., Lucius R., Voelker D. R., and Gupta N. (2014) Phosphatidylethanolamine synthesis in the parasite mitochondrion is required for efficient growth but dispensable for survival of Toxoplasma gondii. J. Biol. Chem. 289, 6809–6824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mazumdar J., H. Wilson E., Masek K., A. Hunter C., and Striepen B. (2006) Apicoplast fatty acid synthesis is essential for organelle biogenesis and parasite survival in Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 103, 13192–13197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramakrishnan S., Docampo M. D., Macrae J. I., Pujol F. M., Brooks C. F., van Dooren G. G., Hiltunen J. K., Kastaniotis A. J., McConville M. J., and Striepen B. (2012) Apicoplast and endoplasmic reticulum cooperate in fatty acid biosynthesis in apicomplexan parasite Toxoplasma gondii. J. Biol. Chem. 287, 4957–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nitzsche R., Zagoriy V., Lucius R., and Gupta N. (2016) Metabolic cooperation of glucose and glutamine is essential for the lytic cycle of obligate intracellular parasite Toxoplasma gondii. J. Biol. Chem. 291, 126–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Janouskovec J., Horák A., Oborník M., Lukes J., and Keeling P. J. (2010) A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc. Natl. Acad. Sci. U.S.A. 107, 10949–10954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Athenstaedt K., and Daum G. (1999) Phosphatidic acid, a key intermediate in lipid metabolism. Eur. J. Biochem. 266, 1–16 [DOI] [PubMed] [Google Scholar]
- 13. Vance J. E., and Vance D. E. (2004) Phospholipid biosynthesis in mammalian cells. Biochem. Cell Biol. 82, 113–128 [DOI] [PubMed] [Google Scholar]
- 14. van Dooren G. G., and Striepen B. (2013) The algal past and parasite present of the apicoplast. Annu. Rev. Microbiol. 67, 271–289 [DOI] [PubMed] [Google Scholar]
- 15. Agrawal S., van Dooren G. G., Beatty W. L., and Striepen B. (2009) Genetic evidence that an endosymbiont-derived endoplasmic reticulum-associated protein degradation (ERAD) system functions in import of apicoplast proteins. J. Biol. Chem. 284, 33683–33691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vollmer M., Thomsen N., Wiek S., and Seeber F. (2001) Apicomplexan parasites possess distinct nuclear-encoded, but apicoplast-localized, plant-type ferredoxin-NADP+ reductase and ferredoxin. J. Biol. Chem. 276, 5483–5490 [DOI] [PubMed] [Google Scholar]
- 17. Meissner M., Schlüter D., and Soldati D. (2002) Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science 298, 837–840 [DOI] [PubMed] [Google Scholar]
- 18. Shen H., Heacock P. N., Clancey C. J., and Dowhan W. (1996) The CDS1 gene encoding CDP-diacylglycerol synthase in Saccharomyces cerevisiae is essential for cell growth. J. Biol. Chem. 271, 789–795 [DOI] [PubMed] [Google Scholar]
- 19. Zhou Y., Peisker H., Weth A., Baumgartner W., Dörmann P., and Frentzen M. (2013) Extraplastidial cytidine diphosphate diacylglycerol synthase activity is required for vegetative development in Arabidopsis thaliana. Plant J. 75, 867–879 [DOI] [PubMed] [Google Scholar]
- 20. Halford S., Dulai K. S., Daw S. C., Fitzgibbon J., and Hunt D. M. (1998) Isolation and chromosomal localization of two human CDP-diacylglycerol synthase (CDS) genes. Genomics 54, 140–144 [DOI] [PubMed] [Google Scholar]
- 21. Inglis-Broadgate S. L., Ocaka L., Banerjee R., Gaasenbeek M., Chapple J. P., Cheetham M. E., Clark B. J., Hunt D. M., and Halford S. (2005) Isolation and characterization of murine Cds (CDP-diacylglycerol synthase) 1 and 2. Gene 356, 19–31 [DOI] [PubMed] [Google Scholar]
- 22. Martin D., Gannoun-Zaki L., Bonnefoy S., Eldin P., Wengelnik K., and Vial H. (2000) Characterization of Plasmodium falciparum CDP-diacylglycerol synthase, a proteolytically cleaved enzyme. Mol. Biochem. Parasitol. 110, 93–105 [DOI] [PubMed] [Google Scholar]
- 23. Shastri S., Zeeman A. M., Berry L., Verburgh R. J., Braun-Breton C., Thomas A. W., Gannoun-Zaki L., Kocken C. H., and Vial H. J. (2010) Plasmodium CDP-DAG synthase: an atypical gene with an essential N-terminal extension. Int. J. Parasitol. 40, 1257–1268 [DOI] [PubMed] [Google Scholar]
- 24. Lilley A. C., Major L., Young S., Stark M. J., and Smith T. K. (2014) The essential roles of cytidine diphosphate-diacylglycerol synthase in bloodstream form Trypanosoma brucei. Mol. Microbiol. 92, 453–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. D'Souza K., Kim Y. J., Balla T., and Epand R. M. (2014) Distinct properties of the two isoforms of CDP-diacylglycerol synthase. Biochemistry 53, 7358–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tamura Y., Harada Y., Nishikawa S., Yamano K., Kamiya M., Shiota T., Kuroda T., Kuge O., Sesaki H., Imai K., Tomii K., and Endo T. (2013) Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 17, 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McFadden G. I., Reith M. E., Munholland J., and Lang-Unnasch N. (1996) Plastid in human parasites. Nature 381, 482–482 [DOI] [PubMed] [Google Scholar]
- 28. Wilson R. J., Denny P. W., Preiser P. R., Rangachari K., Roberts K., Roy A., Whyte A., Strath M., Moore D. J., Moore P. W., and Williamson D. H. (1996) Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J. Mol. Biol. 261, 155–172 [DOI] [PubMed] [Google Scholar]
- 29. Köhler S., Delwiche C. F., Denny P. W., Tilney L. G., Webster P., Wilson R. J., Palmer J. D., and Roos D. S. (1997) A plastid of probable green algal origin in Apicomplexan parasites. Science 275, 1485–1489 [DOI] [PubMed] [Google Scholar]
- 30. Sheiner L., Demerly J. L., Poulsen N., Beatty W. L., Lucas O., Behnke M. S., White M. W., and Striepen B. (2011) A systematic screen to discover and analyze apicoplast proteins identifies a conserved and essential protein import factor. PLoS Pathog. 7, e1002392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waller R. F., Keeling P. J., Donald R. G., Striepen B., Handman E., Lang-Unnasch N., Cowman A. F., Besra G. S., Roos D. S., and McFadden G. I. (1998) Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 95, 12352–12357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cilingir G., Broschat S. L., and Lau A. O. (2012) ApicoAP: the first computational model for identifying apicoplast-targeted proteins in multiple species of Apicomplexa. PLoS ONE 7, e36598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DeRocher A., Gilbert B., Feagin J. E., and Parsons M. (2005) Dissection of brefeldin A-sensitive and -insensitive steps in apicoplast protein targeting. J. Cell Sci. 118, 565–574 [DOI] [PubMed] [Google Scholar]
- 34. Tonkin C. J., Struck N. S., Mullin K. A., Stimmler L. M., and McFadden G. I. (2006) Evidence for Golgi-independent transport from the early secretory pathway to the plastid in malaria parasites. Mol. Microbiol. 61, 614–630 [DOI] [PubMed] [Google Scholar]
- 35. Bouchut A., Geiger J. A., DeRocher A. E., and Parsons M. (2014) Vesicles bearing Toxoplasma apicoplast membrane proteins persist following loss of the relict plastid or Golgi body disruption. PLoS ONE 9, e112096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heiny S. R., Pautz S., Recker M., and Przyborski J. M. (2014) Protein traffic to the Plasmodium falciparum apicoplast: evidence for a sorting branch point at the Golgi. Traffic 15, 1290–1304 [DOI] [PubMed] [Google Scholar]
- 37. Stewart R. J., Ferguson D. J., Whitehead L., Bradin C. H., Wu H. J., and Tonkin C. J. (2016) Phosphorylation of SNAP is required for secretory organelle biogenesis in Toxoplasma gondii. Traffic 17, 102–116 [DOI] [PubMed] [Google Scholar]
- 38. Tawk L., Dubremetz J. F., Montcourrier P., Chicanne G., Merezegue F., Richard V., Payrastre B., Meissner M., Vial H. J., Roy C., Wengelnik K., and Lebrun M. (2011) Phosphatidylinositol 3-monophosphate is involved in Toxoplasma apicoplast biogenesis. PLoS Pathog. 7, e1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Daher W., Morlon-Guyot J., Sheiner L., Lentini G., Berry L., Tawk L., Dubremetz J. F., Wengelnik K., Striepen B., and Lebrun M. (2015) Lipid kinases are essential for apicoplast homeostasis in Toxoplasma gondii. Cell Microbiol. 17, 559–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wichroski M. J., and Ward G. E. (2003) Biosynthesis of glycosylphosphatidylinositol is essential to the survival of the protozoan parasite Toxoplasma gondii. Eukaryot. Cell 2, 1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Debierre-Grockiego F., and Schwarz R. T. (2010) Immunological reactions in response to apicomplexan glycosylphosphatidylinositols. Glycobiology 20, 801–811 [DOI] [PubMed] [Google Scholar]
- 42. Serricchio M., and Bütikofer P. (2012) An essential bacterial-type cardiolipin synthase mediates cardiolipin formation in a eukaryote. Proc. Natl. Acad. Sci. U.S.A. 109, E954–E961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Serricchio M., and Bütikofer P. (2013) Phosphatidylglycerophosphate synthase associates with a mitochondrial inner membrane complex and is essential for growth of Trypanosoma brucei. Mol. Microbiol. 87, 569–579 [DOI] [PubMed] [Google Scholar]
- 44. Amiar S., MacRae J. I., Callahan D. L., Dubois D., van Dooren G. G., Shears M. J., Cesbron-Delauw M. F., Maréchal E., McConville M. J., McFadden G. I., Yamaryo-Botté Y., and Botté C. Y. (2016) Apicoplast-localized lysophosphatidic acid precursor assembly is required for bulk phospholipid synthesis in Toxoplasma gondii and relies on an algal/plant-like glycerol-3-phosphate acyltransferase. PLoS Pathog. 12, e1005765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Holthuis J. C., and Levine T. P. (2005) Lipid traffic: floppy drives and a superhighway. Nat. Rev. Mol. Cell Biol. 6, 209–220 [DOI] [PubMed] [Google Scholar]
- 46. Levine T., and Loewen C. (2006) Inter-organelle membrane contact sites: through a glass, darkly. Curr. Opin. Cell Biol. 18, 371–378 [DOI] [PubMed] [Google Scholar]
- 47. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Dooren G. G., Marti M., Tonkin C. J., Stimmler L. M., Cowman A. F., and McFadden G. I. (2005) Development of the endoplasmic reticulum, mitochondrion, and apicoplast during the asexual life cycle of Plasmodium falciparum. Mol. Microbiol. 57, 405–419 [DOI] [PubMed] [Google Scholar]
- 49. Tomova C., Humbel B. M., Geerts W. J., Entzeroth R., Holthuis J. C., and Verkleij A. J. (2009) Membrane contact sites between apicoplast and ER in Toxoplasma gondii revealed by electron tomography. Traffic 10, 1471–1480 [DOI] [PubMed] [Google Scholar]
- 50. Huynh M. H., and Carruthers V. B. (2009) Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Donald R. G., Carter D., Ullman B., and Roos D. S. (1996) Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J. Biol. Chem. 271, 14010–14019 [DOI] [PubMed] [Google Scholar]
- 52. Donald R. G., and Roos D. S. (1995) Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 92, 5749–5753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Donald R. G., and Roos D. S. (1993) Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc. Natl. Acad. Sci. U.S.A. 90, 11703–11707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim K., Soldati D., and Boothroyd J. C. (1993) Gene replacement in Toxoplasma gondii with chloramphenicol acetyltransferase as selectable marker. Science 262, 911–914 [DOI] [PubMed] [Google Scholar]
- 55. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 56. Brouwers J. F., Aalberts M., Jansen J. W., van Niel G., Wauben M. H., Stout T. A., Helms J. B., and Stoorvogel W. (2013) Distinct lipid compositions of two types of human prostasomes. Proteomics 13, 1660–1666 [DOI] [PubMed] [Google Scholar]
- 57. Smith C. A., Want E. J., O'Maille G., Abagyan R., and Siuzdak G. (2006) XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.