

Abstract
Inflammatory signals induce feedback and feedforward systems that provide temporal control. Although glucocorticoids can repress inflammatory gene expression, glucocorticoid receptor recruitment increases expression of negative feedback and feedforward regulators, including the phosphatase, DUSP1, the ubiquitin-modifying enzyme, TNFAIP3, or the mRNA-destabilizing protein, ZFP36. Moreover, glucocorticoid receptor cooperativity with factors, including nuclear factor-κB (NF-κB), may enhance regulator expression to promote repression. Conversely, MAPKs, which are inhibited by glucocorticoids, provide feedforward control to limit expression of the transcription factor IRF1, and the chemokine, CXCL10. We propose that modulation of feedback and feedforward control can determine repression or resistance of inflammatory gene expression toglucocorticoid.
Keywords: gene expression, gene regulation, glucocorticoid, glucocorticoid receptor, inflammation
Introduction
Acting on the glucocorticoid receptor (GR2: NR3C1), glucocorticoids reduce the expression of many genes induced in inflammatory conditions in vivo or by pro-inflammatory stimuli in vitro (1, 2). Although glucocorticoids are effective in the treatment of chronic inflammation, including mild/moderate asthma, not all inflammatory conditions respond well to glucocorticoid therapies (1, 2). In severe neutrophilic asthma, asthmatics who smoke, chronic obstructive pulmonary disease (COPD), and during exacerbations of asthma and chronic obstructive pulmonary disease, glucocorticoids may show reduced efficacy (3). Thus, pro-inflammatory cytokines, growth factors, viruses, double-stranded RNA, bacterial products, and oxidative stress reduce GR function, and terms such as glucocorticoid “resistance” or “insensitivity” are used to describe this clinical problem (4, 5). As such patients are major healthcare utilizers, solving the problem of glucocorticoid resistance would be of immense societal benefit. For example, pharmacological targeting of signaling pathways, such as mitogen-activated protein kinase (MAPK) or phosphatidylinositol-3-kinase (PI3K) pathways, which are implicated in GR resistance, represents a logical approach to restore sensitivity (3–5).
Despite many mRNAs that are induced by pro-inflammatory stimuli being profoundly repressed by glucocorticoids, many others show partial repression, no repression, or even enhancement by glucocorticoids (6–11). Importantly, it is pertinent to consider that genes induced by pro-inflammatory stimuli, or during inflammation, are not necessarily “pro-inflammatory,” but may exert pro- or anti-inflammatory, or even mixed functional effects. Thus, differential responsiveness to glucocorticoids may be desirable. Nevertheless, although many inflammation-induced mRNAs are resistant to glucocorticoid-mediated repression, this is not a generalized defect in glucocorticoid signaling as repression and glucocorticoid-mediated gene induction occur in the same system (Fig. 1). This fits with the notion that glucocorticoids selectively regulate gene expression, as exemplified by observations that glucocorticoids spare, and even augment, innate immune responses (12). Indeed, the ability of glucocorticoids to induce and/or enhance the expression of inflammatory response genes, including cytokines, chemokines, receptors, and signaling components, is widely reported (6, 7, 9–11, 13–15). For example, in human volunteers, budesonide inhalation not only induced the expression of numerous anti-inflammatory genes, but also enhanced the expression of genes apparently involved in pro-inflammatory, pro-proliferative, and migratory responses (16). Thus, glucocorticoids are not simply anti-inflammatory, but exert a spectrum of effects that may include promoting pro-inflammatory pathways (12).
Figure 1.
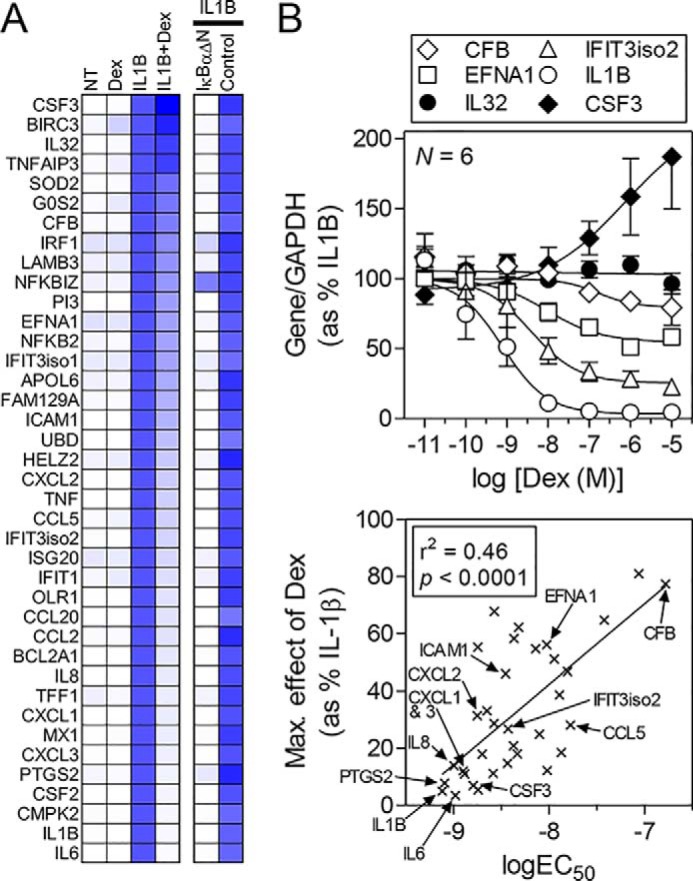
Differential effects of dexamethasone on IL1B-induced mRNAs. Data are derived from King et al. (11) where full details can be found. A, expression of 39 of the most highly IL1B-induced mRNAs in A549 cells was examined by qPCR following no treatment (NT) or treatment with dexamethasone (1 μm) (Dex), IL1B (1 ng/ml), or IL1B + Dex for 6 h. Data for each mRNA were normalized to GAPDH and are expressed as a percentage of IL1B treated, which is set to 100% (blue), and are presented as a heat map (white = 0%). IL1B-induced mRNAs are ranked according to the effect of dexamethasone with IL6 being the most repressed and CSF3 (G-CSF) being enhanced. The effects of NF-κB inhibition, using adenoviral overexpression of the dominant inhibitor, IκBαΔN, or a control virus (Control), are also expressed as a percentage of IL1B-treated and are shown as a heat map. B, A549 cells were stimulated with IL1B (1 ng/ml) in the absence or presence of the indicated concentrations of dexamethasone prior to harvesting at 6 h for qPCR analysis of the 39 mRNAs in A. Data (n = 6) for each mRNA (indicated by an x) were plotted as a percentage of IL1B treatment. The most potently repressed mRNA (IL1B) along with mRNAs showing intermediate (IFIT3 isoform 2, EFNA1, CFB) and no (IL32) repression or enhancement (CSF3) by dexamethasone are shown (upper panel). The maximal effect (Max. effect) of dexamethasone (i.e. at 1 μm) and the EC50 were calculated for each mRNA that showed significant repression. These are plotted (lower panel) to show the correlation between potency and repression by dexamethasone. Error bars indicate ± S.E.
Repression versus a failure to repress inflammatory gene expression
There are arguably three core mechanisms by which glucocorticoids repress inflammatory gene expression (17, 18). 1) Activated GR is recruited to cis-operating DNA sites, for example simple glucocorticoid response elements (GREs), and, acting in trans, GR directly induces the expression of genes that reduce inflammatory gene expression (17, 19). This is repression occurring via GR trans-activation. 2) Without itself binding to DNA, GR interacts with, or tethers to, the inflammatory transcription factors, such as nuclear factor (NF)-κB, that are responsible for the induction of inflammatory mRNAs (18). Commonly referred to as trans-repression, this dampens transcriptional activity at targeted promoters and may involve GR SUMOylation along with nuclear receptor co-repressors and histone deacetylase (HDAC) recruitment (20, 21). 3) GR may bind directly to non-consensus cis-acting DNA elements, or negative GREs, to elicit transcriptional repression of the associated gene (22). Again, referred to as trans-repression, GR binding occurs with a lower affinity when compared with classical GRE sites, and repression may also involve nuclear receptor co-repressor and HDAC recruitment (23, 24). Although a detailed consideration of the mechanisms underlying the repression of gene expression by GR is not the current purpose, it is salient that repression is gene-specific and can involve multiple mechanisms (17). In human A549 pulmonary epithelial cells, 39 IL1B-induced mRNAs were largely NF-κB-dependent, yet varied from profound repression to enhancement by dexamethasone (11) (Fig. 1A). Indeed, the potency of repression correlated with the efficacy of repression (Fig. 1B), with high potency/high efficacy repression of acute phase genes being attributed to the involvement of GR-dependent gene induction (11). Likewise, transcriptomic studies not only show variable glucocorticoid-mediated repression, but confirm that many inflammatory genes do genuinely escape repression (10, 11, 13, 14). Furthermore, such studies also highlight glucocorticoid cooperation with inflammatory stimuli to enhance gene expression. Indeed, whole genome chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) data reveal that GR-binding sites commonly co-localize with sites for inflammatory transcription factors, including activator protein (AP)-1 and NF-κB (13, 15, 25, 26). Similarly, CCAAT/enhancer-binding proteins or signal transducers and activators of transcription (STATs) also interact with GR to promote transcriptional activation (8, 27, 28). Thus, the key inflammatory transcription factors, which up-regulate cytokines and other inflammatory genes that are repressed by glucocorticoid, also interact productively with GR to enhance transcription at a subset of co-regulated targets.
Glucocorticoids maintain and enhance feedback control
MAPK cascades are central to inflammatory gene expression and are regulated by dual-specificity phosphatases (DUSPs), such as DUSP1, which targets the p38, c-Jun N-terminal kinase (JNK), and extracellular-regulated kinase (ERK) pathways (29). In the context of inflammatory stimuli, DUSP1 expression is rapidly induced via MAPK-dependent mechanisms to provide feedback inhibition and limit inflammatory gene expression (29) (Fig. 2A). However, DUSP1 expression is also increased by glucocorticoids and, with a pro-inflammatory stimulus plus glucocorticoid, feedback inhibition of MAPKs is enhanced (30, 31) (Fig. 2A). This may reduce transcriptional activation, for example of AP-1 and NF-κB, and prevent mRNA stabilization and/or translation of target genes (30, 32–35). Thus, DUSP1 contributes, often transiently and redundantly, to the repression of inflammatory gene expression by glucocorticoids (31, 36, 37). However, the relationship between pro-inflammatory stimuli and glucocorticoids with respect to inducing DUSP1 expression is central to this outcome. The inductive effects of pro-inflammatory stimuli and the glucocorticoid summate, or synergize, to increase DUSP1 expression (35, 38–40). Mechanistically, although transcriptional induction of DUSP1 involves GR recruitment (41–45), further up-regulation of DUSP1 expression remains achievable by pro-inflammatory stimuli.
Figure 2.

Regulatory loops controlling inflammatory gene expression. A, schematic showing activation of MAPK pathways and NF-κB by IL1B or TNF leading to the expression of inflammatory genes. MAPKs induce the expression of the phosphatase, DUSP1, which provides feedback control to switch off MAPK activity. NF-κB binds κB sites in promoters of target genes. This activates transcription of NFKBIA, TNFAIP3, and IRAK3 to increase their expression and leads to feedback inhibition of NF-κB or IL1B/TNF signaling. Expression of DUSP1, NFKBIA, TNFAIP3, and IRAK3 can also be enhanced by glucocorticoids (GC). B, type I coherent and incoherent feedforward loops are depicted. In the type I coherent feedforward loop (panel i), X positively regulates Y, and Z is positively regulated by both X and Y. In the type I incoherent feedforward loop (panel ii), X positively regulates both Y and Z, but Y negatively regulates Z. C, schematic showing how feedback and feedforward regulation may interplay to regulate AU-rich element (ARE)-containing inflammatory mRNAs. Panel i, pro-inflammatory stimuli, here IL1B, activate MAPK pathways, leading to the expression of ARE-containing mRNAs, such as TNF. MAPK activation not only also induces expression of the feedback regulator, DUSP1, but also promotes expression of ZFP36. ZFP36 is a feedforward regulator that leads to mRNA destabilization of ARE-containing mRNAs, such as TNF. Thus, the MAPK-dependent induction of ZFP36 leads to repression of ARE-containing mRNAs and constitutes a classic type I incoherent feedforward loop. Note that expression of ARE-containing mRNAs and ZFP36 is also likely to involve NF-κB, and this is not depicted. Panel ii, Following loss, or silencing, of DUSP1, MAPK activity is enhanced and leads to increased expression of downstream genes. However, expression of ZFP36 is also enhanced, and this acts to reduce expression of ARE-containing mRNAs, such as TNF. Panel iii, ZFP36 expression is up-regulated by glucocorticoids alone, but ZFP36 expression induced by the inflammatory stimulus is reduced by glucocorticoid, in part due to reduced MAPK activity following the induction of DUSP1. Although these effects may combine to promote expression of the hypo-phosphorylated and more active, mRNA-destabilizing form of ZFP36, silencing of both DUSP1 and ZFP36 showed little effect on the repression of TNF by glucocorticoid. Additional, glucocorticoid-induced effector processes are therefore likely to play additional repressive roles.
Similarly, the inflammatory gene, TNFAIP3, or A20, has dual ubiquitin-modifying activities that: 1) remove Lys-63-linked polyubiquitin chains from the upstream receptor-interacting protein kinase, RIPK1, in the TNF-induced NF-κB activation pathway; and 2) by conjugating Lys-48 ubiquitin chains, target RIPK1 for proteasomal degradation (46, 47). Because Lys-63 polyubiquitination of RIPK1 is necessary for IκB kinase (IKK) activation, TNFAIP3 profoundly inhibits signaling to NF-κB. As TNFAIP3 expression is NF-κB-dependent and induced by Toll-like receptors (TLRs) or cytokines, such as IL1B (Fig. 1A) or TNF, this provides robust feedback control (Fig. 2A). However, unlike glucocorticoid-repressed, NF-κB-dependent mRNAs, such as IL8 or CSF2 (GM-CSF), TNFAIP3 expression, as induced by pro-inflammatory stimuli, is maintained or enhanced with the addition of glucocorticoid (11, 48, 49) (Fig. 1). This may be explained by the fact that glucocorticoids independently induce TNFAIP3 expression (11, 48). Indeed, in bronchial airway epithelial BEAS-2B cells, GR binds and activates transcription from an intronic enhancer within the TNFAIP3 gene (48) (Fig. 3A, panel i). This region also recruits the NF-κB subunit, RELA, and in combination with glucocorticoid, NF-κB and GR may cooperate to enhance transcription. Indeed NF-κB/GR transcriptional synergy has been reported previously (50), and provides a mechanism by which glucocorticoids may enhance, or maintain, inflammatory gene expression.
Figure 3.

GR and NF-κB (RELA) recruitment to inflammatory gene loci. Data from a ChIP-seq analysis by Kadiyala et al. (15) are shown. BEAS-2B cells were treated for 1 h with Dex (1 μm), TNF (20 ng/ml), or Dex plus TNF for 1 h prior to ChIP-seq analysis. Occupancy of GR and RELA at genomic loci in the vicinity of six genes is shown. A, inflammatory feedback control genes, where GR and RELA may cooperate to enhance or maintain the expression of TNFAIP3 (panel i); NFKBIA (panel ii); and IRAK3 (panel iii). In each case, GR and RELA are recruited to the gene loci following dexamethasone or TNF treatment, respectively. In the context of dexamethasone plus TNF, both GR and RELA are both recruited to at least one DNA region in common (red arrow). Although overall GR occupancy at each gene locus was largely unaffected by TNF, site-specific differences are apparent. Conversely, co-treatment differentially affected RELA occupancy, which was increased at an intronic region for TNFAIP3, slightly decreased on NFKBIA, and markedly increased at the IRAK3 promoter. B, GR and RELA co-recruitment to the SOD2 (panel i), ZCH12A (panel ii), and IL32 (panel iii) gene loci. SOD2 and IL32 are induced by inflammatory stimuli, and here TNF induces RELA binding to each gene locus. In the additional presence of dexamethasone, RELA binding is slightly reduced (red arrow). However, although GR occupancy at this same region was not readily apparent with dexamethasone alone, with TNF plus dexamethasone, GR recruitment is induced (red arrow). GR occupancy at regions that either did not show GR binding, or only showed weak GR binding, in the presence of dexamethasone alone were also enhanced for both SOD2 and IL32 with dexamethasone plus TNF (black arrows). These regions did not show material RELA occupancy. With ZC3H12A, RELA occupancy was induced to multiple intronic regions by TNF. Although dexamethasone reduced RELA occupancy, GR was recruited to these same regions with TNF plus dexamethasone (red arrows). Binding of RELA and GR at a 5′ region was markedly enhanced by TNF plus dexamethasone (black arrows), whereas neither TNF nor dexamethasone alone showed any marked effect on occupancy.
This concept of feedback control in inflammation and enhancement, or maintenance, by glucocorticoid is likely to be relevant to multiple regulatory genes. The cytoplasmic inhibitor of NF-κB, inhibitor of κBα (NFKBIA), is induced by pro-inflammatory cytokines via NF-κB sites in the NFKBIA promoter (51). This allows rapid NFKBIA resynthesis following signal-induced loss and limits NF-κB activity (Fig. 2A). Like DUSP1 and TNFAIP3, NFKBIA expression is also induced by glucocorticoids in multiple cell types, and this transcriptional drive may occur in the presence of inflammatory stimuli (52). In BEAS-2B cells, TNF-induced NFKBIA expression is enhanced by glucocorticoid and GR binds the promoter and genic regions of NFKBIA along with RELA/NF-κB (Fig. 3A, panel ii) (15, 48). Similarly, in A549 cells, glucocorticoids promote GR binding to the NFKBIA gene and IL1B-induced NFKBIA expression is largely unaffected by dexamethasone, such that resynthesis of NFKBIA is maintained (53, 54). IRAK3, or IRAK-M, is a further feedback regulator of NF-κB and MAPKs and is up-regulated by TLRs and the pro-inflammatory cytokines, IL1B and TNF (55) (Fig. 2A). Being kinase-defective, IRAK3 acts as a dominant-negative inhibitor of the upstream kinases, IRAK1/4, that are central to TLR and IL1-like receptor signaling. However, IRAK3 is glucocorticoid-induced and, with pro-inflammatory stimuli, expression is synergistically enhanced via a GR- and NF-κB-dependent mechanism that is consistent with the recruitment of both factors to the IRAK3 promoter (56) (Fig. 3A, panel iii).
Feedforward control and regulation by glucocorticoids
A common regulatory circuit in the control of signal transduction and gene expression is the feedforward loop (57). Such motifs are widespread in biological systems and apply to transcriptional regulation by nuclear hormone receptors (58). These loops are characterized by a three-node structure (X, Y, and Z) in which regulator X controls both Y and Z, and Y also controls Z (Fig. 2B). In the simplest configuration, X and Y are positive regulators, for example, transcription factors, and the unit is described as a coherent feedforward loop (Fig. 2B, panel i). However, if Y were to negatively regulate Z, the effect of the two arms (i.e. X on Z, and X via Y on Z) would be opposed and the circuit would be described as incoherent (Fig. 2B, panel ii). Although eight different configurations, dependent on the sign (+/−) for each interaction, are possible, we now focus on type I incoherent feedforward control (Fig. 2B, panel ii). Thus, X leads to activation of Y and Z. However, increasing levels of the negative regulator, Y, progressively switch off Z and produces pulsed, or spike-like, dynamics for Z (57).
Incoherent feedforward control by ZFP36
For the current illustration, we condense the MAPK cascades to node X, and Z is represented by the mRNA expression of TNF (Fig. 2C). Like many inflammatory mRNAs, TNF expression involves transcriptional and, by virtue of multiple AU-rich elements (AREs) in the 3′-UTR, post-transcriptional control process that are regulated by MAPKs (59). Typically, AREs occur in the 3′-UTRs of cytokines, chemokines, and other inflammatory genes and bind RNA-binding proteins to regulate mRNA stability and translation (59). Although many RNA-binding proteins exist and modulate mRNA stability and/or translation, the zinc finger protein, ZFP36, also known as tristetraprolin, is rapidly induced by pro-inflammatory stimuli and is critical for stimulus-dependent down-regulation of ARE-containing mRNAs (60). ZFP36 induction occurs in many cells, including A549 epithelial and primary human airway smooth muscle cells (61, 62). This requires p38 MAPK to constitute an incoherent feedforward loop with ZFP36 as a negative regulator of ARE-containing mRNAs, such as TNF (60, 63) (Fig. 2C, panel i). Thus, in A549 cells, DUSP1 silencing transiently hyper-activates IL1B-induced MAPKs and correspondingly increases ZFP36 expression (63) (Fig. 2C, panel ii). Similar effects occur in human and mouse macrophages, and there is enhanced ZFP36 expression in LPS-treated Dusp1−/− cells (64–66). However, although DUSP1 silencing transiently increased expression of inflammatory mRNAs induced by IL1B, at longer times (6 h after IL1B), the expression of many of these same mRNAs was reduced relative to control (31). Enhanced ZFP36-dependent (incoherent) feedforward control, consequent to DUSP1 silencing, was shown to contribute to this delayed loss of TNF mRNA (63). Thus, inflammatory mRNAs subject to control by MAPKs and ZFP36-dependent feedforward regulation reveal complex kinetics due to interplay between feedback and incoherent feedforward control.
Newly synthesized ZFP36 is rapidly phosphorylated by MAPKAPK2 to promote protection from proteolytic degradation and reduce, or prevent, ARE-dependent destabilizing activity (60). Thus, in LPS-treated Dusp−/− mice, there was enhanced MAPK activation and increased inflammatory gene expression despite elevated ZFP36 expression (66). However, these increases were blocked by a dominant Zfp36 mutation (Zfp36aa) in which the two main MAPKAPK2 phosphorylation sites (Ser-53 and Ser-178) were modified from serine to alanine (66). Although this effect may be consequent to the dominant ARE-dependent destabilizing effect of the Zfp36aa mutation, it is suggested that enhanced ZFP36 phosphorylation was central to the increases in ARE-containing mRNA expression in the Dusp1−/− mice (66). Thus, ZFP36 was present in the phosphorylated form where destabilization activity is low and ARE-containing mRNA translation may actively occur (67). Although a switch to unphosphorylated, and therefore mRNA degradation-active, ZFP36 is driven by the serine/threonine protein phosphatase, PP2A (68, 69), this appears to occur later after stimulation, when p38 activity is reduced (60, 67). This delay in ZFP36 activation, combined with increased total ZFP36, may therefore explain the enhanced loss of ARE-containing transcripts that occurred at longer IL1B-treatment times after DUSP1 silencing in A549 cells (31, 60, 63).
The above data produce various complications in the context of active glucocorticoid signaling. The induction of ZFP36 expression by pro-inflammatory stimuli requires MAPKs and is decreased via the ability of glucocorticoids to reduce MAPK activity (61, 70, 71) (Fig. 2C). Thus, glucocorticoids reduce pro-inflammatory stimulus-induced expression of a protein that destabilizes those inflammatory mRNAs, TNF, CSF2 (GM-CSF), PTGS2, and others, that are in fact repressed by the glucocorticoid. Conversely, in both in vivo inhaled glucocorticoid and cell culture, glucocorticoids alone up-regulate ZFP36 expression (16, 72–74). This is consistent with GR recruitment to the ZFP36 gene (75). Therefore, although glucocorticoids inhibit MAPK activity and reduce inflammation-induced ZFP36, the presence of active GR at the ZFP36 gene locus may act to counteract this by helping to maintain ZFP36 expression. Furthermore, in the presence of pro-inflammatory stimulus plus glucocorticoid, the glucocorticoid-mediated loss of p38 MAPK activity may result in elevated levels of unphosphorylated ZFP36 that displays the greatest ARE-destabilizing activity (60). Indeed, although this effect is described in airway smooth muscle cells (71), a detailed analysis is required to confirm functionality. Nevertheless, simultaneous silencing of DUSP1 and ZFP36 in A549 cells revealed little effect on the dexamethasone-dependent repression of inflammatory mRNAs and suggests that other glucocorticoid-induced, or activated, effectors are important for repression (63). Furthermore, as feedback regulators, including DUSP1 and TNFAIP3, may be targets of ZFP36 (76), a detailed temporal assessment of feedforward control by ZFP36 and the corresponding effects on the expression dynamics of pro- and anti-inflammatory regulators is necessary.
Reduced feedforward control promotes resistance to glucocorticoid
As noted, glucocorticoids induce DUSP1 expression, down-regulate MAPK activity, and promote repression of inflammatory genes. Thus, DUSP1 overexpression reduced expression of 19 out of 46 IL1B-induced mRNAs tested in A549 cells, and in mRNAs tested with DUSP1 silencing, or in cells from Dusp1−/− mice, many of these same genes showed enhanced expression (31, 36, 77). However, overexpression of DUSP1 also increased, often quite dramatically, the expression of 14 of 46 mRNAs induced by IL1B (77). These included the inflammatory transcription factor, IRF1, as well as IRF1-dependent genes, such as the chemokine, CXCL10 (77). Conversely, in bone marrow-derived macrophage from Dusp1 knock-out mice, IRF1 expression, and some 20% of transcripts that were induced ≥3-fold by LPS, showed lower expression when compared with cells from wild-type animals (66). Similarly, i.p. LPS induced the expression of numerous inflammatory genes, many of which revealed reduced expression in Dusp1−/− animals (78). Thus, in vitro and in vivo, DUSP1 maintains expression of inflammatory genes! Such effects are not isolated, and negative regulation of IRF1 and/or IRF1-dependent gene expression by MAPKs is reported in A549 cells, primary human bronchial epithelial cells, bronchial epithelial BEAS-2B cells, mouse macrophages, and other cell lines (66, 77, 79–81).
IRF1 expression is induced by viral infections, interferons, and cytokines, such as TNF, IL6, and IL1B, via transcription factors that include STAT1, STAT2, and NF-κB (82). Furthermore, expression of late-phase genes, for example, the retinoic acid-induced gene I (DDX58) or CXCL10 (77, 83, 84), that are central in innate immune and antiviral responses are induced by IRF1 (82, 85) (Fig. 4A). Indeed, mice lacking Irf1 are susceptible to death during viral infections (86), whereas ubiquitin-mediated proteasomal degradation of IRF1 allows viral suppression of the immune response (87). In terms of IRF1 mRNA and protein induced by TLR or IL1 type receptors, these follow spike-like kinetics and are negatively regulated by MAPKs (66, 77, 80) (Fig. 4). However, although the rapid induction of IRF1 mRNA and protein by IL1B was largely unaffected by MAPK inhibition, their precipitous loss, after peak expression, was significantly attenuated due to a failure to terminate IRF1 expression (77, 80). This constitutes a classical incoherent feedforward circuit in which MAPK, primarily p38, inhibition not only prolonged IRF1 transcription and enhanced IRF1 mRNA stability, but also reduced IRF1 degradation (Fig. 4A) (77). Such data are consistent with the phosphorylation and ubiquitination of IRF1 coupled with a half-life of just 30 min following induction by IL1B (77, 88, 89).
Figure 4.
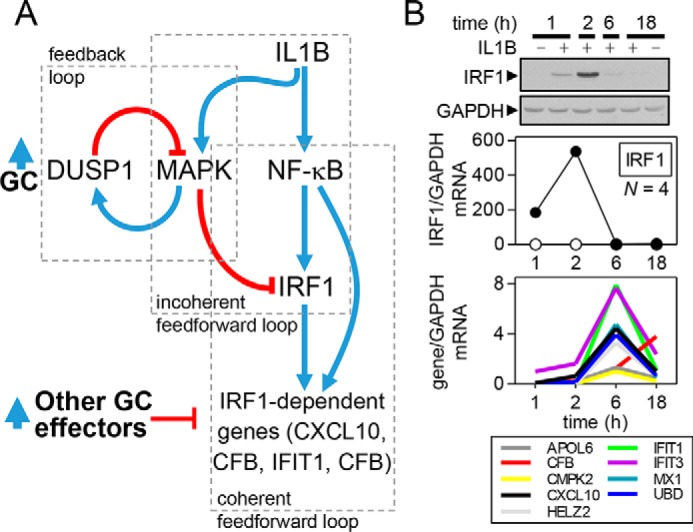
Loss of feedforward control may promote glucocorticoid resistance. A, schematic showing the regulation of IRF1, as well as IRF1-dependent gene expression. Pro-inflammatory stimuli (IL1B) induce NF-κB activity, leading to the transcriptional activation of IRF1. IRF1 expression is rapidly induced to promote expression of downstream IRF1-dependent genes. Many IRF1-depedent genes, for example CXCL10, are also directly regulated by NF-κB to constitute a type I coherent feedforward loop. Pro-inflammatory stimuli, such as IL1B, promote activation of MAPK pathways. Acting via multiple mechanisms, MAPKs promote the switching off and/or loss of IRF1 expression. This terminates IRF1 expression and prevents continued expression of IRF1-dependent genes. In the presence of glucocorticoid (GC), DUSP1 expression is enhanced. By reducing MAPK activity, glucocorticoids reduce incoherent feedforward control of IRF1, and this promotes IRF1 expression. This effect also occurs following MAPK inhibition. Maintenance of IRF1 expression helps CXCL10 to escape the otherwise repressive effects of the glucocorticoid. However, many other IRF1-dependent genes (for examples, see CMPK2, MX1, IFIT1, and others on Fig. 1A) show significant repression by glucocorticoids. Therefore, the existence of additional mechanisms of repression must be invoked. B, data are modified from Shah et al. (77) where full details can be found. A549 cells were either not treated, or treated with IL1B (1 ng/ml), for the times indicated prior to Western blotting and qPCR analysis of IRF1 and GAPDH (upper and middle panels) or qPCR of established IRF1-dependent genes. Spike kinetics for IFR1 mRNA and protein, as well as late-phase kinetics for IRF1-dependent genes, is shown.
The above network, whereby DUSP1 reduces MAPK activity to maintain IRF1 and IRF1-dependent gene expression, has potentially profound implications in the context of a glucocorticoid (Fig. 4A). Glucocorticoids up-regulate DUSP1 and reduce MAPK activity, which, in the absence of any further control mechanisms, would be predicted to maintain IRF1 expression. Indeed, with IL1B plus dexamethasone, DUSP1 silencing increased MAPK activity and modestly reduced expression of IRF1 and the IRF1-dependent gene, CXCL10 (31, 77). Thus, glucocorticoid-induced DUSP1 may help to maintain IRF1 and CXCL10 expression. This is consistent with the poor response of CXCL10 to glucocorticoids and essential roles for IRF1 and CXCL10 in host defense (77, 90–92). Indeed, IRF1 is implicated in glucocorticoid resistance (93), potentially via the steroid receptor co-activator, NCOA2, which may be required for DUSP1 up-regulation by glucocorticoids (94). Although the generalizability of these data remains to be explored, we speculate that the maintenance of IRF1, as well as the coupled expression of key immune genes, for example CXCL10, is desirable during infections and could confer an advantage to the host (85). Indeed, although glucocorticoids dampen inflammation to promote healing, their ability to maintain select immune responses may also represent a key function of GR. Nevertheless, although in A549 cells the maintenance of IRF1 allows expression of CXCL10 in the presence of glucocorticoid, other IRF1-dependent genes were profoundly repressed (77) (see CPMK2 and MX1 on Fig. 1A). This implicates independent mechanisms of glucocorticoid repression that allow differential repression of IRF1-dependent genes (Fig. 4A).
Incoherent feedforward control by MAPK pathways is not without precedent. MAPKs not only activate transcription, but also target downstream proteins, including transcription factors, such as ELK1 or c-Jun (JUN), for inactivation via mechanisms that may involve phosphorylation and/or ubiquitination and degradation (95, 96). Similarly, HDAC4 is activated by ERK and should reduce target gene transcription (97). Likewise, the ubiquitin ligase, ITCH, is a substrate for JNK and promotes ubiquitination and proteolysis of signaling molecules and transcription factors necessary to activate pro-apoptotic gene expression (98). Thus, the transcription of many genes is inhibited by MAPKs (99). Furthermore, as suggested for IRF1 (63), the ability of glucocorticoids to induce DUSP1 expression and reduce MAPK activity should contribute toward maintaining, even enhancing, the expression of such genes. This concept is supported by the large number of LPS-induced transcripts that show reduced expression following knock-out of Dusp1 (66, 78). Although we suggest a role in mediating resistance of CXCL10 to glucocorticoid treatment, the wider implication of genes that are negatively regulated by MAPKs is currently underappreciated.
Other inflammatory genes not repressed by glucocorticoid may show GR recruitment
As discussed above, inflammatory signaling is subject to intense negative control that may be enhanced by glucocorticoids. Moreover, inflammatory genes, whether of anti- or pro-inflammatory effect, are regulated through multiple enhancers that are occupied by NF-κB or other inflammation-activated transcription factors. Accordingly, the net effect of this repression when combined with cooperation between GR and inflammatory transcription factors, such as NF-κB, at specific enhancers may result in only modest or no increase in mRNA of the associated target gene, as exemplified by TNFAIP3. Similarly, the effect of glucocorticoid stimulation alone on expression of any specific gene may reveal only weak induction, and such targets may thus appear unlikely to be important effectors of glucocorticoid action. However, integrated analysis of ChIP-seq and expression data can reveal patterns that suggest cooperation between GR and inflammatory transcription factors at specific enhancers (Fig. 3). These are therefore revealed as potentially glucocorticoid-regulated, despite an apparently minimal effect of glucocorticoid on expression of the associated gene. For example, SOD2, which protects against oxidative stress, is very weakly induced by glucocorticoid in airway epithelial cells, but when induced by IL1B, it is only modestly repressed (Fig. 1A) (11). However, ∼20 kb upstream of the SOD2 transcription start site, there is a site of robust GR-RELA co-occupancy (Fig. 3B, panel i) that appears to mediate cooperative recruitment of RNA polymerase II upon co-stimulation with TNF and glucocorticoid (not shown). Likewise, TNF-induced expression of ZC3H12A, which is implicated in promoting degradation of cytokine mRNAs, is maintained with glucocorticoid treatment (13),3 likely through the activity of 5′ enhancers that exhibit GR and RELA occupancy patterns that are characteristic of cooperation between these two factors (15) (Fig. 3B, panel ii). Intriguingly, other TNF-induced RELA-binding sites within the ZC3H12A gene body appear to be repressed by glucocorticoid, and this highlights the diversity of crosstalk between TNF and glucocorticoid signaling. SOD2 and ZC3H12A not only provide further examples where GR/NF-κB cooperation may maintain or enhance expression of protective genes, but these examples support the concept that maintained expression of specific TNF-target genes is an active GR-dependent regulatory process that is quite prevalent. Importantly, and probably more contentiously, we speculate that this effect may also extend to other, apparently pro-inflammatory, genes. Thus, other maintained genes (Fig. 1A), such as the cytokine, IL32 (Fig. 3B, panel iii), and potentially, the colony-stimulating factor, CSF3 (G-CSF) (not shown), reveal GR recruitment and may therefore represent active regulation by GR.
Summary and conclusions
Pro-inflammatory signals necessarily interact with GR signaling to maintain, or enhance, the expression of regulatory genes. This involves core inflammatory factors, such as NF-κB, and explains why a blanket glucocorticoid-dependent repression of NF-κB is not observed. Rather, many such regulators are specifically targeted by GR, enabling co-regulation and ensuring maintenance of expression in the context of glucocorticoid. Thus, cooperation between GR and inflammatory transcription factors allows GR transactivation to promote repression of inflammation. This raises the question as to the physical determinants of GR cooperation versus direct repression by GR. Certainly, differences in the nature and location of GR-binding sites relative to an inflammatory factor could play a role. However, differential post-translational modification and/or interaction with other factors may also promote differential responsiveness, and these issues require exploration. Therapeutically, this cooperativity with GR to maintain, or induce, regulatory genes suggests a need for caution. When seeking to identify novel GR ligands with reduced side-effect profiles, simply screening for reduced GR transactivation may be quite unhelpful (17). Equally, genes that are co-regulated by GR and factors such as NF-κB are inherently resistant to glucocorticoid repression. This makes biological sense in the context of feedback and feedforward control. However, there are also genes, again induced by factors such as NF-κB, that are not regulators and yet are also not repressed by glucocorticoid. We mention IL32, CSF3, and SOD2. Although SOD2 may be protective against oxidative injury, cytokines, such as IL32 or CSF3, may link to inflammatory responses that are maintained by the glucocorticoid. GR recruitment may, in the same way as the feedback regulator genes, actively maintain expression. This requires careful testing, but the identification of multiple genes with apparent effects on inflammation, proliferation, and cell migration that are all modestly glucocorticoid-induced in vivo raises the prospect that the maintenance by direct GR binding is widespread (16).
Maintenance of inflammatory gene expression may also be achieved by reducing feedforward control. For example, MAPKs actively reduce IRF1 expression, and the glucocorticoid-dependent inhibition of MAPKs lessens feedforward control to promote glucocorticoid insensitivity of the IRF1-dependent gene, CXCL10. This may be advantageous in the context of viral infections, but the maintained expression of such mediators may be undesirable in chronic inflammatory disease. This raises a radical line of thought. Could a failure to inhibit MAPKs actually improve efficacy in cases where the response is maintained due a loss of MAPK-dependent feedforward control? Certainly, reducing glucocorticoid-induced DUSP1 expression can have relatively little effect on inflammatory gene expression (31), presumably due to other glucocorticoid-induced effectors providing redundant actions. Thus, GR ligands that show reduced repression of MAPKs could paradoxically provide superior repression where MAPKs act in feedforward loops to switch off inflammatory processes. Identification of inflammatory genes that evade repression also allows consideration of alternate strategies, for example small molecule inhibitors, to limit expression and would necessarily act as an add-on therapy alongside conventional GR-based approaches. Nevertheless, the above discussion highlights an urgent need for modeling and systems-based approaches to better predict the behavior of inflammatory pathways and gene expression.
This work was supported by Canadian Institutes of Health Research operating grant (MOP 125918) (to R. N.), Alberta Innovates Health Solutions Senior Scholar salary support (to R. N.), and NHLBI, National Institutes of Health Grant R01 HL-109557 (to A. N. G.). R. N. receives research funding from AstraZeneca. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
A. Gerber, unpublished data.
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- HDAC
- histone deacetylase
- DUSP
- dual-specificity phosphatase
- ARE
- AU-rich element
- TLR
- Toll-like receptor
- ChIP-seq
- ChIP sequencing
- qPCR
- quantitative PCR
- Dex
- dexamethasone.
References
- 1. Oakley R. H., and Cidlowski J. A. (2013) The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 132, 1033–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnes P. J. (2011) Glucocorticosteroids: current and future directions. Br. J. Pharmacol. 163, 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnes P. J. (2013) Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 131, 636–645 [DOI] [PubMed] [Google Scholar]
- 4. Keenan C. R., Salem S., Fietz E. R., Gualano R. C., and Stewart A. G. (2012) Glucocorticoid-resistant asthma and novel anti-inflammatory drugs. Drug Discov. Today 17, 1031–1038 [DOI] [PubMed] [Google Scholar]
- 5. Ammit A. J. (2013) Glucocorticoid insensitivity as a source of drug targets for respiratory disease. Curr. Opin. Pharmacol. 13, 370–376 [DOI] [PubMed] [Google Scholar]
- 6. Sukkar M. B., Issa R., Xie S., Oltmanns U., Newton R., and Chung K. F. (2004) Fractalkine/CX3CL1 production by human airway smooth muscle cells: induction by IFN-γ and TNF-α and regulation by TGF-β and corticosteroids. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L1230–L1240 [DOI] [PubMed] [Google Scholar]
- 7. Zhang N., Truong-Tran Q. A., Tancowny B., Harris K. E., and Schleimer R. P. (2007) Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J. Immunol. 179, 578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Langlais D., Couture C., Balsalobre A., and Drouin J. (2008) Regulatory network analyses reveal genome-wide potentiation of LIF signaling by glucocorticoids and define an innate cell defense response. PLoS. Genet. 4, e1000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Busillo J. M., Azzam K. M., and Cidlowski J. A. (2011) Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J. Biol. Chem. 286, 38703–38713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lannan E. A., Galliher-Beckley A. J., Scoltock A. B., and Cidlowski J. A. (2012) Proinflammatory actions of glucocorticoids: glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology 153, 3701–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. King E. M., Chivers J. E., Rider C. F., Minnich A., Giembycz M. A., and Newton R. (2013) Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation- and transrepression-dependent mechanisms. PLoS ONE 8, e53936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Busillo J. M., and Cidlowski J. A. (2013) The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol. Metab. 24, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rao N. A., McCalman M. T., Moulos P., Francoijs K. J., Chatziioannou A., Kolisis F. N., Alexis M. N., Mitsiou D. J., and Stunnenberg H. G. (2011) Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 21, 1404–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van de Garde M. D., Martinez F. O., Melgert B. N., Hylkema M. N., Jonkers R. E., and Hamann J. (2014) Chronic exposure to glucocorticoids shapes gene expression and modulates innate and adaptive activation pathways in macrophages with distinct changes in leukocyte attraction. J. Immunol. 192, 1196–1208 [DOI] [PubMed] [Google Scholar]
- 15. Kadiyala V., Sasse S. K., Altonsy M. O., Berman R., Chu H. W., Phang T. L., and Gerber A. N. (2016) Cistrome-based cooperation between airway epithelial glucocorticoid receptor and NF-κB orchestrates anti-inflammatory effects. J. Biol. Chem. 291, 12673–12687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leigh R., Mostafa M. M., King E. M., Rider C. F., Shah S., Dumonceaux C., Traves S. L., McWhae A., Kolisnik T., Kooi C., Slater D. M., Kelly M. M., Bieda M., Miller-Larsson A., and Newton R. (2016) An inhaled dose of budesonide induces genes involved in transcription and signaling in the human airways: enhancement of anti- and proinflammatory effector genes. Pharmacol. Res. Perspect. 4, e00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newton R., and Holden N. S. (2007) Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol. Pharmacol. 72, 799–809 [DOI] [PubMed] [Google Scholar]
- 18. Petta I., Dejager L., Ballegeer M., Lievens S., Tavernier J., De Bosscher K., and Libert C. (2016) The interactome of the glucocorticoid receptor and its influence on the actions of glucocorticoids in combatting inflammatory and infectious diseases. Microbiol. Mol. Biol. Rev. 80, 495–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clark A. R., and Belvisi M. G. (2012) Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol. Ther. 134, 54–67 [DOI] [PubMed] [Google Scholar]
- 20. Ito K., Yamamura S., Essilfie-Quaye S., Cosio B., Ito M., Barnes P. J., and Adcock I. M. (2006) Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J. Exp. Med. 203, 7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hua G., Ganti K. P., and Chambon P. (2016) Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proc. Natl. Acad. Sci. U.S.A. 113, E635–E643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Surjit M., Ganti K. P., Mukherji A., Ye T., Hua G., Metzger D., Li M., and Chambon P. (2011) Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145, 224–241 [DOI] [PubMed] [Google Scholar]
- 23. Hudson W. H., Youn C., and Ortlund E. A. (2013) The structural basis of direct glucocorticoid-mediated transrepression. Nat. Struct. Mol. Biol. 20, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hua G., Paulen L., and Chambon P. (2016) GR SUMOylation and formation of an SUMO-SMRT/NCoR1-HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc. Natl. Acad. Sci. U.S.A. 113, E626–E634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. So A. Y., Chaivorapol C., Bolton E. C., Li H., and Yamamoto K. R. (2007) Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS. Genet. 3, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Biddie S. C., John S., Sabo P. J., Thurman R. E., Johnson T. A., Schiltz R. L., Miranda T. B., Sung M. H., Trump S., Lightman S. L., Vinson C., Stamatoyannopoulos J. A., and Hager G. L. (2011) Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grøntved L., John S., Baek S., Liu Y., Buckley J. R., Vinson C., Aguilera G., and Hager G. L. (2013) C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 32, 1568–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langlais D., Couture C., Balsalobre A., and Drouin J. (2012) The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell 47, 38–49 [DOI] [PubMed] [Google Scholar]
- 29. Arthur J. S., and Ley S. C. (2013) Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 [DOI] [PubMed] [Google Scholar]
- 30. Clark A. R., Martins J. R., and Tchen C. R. (2008) Role of dual specificity phosphatases in biological responses to glucocorticoids. J. Biol. Chem. 283, 25765–25769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shah S., King E. M., Chandrasekhar A., and Newton R. (2014) Roles for the mitogen-activated protein kinase (MAPK) phosphatase, DUSP1, in feedback control of inflammatory gene expression and repression by dexamethasone. J. Biol. Chem. 289, 13667–13679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diefenbacher M., Sekula S., Heilbock C., Maier J. V., Litfin M., van Dam H., Castellazzi M., Herrlich P., and Kassel O. (2008) Restriction to Fos family members of Trip6-dependent coactivation and glucocorticoid receptor-dependent trans-repression of activator protein-1. Mol. Endocrinol. 22, 1767–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. King E. M., Holden N. S., Gong W., Rider C. F., and Newton R. (2009) Inhibition of NF-κB-dependent transcription by MKP-1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J. Biol. Chem. 284, 26803–26815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bladh L. G., Johansson-Haque K., Rafter I., Nilsson S., and Okret S. (2009) Inhibition of extracellular signal-regulated kinase (ERK) signaling participates in repression of nuclear factor (NF)-κB activity by glucocorticoids. Biochim. Biophys. Acta 1793, 439–446 [DOI] [PubMed] [Google Scholar]
- 35. Issa R., Xie S., Khorasani N., Sukkar M., Adcock I. M., Lee K. Y., and Chung K. F. (2007) Corticosteroid inhibition of growth-related oncogene protein-α via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells. J. Immunol. 178, 7366–7375 [DOI] [PubMed] [Google Scholar]
- 36. Abraham S. M., Lawrence T., Kleiman A., Warden P., Medghalchi M., Tuckermann J., Saklatvala J., and Clark A. R. (2006) Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 203, 1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maier J. V., Brema S., Tuckermann J., Herzer U., Klein M., Stassen M., Moorthy A., and Cato A. C. (2007) Dual specificity phosphatase 1 knockout mice show enhanced susceptibility to anaphylaxis but are sensitive to glucocorticoids. Mol. Endocrinol. 21, 2663–2671 [DOI] [PubMed] [Google Scholar]
- 38. Lasa M., Abraham S. M., Boucheron C., Saklatvala J., and Clark A. R. (2002) Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol. Cell Biol. 22, 7802–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Imasato A., Desbois-Mouthon C., Han J., Kai H., Cato A. C., Akira S., and Li J. D. (2002) Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of Toll-like receptor 2. J. Biol. Chem. 277, 47444–47450 [DOI] [PubMed] [Google Scholar]
- 40. Newton R., King E. M., Gong W., Rider C. F., Staples K. J., Holden N. S., and Bergmann M. W. (2010) Glucocorticoids inhibit IL-1β-induced GM-CSF expression at multiple levels: roles for the ERK pathway and repression by MKP-1. Biochem. J. 427, 113–124 [DOI] [PubMed] [Google Scholar]
- 41. Johansson-Haque K., Palanichamy E., and Okret S. (2008) Stimulation of MAPK-phosphatase 1 gene expression by glucocorticoids occurs through a tethering mechanism involving C/EBP. J. Mol. Endocrinol. 41, 239–249 [DOI] [PubMed] [Google Scholar]
- 42. Tchen C. R., Martins J. R., Paktiawal N., Perelli R., Saklatvala J., and Clark A. R. (2010) Glucocorticoid regulation of mouse and human dual specificity phosphatase 1 (DUSP1) genes: unusual cis-acting elements and unexpected evolutionary divergence. J. Biol. Chem. 285, 2642–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vandevyver S., Dejager L., Van Bogaert T., Kleyman A., Liu Y., Tuckermann J., and Libert C. (2012) Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J. Clin. Invest. 122, 2130–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shipp L. E., Lee J. V., Yu C. Y., Pufall M., Zhang P., Scott D. K., and Wang J. C. (2010) Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS. One 5, e13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jubb A. W., Young R. S., Hume D. A., and Bickmore W. A. (2016) Enhancer turnover is associated with a divergent transcriptional response to glucocorticoid in mouse and human macrophages. J. Immunol. 196, 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wertz I. E., O'Rourke K. M., Zhou H., Eby M., Aravind L., Seshagiri S., Wu P., Wiesmann C., Baker R., Boone D. L., Ma A., Koonin E. V., and Dixit V. M. (2004) De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430, 694–699 [DOI] [PubMed] [Google Scholar]
- 47. Tokunaga F. (2013) Linear ubiquitination-mediated NF-κB regulation and its related disorders. J. Biochem. 154, 313–323 [DOI] [PubMed] [Google Scholar]
- 48. Altonsy M. O., Sasse S. K., Phang T. L., and Gerber A. N. (2014) Context-dependent cooperation between nuclear factor κB (NF-κB) and the glucocorticoid receptor at a TNFAIP3 intronic enhancer: a mechanism to maintain negative feedback control of inflammation. J. Biol. Chem. 289, 8231–8239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sasse S. K., Altonsy M. O., Kadiyala V., Cao G., Panettieri R. A. Jr., and Gerber A. N. (2016) Glucocorticoid and TNF signaling converge at A20 (TNFAIP3) to repress airway smooth muscle cytokine expression. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L421–L432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hofmann T. G., and Schmitz M. L. (2002) The promoter context determines mutual repression or synergism between NF-κB and the glucocorticoid receptor. Biol. Chem. 383, 1947–1951 [DOI] [PubMed] [Google Scholar]
- 51. Le Bail O., Schmidt-Ullrich R., and Israël A. (1993) Promoter analysis of the gene encoding the IκB-α/MAD3 inhibitor of NF-κB: positive regulation by members of the rel/NF-κB family. EMBO J. 12, 5043–5049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Newton R. (2014) Anti-inflammatory glucocorticoids: changing concepts. Eur. J. Pharmacol. 724, 231–236 [DOI] [PubMed] [Google Scholar]
- 53. Reddy T. E., Pauli F., Sprouse R. O., Neff N. F., Newberry K. M., Garabedian M. J., and Myers R. M. (2009) Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 19, 2163–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Newton R., Hart L. A., Stevens D. A., Bergmann M., Donnelly L. E., Adcock I. M., and Barnes P. J. (1998) Effect of dexamethasone on interleukin-1β-(IL-1β)-induced nuclear factor-κB (NF-κB) and κB-dependent transcription in epithelial cells. Eur. J. Biochem. 254, 81–89 [DOI] [PubMed] [Google Scholar]
- 55. Hubbard L. L., and Moore B. B. (2010) IRAK-M regulation and function in host defense and immune homeostasis. Infect. Dis. Rep. 2, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miyata M., Lee J. Y., Susuki-Miyata S., Wang W. Y., Xu H., Kai H., Kobayashi K. S., Flavell R. A., and Li J. D. (2015) Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat. Commun. 6, 6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alon U. (2007) Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461 [DOI] [PubMed] [Google Scholar]
- 58. Sasse S. K., and Gerber A. N. (2015) Feed-forward transcriptional programming by nuclear receptors: regulatory principles and therapeutic implications. Pharmacol. Ther. 145, 85–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anderson P. (2008) Post-transcriptional control of cytokine production. Nat. Immunol. 9, 353–359 [DOI] [PubMed] [Google Scholar]
- 60. Clark A. R., and Dean J. L. (2016) The control of inflammation via the phosphorylation and dephosphorylation of tristetraprolin: a tale of two phosphatases. Biochem. Soc. Trans. 44, 1321–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. King E. M., Kaur M., Gong W., Rider C. F., Holden N. S., and Newton R. (2009) Regulation of tristetraprolin expression by interleukin-1β and dexamethasone in human pulmonary epithelial cells: roles for nuclear factor-κB and p38 mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 330, 575–585 [DOI] [PubMed] [Google Scholar]
- 62. Prabhala P., Bunge K., Rahman M. M., Ge Q., Clark A. R., and Ammit A. J. (2015) Temporal regulation of cytokine mRNA expression by tristetraprolin: dynamic control by p38 MAPK and MKP-1. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L973–L980 [DOI] [PubMed] [Google Scholar]
- 63. Shah S., Mostafa M. M., McWhae A., Traves S. L., and Newton R. (2016) Negative feed-forward control of tumor necrosis factor (TNF) by tristetraprolin (ZFP36) is limited by the mitogen-activated protein kinase phosphatase, dual-specificity phosphatase 1 (DUSP1): implications for regulation by glucocorticoids. J. Biol. Chem. 291, 110–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huotari N., Hömmö T., Taimi V., Nieminen R., Moilanen E., and Korhonen R. (2012) Regulation of tristetraprolin expression by mitogen-activated protein kinase phosphatase-1. APMIS 120, 988–999 [DOI] [PubMed] [Google Scholar]
- 65. Ross E. A., Smallie T., Ding Q., O'Neil J. D., Cunliffe H. E., Tang T., Rosner D. R., Klevernic I., Morrice N. A., Monaco C., Cunningham A. F., Buckley C. D., Saklatvala J., Dean J. L., and Clark A. R. (2015) Dominant suppression of inflammation via targeted mutation of the mRNA destabilizing protein tristetraprolin. J. Immunol. 195, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smallie T., Ross E. A., Ammit A. J., Cunliffe H. E., Tang T., Rosner D. R., Ridley M. L., Buckley C. D., Saklatvala J., Dean J. L., and Clark A. R. (2015) Dual-specificity phosphatase 1 and tristetraprolin cooperate to regulate macrophage responses to lipopolysaccharide. J. Immunol. 195, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kratochvill F., Machacek C., Vogl C., Ebner F., Sedlyarov V., Gruber A. R., Hartweger H., Vielnascher R., Karaghiosoff M., Rülicke T., Müller M., Hofacker I., Lang R., and Kovarik P. (2011) Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol. Syst. Biol. 7, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sun L., Stoecklin G., Van Way S., Hinkovska-Galcheva V., Guo R. F., Anderson P., and Shanley T. P. (2007) Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-α mRNA. J. Biol. Chem. 282, 3766–3777 [DOI] [PubMed] [Google Scholar]
- 69. Rahman M. M., Rumzhum N. N., Morris J. C., Clark A. R., Verrills N. M., and Ammit A. J. (2015) Basal protein phosphatase 2A activity restrains cytokine expression: role for MAPKs and tristetraprolin. Sci. Rep. 5, 10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jalonen U., Lahti A., Korhonen R., Kankaanranta H., and Moilanen E. (2005) Inhibition of tristetraprolin expression by dexamethasone in activated macrophages. Biochem. Pharmacol. 69, 733–740 [DOI] [PubMed] [Google Scholar]
- 71. Prabhala P., Bunge K., Ge Q., and Ammit A. J. (2016) Corticosteroid-induced MKP-1 represses pro-inflammatory cytokine secretion by enhancing activity of tristetraprolin (TTP) in ASM cells. J. Cell Physiol. 231, 2153–2158 [DOI] [PubMed] [Google Scholar]
- 72. Chivers J. E., Gong W., King E. M., Seybold J., Mak J. C., Donnelly L. E., Holden N. S., and Newton R. (2006) Analysis of the dissociated steroid, RU24858, does not exclude a role for inducible genes in the anti-inflammatory actions of glucocorticoids. Mol. Pharmacol. 70, 2084–2095 [DOI] [PubMed] [Google Scholar]
- 73. Smoak K., and Cidlowski J. A. (2006) Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor α inflammatory signaling. Mol. Cell Biol. 26, 9126–9135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kaur M., Chivers J. E., Giembycz M. A., and Newton R. (2008) Long-acting β2-adrenoceptor agonists synergistically enhance glucocorticoid-dependent transcription in human airway epithelial and smooth muscle cells. Mol. Pharmacol. 73, 203–214 [DOI] [PubMed] [Google Scholar]
- 75. So A. Y., Cooper S. B., Feldman B. J., Manuchehri M., and Yamamoto K. R. (2008) Conservation analysis predicts in vivo occupancy of glucocorticoid receptor-binding sequences at glucocorticoid-induced genes. Proc. Natl. Acad. Sci. U.S.A. 105, 5745–5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tiedje C., Diaz-Muñoz M. D., Trulley P., Ahlfors H., Laaß K., Blackshear P. J., Turner M., and Gaestel M. (2016) The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. 44, 7418–7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shah S., King E. M., Mostafa M. M., Altonsy M. O., and Newton R. (2016) DUSP1 maintains IRF1 and leads to increased expression of IRF1-dependent genes: a mechanism promoting glucocorticoid insensitivity. J. Biol. Chem. 291, 21802–21816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hammer M., Mages J., Dietrich H., Servatius A., Howells N., Cato A. C., and Lang R. (2006) Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 203, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zaheer R. S., Koetzler R., Holden N. S., Wiehler S., and Proud D. (2009) Selective transcriptional down-regulation of human rhinovirus-induced production of CXCL10 from airway epithelial cells via the MEK1 pathway. J. Immunol. 182, 4854–4864 [DOI] [PubMed] [Google Scholar]
- 80. Korhonen R., Huotari N., Hömmö T., Leppänen T., and Moilanen E. (2012) The expression of interleukin-12 is increased by MAP kinase phosphatase-1 through a mechanism related to interferon regulatory factor 1. Mol. Immunol. 51, 219–226 [DOI] [PubMed] [Google Scholar]
- 81. AbuSara N., Razavi S., Derwish L., Komatsu Y., Licursi M., and Hirasawa K. (2015) Restoration of IRF1-dependent anticancer effects by MEK inhibition in human cancer cells. Cancer Lett. 357, 575–581 [DOI] [PubMed] [Google Scholar]
- 82. Taniguchi T., Ogasawara K., Takaoka A., and Tanaka N. (2001) IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19, 623–655 [DOI] [PubMed] [Google Scholar]
- 83. Soye K. J., Trottier C., Richardson C. D., Ward B. J., and Miller W. H. Jr. (2011) RIG-I is required for the inhibition of measles virus by retinoids. PLoS. One 6, e22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zaheer R. S., and Proud D. (2010) Human rhinovirus-induced epithelial production of CXCL10 is dependent upon IFN regulatory factor-1. Am. J. Respir. Cell Mol. Biol. 43, 413–421 [DOI] [PubMed] [Google Scholar]
- 85. Leigh R., and Proud D. (2015) Virus-induced modulation of lower airway diseases: pathogenesis and pharmacologic approaches to treatment. Pharmacol. Ther. 148, 185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nair S., Michaelsen-Preusse K., Finsterbusch K., Stegemann-Koniszewski S., Bruder D., Grashoff M., Korte M., Köster M., Kalinke U., Hauser H., and Kröger A. (2014) Interferon regulatory factor-1 protects from fatal neurotropic infection with vesicular stomatitis virus by specific inhibition of viral replication in neurons. PLoS. Pathog. 10, e1003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Remoli A. L., Marsili G., Perrotti E., Acchioni C., Sgarbanti M., Borsetti A., Hiscott J., and Battistini A. (2016) HIV-1 Tat recruits HDM2 E3 ligase to target IRF-1 for ubiquitination and proteasomal degradation. MBio. 7, e01528–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lin R., and Hiscott J. (1999) A role for casein kinase II phosphorylation in the regulation of IRF-1 transcriptional activity. Mol. Cell. Biochem. 191, 169–180 [PubMed] [Google Scholar]
- 89. Nakagawa K., and Yokosawa H. (2000) Degradation of transcription factor IRF-1 by the ubiquitin-proteasome pathway. The C-terminal region governs the protein stability. Eur. J. Biochem. 267, 1680–1686 [DOI] [PubMed] [Google Scholar]
- 90. Sauty A., Dziejman M., Taha R. A., Iarossi A. S., Neote K., Garcia-Zepeda E. A., Hamid Q., and Luster A. D. (1999) The T cell-specific CXC chemokines IP-10, Mig, and I-TAC are expressed by activated human bronchial epithelial cells. J. Immunol. 162, 3549–3558 [PubMed] [Google Scholar]
- 91. Wark P. A., Bucchieri F., Johnston S. L., Gibson P. G., Hamilton L., Mimica J., Zummo G., Holgate S. T., Attia J., Thakkinstian A., and Davies D. E. (2007) IFN-γ-induced protein 10 is a novel biomarker of rhinovirus-induced asthma exacerbations. J. Allergy Clin. Immunol. 120, 586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Medoff B. D., Sauty A., Tager A. M., Maclean J. A., Smith R. N., Mathew A., Dufour J. H., and Luster A. D. (2002) IFN-γ-inducible protein 10 (CXCL10) contributes to airway hyperreactivity and airway inflammation in a mouse model of asthma. J. Immunol. 168, 5278–5286 [DOI] [PubMed] [Google Scholar]
- 93. Tliba O., Damera G., Banerjee A., Gu S., Baidouri H., Keslacy S., and Amrani Y. (2008) Cytokines induce an early steroid resistance in airway smooth muscle cells: novel role of interferon regulatory factor-1. Am. J. Respir. Cell Mol. Biol. 38, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bhandare R., Damera G., Banerjee A., Flammer J. R., Keslacy S., Rogatsky I., Panettieri R. A., Amrani Y., and Tliba O. (2010) Glucocorticoid receptor interacting protein-1 restores glucocorticoid responsiveness in steroid-resistant airway structural cells. Am. J. Respir. Cell Mol. Biol. 42, 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mylona A., Theillet F. X., Foster C., Cheng T. M., Miralles F., Bates P. A., Selenko P., and Treisman R. (2016) Opposing effects of Elk-1 multisite phosphorylation shape its response to ERK activation. Science 354, 233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Laine A., and Ronai Z. (2005) Ubiquitin chains in the ladder of MAPK signaling. Sci. STKE. 2005, re5. [DOI] [PubMed] [Google Scholar]
- 97. Zhou X., Richon V. M., Wang A. H., Yang X. J., Rifkind R. A., and Marks P. A. (2000) Histone deacetylase 4 associates with extracellular signal-regulated kinases 1 and 2, and its cellular localization is regulated by oncogenic Ras. Proc. Natl. Acad. Sci. U.S.A. 97, 14329–14333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Nguyen L. K., Kolch W., and Kholodenko B. N. (2013) When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun. Signal. 11, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yang S. H., Sharrocks A. D., and Whitmarsh A. J. (2013) MAP kinase signalling cascades and transcriptional regulation. Gene. 513, 1–13 [DOI] [PubMed] [Google Scholar]