

Abstract
Pyrroline-5-carboxylate reductase (PYCR) is the final enzyme in proline biosynthesis, catalyzing the NAD(P)H-dependent reduction of Δ1-pyrroline-5-carboxylate (P5C) to proline. Mutations in the PYCR1 gene alter mitochondrial function and cause the connective tissue disorder cutis laxa. Furthermore, PYCR1 is overexpressed in multiple cancers, and the PYCR1 knock-out suppresses tumorigenic growth, suggesting that PYCR1 is a potential cancer target. However, inhibitor development has been stymied by limited mechanistic details for the enzyme, particularly in light of a previous crystallographic study that placed the cofactor-binding site in the C-terminal domain rather than the anticipated Rossmann fold of the N-terminal domain. To fill this gap, we report crystallographic, sedimentation-velocity, and kinetics data for human PYCR1. Structures of binary complexes of PYCR1 with NADPH or proline determined at 1.9 Å resolution provide insight into cofactor and substrate recognition. We see NADPH bound to the Rossmann fold, over 25 Å from the previously proposed site. The 1.85 Å resolution structure of a ternary complex containing NADPH and a P5C/proline analog provides a model of the Michaelis complex formed during hydride transfer. Sedimentation velocity shows that PYCR1 forms a concentration-dependent decamer in solution, consistent with the pentamer-of-dimers assembly seen crystallographically. Kinetic and mutational analysis confirmed several features seen in the crystal structure, including the importance of a hydrogen bond between Thr-238 and the substrate as well as limited cofactor discrimination.
Keywords: analytical ultracentrifugation, enzyme kinetics, nicotinamide adenine dinucleotide (NADH), reductase, site-directed mutagenesis, substrate specificity, X-ray crystallography, NAD(P)H-dependent reductase, Rossmann fold
Introduction
Proline, a unique amino acid that lacks a primary amino group, is a key building block of proteins and plays an important role in stress protection and redox balance of cells across multiple kingdoms (1–3). The biosynthesis of proline occurs via two pathways to produce the intermediate Δ1-pyrroline-5-carboxylate (P5C)2 from glutamate or ornithine (Fig. 1). The glutamate route involves two enzymatic steps catalyzed sequentially by glutamate-5-kinase and γ-glutamate phosphate reductase, producing l-glutamate γ-semialdehyde, which forms a nonenzymatic equilibrium with P5C. In later evolved eukaryotes, this transformation is catalyzed by P5C synthetase, a bifunctional enzyme that possesses both glutamate-5-kinase and γ-glutamate phosphate reductase activities. The route from ornithine is catalyzed by ornithine-γ-aminotransferase, which likewise produces l-glutamate γ-semialdehyde. The glutamate and ornithine pathways converge on P5C reductase (P5CR, EC 1.5.1.2), which catalyzes the final reduction of P5C to proline using NAD(P)H.
Figure 1.

The reactions and enzymes of proline biosynthesis. G5K, glutamate-5-kinase; γ-GPR, γ-glutamate-phosphate reductase; OAT, ornithine-γ-aminotransferase.
In humans, P5CR is known as PYCR. The human genome contains three homologous PYCR genes, PYCR1, PYCR2, and PYCRL (also known as PYCR3), which produce a total of nine PYCR enzyme isoforms. Supplemental Table S1 lists the pairwise sequence identities between the isoforms. Supplemental Fig. S1 shows a global sequence alignment of PYCR enzymes. The various PYCR isoforms have specialized roles in proline biosynthesis and different subcellular localizations. A study of PYCRs in melanoma cells showed that both PYCR1 and PYCR2 function primarily in the glutamate-to-proline pathway, whereas PYCRL functions mainly in the ornithine route to proline (4). PYCR1 and -2 localize to mitochondria, whereas PYCRL is cytosolic (4, 5). Other studies have shown that PYCR1/2 is recoverable from the cytosol, suggesting the possibility that the enzymes associate with the outer mitochondrial membrane (6).
PYCR has been implicated in various cancers and has emerged as a potential therapeutic target. A study of mRNA profiles from 1,981 tumors identified PYCR1 as one of the most consistently overexpressed metabolic genes across 19 different cancer types (7). Additional studies highlight an abundance of PYCR1 in melanoma cells as compared with healthy melanocytes, indicating this enzyme as a potential therapeutic target in skin cancer treatment (8, 9). Similarly, depletion of PYCR1 in different types of cancers, such as breast, prostate, and renal cell carcinoma, is associated with diminished cell proliferation and tumorigenic growth (8, 10–12).
Deficiencies in PYCR are also linked to inherited metabolic disorders. Certain mutations in the PYCR1 gene cause the autosomal recessive connective tissue disorder cutis laxa (5). The decrease in PYCR1 activity is thought to impair mitochondrial function, leading to developmental defects through increased apoptosis (5). Mutations in the PYCR2 gene cause a developmental disorder of the brain characterized by microcephaly, hypomyelination, and reduced white matter (13). As with cutis laxa, the pathogenicity of the disorder involves mitochondria-induced apoptosis in the central nervous system (13).
Although crystal structures of P5CRs from microorganisms and a plant species have been described (14–16), only one structural study has been reported on human P5CR. Ten years ago, Meng et al. (17) reported 3.1 Å crystal structures of PYCR1 complexed with NADPH and NADH. A major conclusion of that study was that the NAD(P)H cofactor binds to the C-terminal domain rather than in the canonical site within the N-terminal Rossmann fold domain (17). This result is notable because it contradicts other P5CR structures and departs from the accepted structure-function paradigm of Rossmann fold enzymes, which asserts that the cofactor binds to the N termini of the strands of the Rossmann dinucleotide-binding fold.
Herein we reexamine the structure of PYCR1 at high resolution to better establish the location of the active site and elucidate the interactions responsible for cofactor and substrate recognition. Crystal structures of binary complexes of PYCR1 with NADPH and proline have been determined at 1.9 Å resolution. The structure of a ternary complex with NADPH and the P5C/proline analog l-tetrahydrofuroic acid (THFA) has been obtained at 1.85 Å resolution. Furthermore, we present analytical ultracentrifugation analysis of PYCR1 in solution and kinetic data for the forward reaction of P5C reduction to proline. In contrast to the previous study (17), our electron density maps unequivocally identify the location of the cofactor-binding site and show that PYCR1 follows the canonical structure-function paradigm of Rossmann fold enzymes.
Results
Tertiary and quaternary structure of PYCR1
High-resolution crystal structures of PYCR1 were determined in space groups P21212 and C2 (Table 1). The protomer consists of an N-terminal α/β domain and a C-terminal α domain (Fig. 2A). The N-terminal domain features the classic Rossmann dinucleotide-binding fold, which consists of a pair of βαβαβ motifs that interact across a pseudo-2-fold axis to form a six-stranded parallel β-sheet with strand order 321456 (18, 19). An additional βαβ substructure follows strand 6 such that the full β-sheet of the N-terminal domain has eight β-strands, with the last two strands antiparallel to the Rossmann strands. β-Strand 8 of the N-terminal domain connects to the C-terminal domain, which consists of a bundle of six α-helices. The C-terminal domain plays a major role in oligomerization. For example, the C-terminal domain mediates dimerization (Fig. 2B). In the dimer, the C-terminal domains of two protomers form an interlocking bundle of helices. Overall, the domain architecture of PYCR1 is typical for P5CRs (14).
Table 1.
Data collection and refinement statistics
The values for the outer resolution shell of data are given in parenthesis.
| Parameter | NADPH | Pro | NADPH/THFA | Ligand-free P21212 | Ligand-free C2 |
|---|---|---|---|---|---|
| Space group | P21212 | P21212 | P21212 | P21212 | C2 |
| Unit cell parameters | |||||
| a (Å) | 162.1 | 165.1 | 161.5 | 163.03 | 183.8 |
| b (Å) | 87.8 | 87.7 | 87.7 | 88.1 | 120.7 |
| c (Å) | 115.9 | 117.3 | 115.6 | 117.3 | 88.1 |
| β (degrees) | 109.2 | ||||
| Wavelength | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| Resolution (Å) | 64.24–1.92 (1.95–1.92) | 64.63–1.90 (1.93–1.90) | 64.10–1.85 (1.88–1.85) | 64.68–1.85 (1.88–1.85) | 60.34–1.85 (1.88–1.85) |
| Observations | 879,308 (35,441) | 993,789 (45,238) | 978,352 (38,421) | 1,301,444 (45,882) | 678,751 (21,389) |
| Unique reflections | 126,545 (6,217) | 133,932 (6,541) | 139,847 (6,540) | 144,477 (7,100) | 153,414 (7,335) |
| Rmerge(I) | 0.094 (0.965) | 0.105 (0.899) | 0.080 (0.858) | 0.075 (0.902) | 0.055 (0.672) |
| Rmeas(I) | 0.101 (1.065) | 0.113 (0.971) | 0.087 (0.941) | 0.079 (0.981) | 0.062 (0.821) |
| Rpim(I) | 0.038 (0.443) | 0.040 (0.366) | 0.033 (0.380) | 0.024 (0.384) | 0.027 (0.462) |
| Mean I/σ | 14.9 (1.7) | 10.6 (1.5) | 19.3 (1.9) | 18.7 (2.0) | 16.1 (1.4) |
| Mean CC½ | 0.999 (0.720) | 0.997 (0.846) | 0.999 (0.699) | 0.999 (0.789) | 0.999 (0.688) |
| Completeness (%) | 100.0 (99.9) | 99.7 (99.9) | 99.7 (95.0) | 100.0 (99.0) | 99.3 (95.9) |
| Multiplicity | 6.9 (5.7) | 7.4 (6.9) | 7.0 (5.9) | 9.0 (6.5) | 4.4 (2.9) |
| No. of atoms | 10,557 | 10,250 | 11,035 | 10,567 | 10,002 |
| Protein | 9,877 | 9,712 | 10,089 | 9,974 | 9,495 |
| NADPH | 96 | NAa | 240 | NA | NA |
| Pro or THFA | NA | 40 | 40 | NA | NA |
| Water | 557 | 438 | 659 | 568 | 502 |
| Rcryst | 0.174 (0.248) | 0.196 (0.315) | 0.172 (0.244) | 0.180 (0.266) | 0.172 (0.269) |
| Rfreeb | 0.211 (0.274) | 0.228 (0.362) | 0.200 (0.261) | 0.2157 (0.295) | 0.200 (0.279) |
| RMSDc bond lengths (Å) | 0.011 | 0.005 | 0.007 | 0.012 | 0.014 |
| RMSD bond angles (degrees) | 1.099 | 0.699 | 0.907 | 1.157 | 1.273 |
| Ramachandran plotd | |||||
| Favored (%) | 97.57 | 98.08 | 97.97 | 97.94 | 97.75 |
| Outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Clashscore (PR)d | 1.86 (100) | 1.60 (100) | 1.35 (100) | 3.39 (98) | 2.13 (99) |
| MolProbity score (PR)d | 1.10 (100) | 1.03 (100) | 0.92 (100) | 1.54 (93) | 1.36 (98) |
| Average B-factor (Å2) | |||||
| Protein | 32.2 | 43.3 | 26.2 | 38.8 | 35.5 |
| NADPH | 32.5 | NA | 25.8 | NA | NA |
| Pro or THFA | NA | 40.2 | 26.6 | NA | NA |
| Water | 33.2 | 38.4 | 30.3 | 34.5 | 36.3 |
| Coordinate error (Å)e | 0.19 | 0.23 | 0.18 | 0.20 | 0.20 |
| PDB entry | 5UAT | 5UAU | 5UAV | 5UAW | 5UAX |
aNA, not applicable.
b5% test set.
cRMSD, root mean square deviation.
dFrom MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parenthesis.
eMaximum likelihood-based coordinate error estimate reported by phenix.refine.
Figure 2.
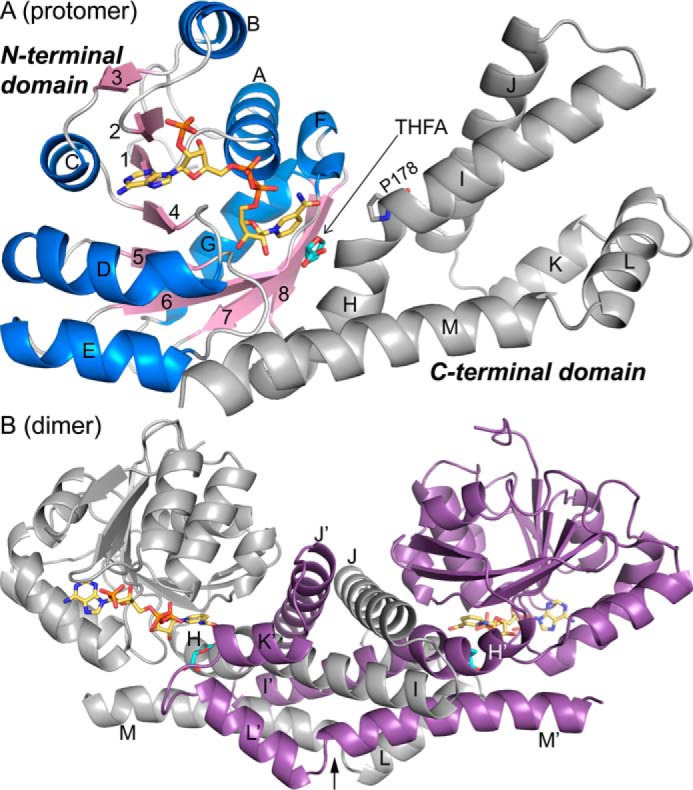
Structures of the PYCR1 protomer and dimer. A, structure of the protomer of the ternary complex with NADPH and the proline/P5C analog THFA. The N-terminal NAD(P)H-binding domain is colored according to secondary structure, with β-strands in pink and α-helices in blue. The C-terminal oligomerization domain is colored gray. NADPH appears in gold sticks. THFA is shown as cyan sticks. β-Strands are labeled 1–8; α-helices are labeled A–M. Helix-disrupting Pro-178 is shown. B, structure of the dimer. The α-helices of the C-terminal domain are labeled H–M for the gray protomer and H′–M′ for the purple protomer. NADPH and THFA are colored gold and cyan, respectively. The arrow represents the 2-fold axis of the dimer.
The oligomeric state of PYCR1 in solution was determined with analytical ultracentrifugation using sedimentation velocity. Initial studies performed at 0.8 mg/ml (24 μm) revealed a distribution of apparent sedimentation coefficient that exhibits a series of peaks in the range 1–7.5 S (Fig. 3A). The corresponding distribution of molecular masses reveals multiple species in solution spanning from ∼30 to 350 kDa (Fig. 3B). Because the theoretical molecular mass of a monomer is 34 kDa, the c(M) distribution is consistent with the presence of multiple oligomeric states ranging from monomer to decamer. To further investigate the possibility that decamer formation is concentration-dependent, sedimentation velocity was also performed at the higher concentration of 6 mg/ml (180 μm). This experiment yielded a sedimentation coefficient distribution with a single peak at 7.47 S (Fig. 3A), corresponding to a molecular mass of 350 kDa (Fig. 3B). Thus, at a concentration of 180 μm, PYCR1 is almost entirely decameric. These results are consistent with P5CR forming a self-association equilibrium in solution.
Figure 3.

Oligomerization of PYCR1 in solution. A, apparent sedimentation coefficient distribution determined at 6 mg/ml (solid line) and 0.8 mg/ml (dashed line). B, molecular mass distribution determined at 6 mg/ml (solid line) and 0.8 mg/ml (dashed line).
Analysis of crystal packing with PDBePISA (20) reveals a pentamer-of-dimers assembly in both crystal forms (Fig. 4), consistent with the decamer observed in sedimentation-velocity experiments. This assembly is also present in other P5CR crystal structures from a variety of organisms (14–16). The consistency of this assembly across space groups and kingdoms suggests the decamer is a fundamental structural property of P5CRs.
Figure 4.
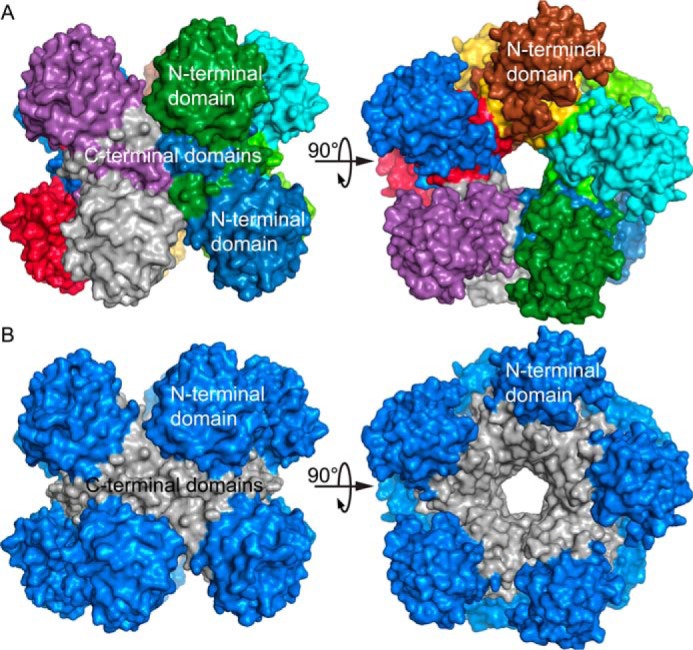
The PYCR1 pentamer-of-dimers decamer. A, two orthogonal views of the decamer, with each chain differently colored. B, two orthogonal views of the decamer, with the N-terminal domains colored blue and the C-terminal domains colored gray. Note that the C-terminal domains mediate all protein-protein interactions in the decamer.
The P5CR decamer has been described in detail elsewhere, so only a summary is provided here (14, 15, 17). The decamer is a pentamer-of-dimers assembly that resembles an “hourglass-shaped barrel” (14) when viewed perpendicular to the 5-fold axis (Fig. 4). The dimers are arranged such that the N-terminal domains form pentameric rings at the top and bottom of the barrel, and the C-terminal domains form a cylinder that connects the two rings. A notable feature of the decamer is that most of the interfacial surface area results from the interaction between C-terminal domains, whereas the N-terminal domains are relatively isolated from other protomers (Fig. 4B).
Identification of the NAD(P)H-binding site
Electron density maps clearly revealed the location and conformation of NADPH bound to PYCR1 (Fig. 5). In the binary enzyme-NADPH complex structure, the density is very strong in two of the five chains in the asymmetric unit, allowing modeling of the complete cofactor with occupancy of 1.0 (chains C and E). The cofactor was not modeled in the other three chains (A, B, and D). We note that high occupancy of NADPH appears to be correlated with stabilization of residues 34–40, which form a loop that interacts with the adenosine portion of NADPH. Density for this loop is very strong in chains C and E but much weaker in chains A, B, and D. The loop was omitted in chain A.
Figure 5.
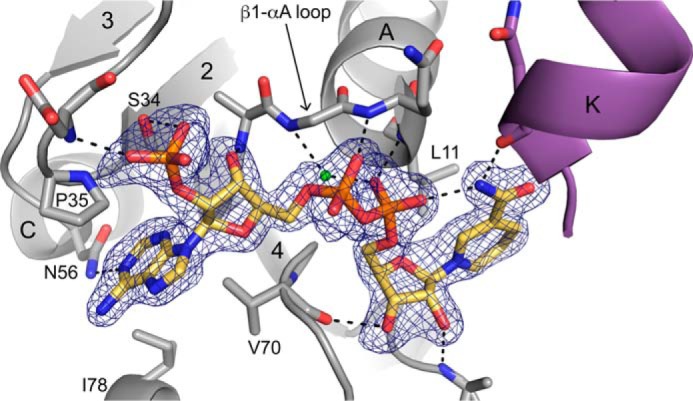
Electron density and interactions for NADPH bound to PYCR1. The cage represents a simulated annealing Fo − Fc map contoured at 3σ. Selected α-helices and β-strands are labeled in accordance with Fig. 2A. Helix K (purple) is from the opposite protomer of the dimer. The conserved water molecule of the Rossmann dinucleotide-binding fold is colored green (21).
NADPH binds at the C termini of the strands of the Rossmann dinucleotide-binding fold (Fig. 5). The cofactor adopts an extended conformation with the pyrophosphate poised above the N terminus of the first helix of the Rossmann fold (αA). The adenosine group interacts with the first βαβαβ motif, whereas the nicotinamide riboside contacts the second βαβαβ motif. In summary, NADPH adopts the canonical pose expected for nicotinamide adenine dinucleotides bound to Rossmann fold domains (21).
NADPH forms several noncovalent interactions with the enzyme (Fig. 5). The ribose hydroxyls and pyrophosphate hydrogen bond exclusively with protein backbone atoms. Only two side chains participate in hydrogen bonding with NADPH. Asn-56 hydrogen-bonds with the adenine ring, whereas Ser-34 interacts with the 2′-phosphoryl. Note that Asn-56 is present in all PYCR isoforms (supplemental Fig. S1). Ser-34 is present in all of the isoforms except PYCR3, which has Ala in this position. Nonpolar side chains help position the bases of NADPH, with Pro-35, Val-70, and Ile-78 contacting the adenine and Leu-11 packing against the nicotinamide. Pro-35 is present all human PYCR isoforms, whereas Val-70 and Ile-78 are replaced by Thr and Val in PYCR3 (supplemental Fig. S1).
The carboxamide of NADPH has a 2-fold rotational degeneracy at the resolution of our structures; however, hydrogen bonding can be used to deduce the correct orientation of this group. The carboxamide amino group has been modeled so it forms an intramolecular hydrogen bond with the pyrophosphate and another hydrogen bond with the backbone carbonyl of Asn-230 (Fig. 5). In contrast, rotation by 180° places the carboxamide carbonyl next to obligate hydrogen bond acceptors, resulting in no hydrogen bonds.
The proline-binding site
The structure of PYCR1 complexed with the product l-proline was determined at 1.90 Å resolution from a crystal that had been soaked in 1.8 m proline (Table 1 and Fig. 6). The electron density allowed modeling of proline in the active site of all five chains with occupancy of 1.0. We note that a sulfate ion occupies the proline site in the ligand-free orthorhombic structure. Proline was also modeled into an election density feature on the surface of the protein near Thr-137. The adventitious binding of proline to the protein surface is common when used in high concentration as a cryoprotectant, as is the case here (22).
Figure 6.
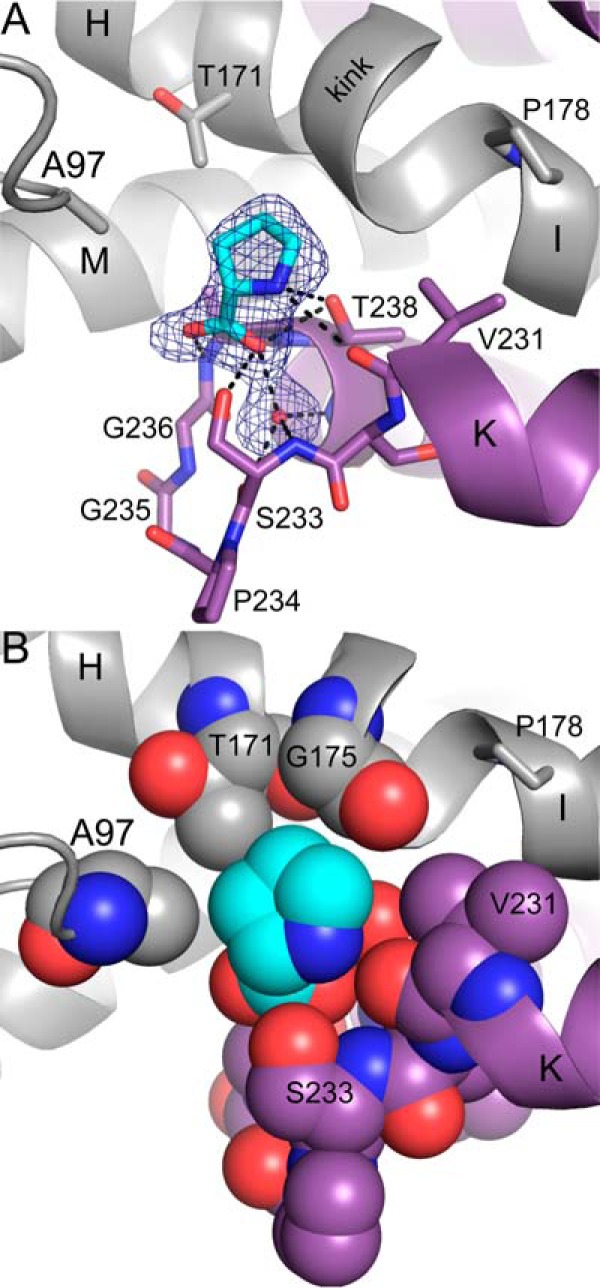
The proline-binding site. A, electron density and interactions for proline (cyan) bound to PYCR1. The cage represents a simulated annealing Fo − Fc map contoured at 3σ. The two protomers of the dimer are colored purple and gray. Note that proline binds in the dimer interface. Selected α-helices are labeled in accordance with Fig. 2A. B, space-filling representation of the proline-binding site highlighting nonpolar residues that contact the methylene groups of proline.
Proline binds in a section of the dimer interface where the αK-αL loop of one protomer meets α-helices H, I, and M of the other protomer (Figs. 2B and 6A). The carboxylate and amino groups of proline are anchored to the αK-αL loop via several hydrogen bonds, some of which are mediated by a water molecule (Fig. 6A). Of note is the hydrogen bond between Thr-238 and the amino group of proline. It has been proposed that this conserved residue functions in catalysis by donating a proton to the imine nitrogen atom of P5C (14).
The roof of the binding site provides nonpolar contacts for the methylene groups of proline. These contacts include Ala-97 of the Rossmann fold and the kink between helices αH and αI (Fig. 6B). The kink is caused by the presence of conserved Pro-178 and appears to be a common feature of P5CRs, because it is also present in structures of P5CR from microorganisms (14). Within the kink, the carbonyls of residues 174 and 175 splay outward to avoid steric clash with the Cδ of Pro-178, and as a result, these groups do not form i to i + 4 hydrogen bonds, and the helical structure is disrupted. Thr-171 and Gly-175 of the kink region provide nonpolar contacts with the methylene groups of proline. These interactions bury the carboxylate face of the proline, leaving the other face solvent-exposed and available for catalysis (Fig. 6B).
The ternary complex with NADPH and THFA
The structure of PYCR1 complexed with NADPH and THFA was determined at 1.85 Å resolution (Table 1 and Fig. 7). The electron density is very strong for both ligands in all five chains, allowing inclusion of the ligands in the model at an occupancy of 1.0. This is the first structure of any P5CR that mimics the enzyme-NADPH-P5C Michaelis complex.
Figure 7.

Structure of the ternary complex of PYCR1 with NADPH and THFA. The cage represents a simulated annealing Fo − Fc map contoured at 3σ. NADPH and THFA are colored gold and cyan, respectively. The two protomers of the dimer are colored purple and gray. Two orthogonal views are shown. Selected α-helices are labeled in accordance with Fig. 2A.
The structure shows that dimerization is essential for catalytic activity. NADPH interacts with the Rossmann fold of one protomer, and THFA binds in the αK-αL loop of the opposite protomer of the dimer (Fig. 2B). NADPH adopts the same conformation as in the binary enzyme-NADPH complex, whereas THFA occupies the proline site described under “The proline-binding site.” Water-mediated hydrogen bonding appears to play a role in stabilizing the ternary complex, because five water molecules with strong electron density connect THFA, NADPH, and the protein (Fig. 7). The ring of THFA stacks in parallel with the nicotinamide such that the C5 of THFA, which represents the hydride acceptor atom of P5C, is 3.7 Å from the C4 of the nicotinamide. The structure is consistent with a direct hydride-transfer mechanism.
Comparison with previous human PYCR1 structures
As noted in the Introduction, the first report of the human PYCR1 structure appeared in 2006 from Meng et al. (17). This paper described structures of the ligand-free enzyme (PDB code 2GER), a ternary complex with NADH and the “substrate analog” glutamate (PDB code 2GR9), and another ternary complex with NADPH and glutamate (PDB code 2GRA). The structures were solved at 3.1 Å resolution and contained the expected fold as well as the same quaternary arrangement as had been reported a year earlier for prokaryotic P5CR structures (14). However, Meng et al. (17) reported an unexpected binding site for NAD(P)H located in the dimer interface (supplemental Fig. S2A). Although the paper refers to the importance of the Rossmann fold in this structure, their dinucleotide-binding site is over 25 Å away from the canonical NADPH site in our structures. Notably, the NADPH site that we report here is the same as that found in the structures of prokaryotic P5CRs (14). Furthermore, the results of Meng et al. (17) contradict the many structures of other Rossmann fold proteins that have been reported since the discovery of the fold in lactate dehydrogenase in the 1970s (18, 19).
Because the interpretation of Meng et al. (17) contradicts our findings and differs from the conventional dinucleotide-binding model found in other NAD(P) enzymes with a Rossmann fold, it is important to carefully study their binding site. Notably, Meng et al. (17) summarized the basis for modeling the dinucleotide and glutamate as follows: “the entire cofactor and substrate analog molecules could be placed accurately from unambiguous electron density.” If the location of this unusual dinucleotide-binding site is accurate, this result would add dramatically to our knowledge of the structural biology of P5CR and Rossmann fold enzymes in general. On the other hand, if it is in error, it is important that the record be adjusted.
Below, several aspects of the Meng et al. (17) structures are reviewed, including the original electron density, the geometry and close contacts of the modeled dinucleotide, the re-refined structures that we obtained from the PDB_REDO server (23), and finally simulated annealing omit maps calculated with the dinucleotide and glutamate ligands excluded to remove phase bias. In all of this analysis, the same answer has been obtained, namely that the dinucleotide and glutamate ligands in the Meng et al. (17) crystal form were built in error and should be removed from the structure.
The human PYCR1 structures described by Meng et al. (17) have space group C2 with five protomers in the asymmetric unit (different from our C2 lattice; Table 1). A crystallographic 2-fold axis correctly generates the decamer. The fold is consistent with previously reported P5CR structures as well as our structures. Superposition of monomers from our structures with those of Meng et al. (17) results in pairwise root mean square differences of 0.7–1.0 Å, which indicates similar overall folds. This similarity is evident from a superposition of dimers from 2GR9 and our ternary complex (supplemental Fig. S2A). Also, the Rossmann domains of the Meng et al. (17) structures superimpose with our structures with root mean square difference of 0.5–0.9 Å (supplemental Fig. S2B). This low value suggests that the canonical dinucleotide-binding site is available in the Meng et al. (17) structures.
All of the cofactor-binding sites in 2GR9 and 2GRA were reviewed for their steric contacts, molecular geometry, and B-factors and the quality of the electron density. Serious concerns have been identified. For example, although the 2Fo − Fc maps for 2GRA/9 obtained from the Electron Density Server (24) have some coverage of the dinucleotide at 1σ in some of the chains (supplemental Figs. S3A and S4A), all five NAD(P)H-binding locations contain strong negative Fo − Fc density covering the ligand at negative 3σ (supplemental Figs. S3B and S4B). The average B-factors of NAD(P)H are also high: 76–94 Å2 in 2GRA and 84–195 Å2 in 2GR9 (supplemental Table S2). For comparison, the average B-factors of NADPH in our ternary complex are 19–33 Å2 (supplemental Table S2).
The structures also include a glutamate ligand modeled near the nicotinamide. The authors suggest that this ligand is a “substrate analog,” which presumably refers to the similarity between Glu and l-glutamate γ-semialdehyde (Fig. 1). The average B-factors of the Glu ligand are very high: 124–200 Å2 (supplemental Table S2). For comparison, the average B-factors of THFA in our ternary complex are 23–30 Å2 (supplemental Table S2). Furthermore, there is a noticeable lack of 2Fo − Fc density at 1σ for Glu in 9 of the 10 chains of the two structures (supplemental Fig. S5). Although some electron density covers part of the ligand in chain A of 2GRA, it is hard to argue that this feature resembles Glu (supplemental Fig. S5).
Inspection of the cofactor geometry and dinucleotide environment raises additional concerns. For example, Arg-129 makes close contacts of 1.7 and 1.4 Å with NADH in chains B and C, respectively (supplemental Fig. S3). These distances imply covalent bonding, which is impossible. Indeed, the default settings of the graphics program PyMOL (25) interpret these close contacts as covalent bonds and draw connections between the atoms (supplemental Fig. S3). Similar abnormalities are evident in the NADPH modeled in 2GRA. One example is found in chain D, where the carbonyl oxygen of Ser-154 is 1.8 Å from the nicotinamide amino group, close enough for PyMOL to draw a covalent bond (supplemental Fig. S4).
The NADH in chain E of 2GR9 provides one example of severe steric clash and poor geometry. The NADH makes close contacts of 0.6 and 1.5 Å with the modeled Glu ligand (supplemental Fig. S6). The pyrophosphate of NADH deviates substantially from the expected tetrahedral geometry, displaying an O–P–O bond angle of 83° and a P–O–C5′ angle of 92° (supplemental Fig. S6). The distorted pyrophosphate makes a close contact of 2.1 Å with the adenosine ribose. Finally, the vector of the N-glycosidic bond to adenine is not in the plane of the nucleobase, which is unexpected (supplemental Fig. S6). All of these findings (lack of electron density, impossibly close contacts, and bad geometry) are consistent with inaccurate modeling and poor refinement of ligands into electron density maps.
The results reported here from studying the Electron Density Server maps contrast with the density described by Meng et al. (17). They report full coverage of both NADH and NADPH in Fo − Fc omit maps calculated at 1.7σ, but details of these calculations were not provided. To help clarify the nature of the dinucleotide ligand density, we utilized the PDB_REDO server (23) to review the density at these key sites in 2GR9 and 2GRA. PDB_REDO maps generated using the conservative protocol (besttls coordinate file) improved the geometry for the bound cofactors, and the maps exhibited less negative Fo − Fc density surrounding the ligands. However, this apparent improvement was accompanied by a substantial increase of the B-factors of the bound ligands to values physically inconsistent with significant binding. For example, the average B-factors for NADH and NADPH in the PDB_REDO structures are 198–271 Å2 (supplemental Table S2). The average B-factors for the glutamate ligand increased to 169–229 Å2 (supplemental Table S2). Consistent with the very high B-factors, the PDB_REDO structures have a conspicuous lack of 2Fo − Fc density for the modeled ligands.
All of the observations described above point to the NAD(P)H-binding site in Meng et al. (17) being in error. To explore what the density may have resembled before including the ligands in the model, we used phenix.refine to calculate simulated annealing omit maps. The Fo − Fc omit maps lacked density consistent with bound NAD(P)H at any of the five sites. For example, Fo − Fc density for NADH is essentially absent at 3σ (supplemental Figs. S7A and S8A). There were patches of positive Fo − Fc density at 2σ in some chains, but none resembled a dinucleotide (supplemental Figs. S7B and S8B). As a positive control, we omitted the side chain of Phe-250 in addition to the dinucleotide. In the omit map, Phe-250 is fully covered with positive Fo − Fc density at 3σ (supplemental Fig. S7C). This finding is consistent with the omit maps being able to effectively return density for an important feature that was left out of the model. The lack of convincing positive Fo − Fc omit density also supports the conclusion that the modeling of the dinucleotide and glutamate ligands in 2GR9 and 2GRA as reported by Meng et al. (17) must be incorrect.
Kinetic measurements
Steady-state kinetics assays showed that PYCR1 utilizes both NADH and NADPH cofactors (supplemental Fig. S9, A and B). When keeping P5C fixed (3.5 mm) while varying the dinucleotide concentration, Km values of 70 and 283 μm for NADH and NADPH were obtained (Table 2). The kcat value was nearly 3-fold higher with NADH, resulting in a 12-fold greater catalytic efficiency (kcat/Km) relative to NADPH (Table 2). A previous study reported a 6.5-fold higher catalytic efficiency of PYCR1 with NADH over NADPH (8). A kcat of 31 s−1 and Km of 667 μm l-P5C were determined for wild-type PYCR1 from assays varying P5C while keeping NADPH constant (Table 2, supplemental Fig. S9C).
Table 2.
Steady-state kinetic parameters for PYCR1
Values reported are the best fit parameters from non-linear least squares fit to the Michaelis-Menten equation (supplemental Fig. S9).
| Enzyme | Variable substrate | Fixed substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|---|
| μm | s−1 | m−1 s−1 | |||
| Wild type | NADH | P5Ca | 70 ± 11 | 218 ± 8 | 31 ± 5. × 105 |
| Wild type | NADPH | P5Ca | 283 ± 119 | 74 ± 8 | 2.6 ± 1.1 × 105 |
| Wild type | P5C | NADPHb | 667 ± 88c | 31 ± 2 | 0.47 ± 0.07 × 105 |
| T238A | NADPH | P5Ca | 159 ± 47 | 23 ± 2 | 1.4 ± 0.4 × 105 |
| T238A | P5C | NADPHb | 2,887 ± 774c | 14 ± 3 | 0.05 ± 0.02 × 105 |
a Fixed dl-P5C concentration of 3.5 mm.
b Fixed NADPH concentration of 0.5 mm.
c Concentration of l-P5C, which is considered to be half the total dl-P5C concentration.
The importance of Thr-238 was tested with the site-directed mutation T238A. It has been proposed that this universally conserved residue functions in catalysis by donating a proton to the imine nitrogen of P5C (14). This proposition was based on the observation of a hydrogen bond between the hydroxyl group of the conserved Thr and the amino group of the product proline in P5CR structures from microorganisms (14), an interaction that is also seen in human PYCR1 (Fig. 6A). The analogous hydrogen bond to THFA is also present in the ternary complex (Fig. 7).
Kinetic parameters for T238A were determined by varying NADPH and P5C (supplemental Fig. S9, D and E). The estimates of kcat were 23 and 14 s−1 with Km values of 159 and 2887 μm for NADPH and l-P5C, respectively (Table 2). Because the estimated Km is near the upper concentration of P5C available, a kcat/Km value of 16,700 m−1 s−1 was also estimated from the slope of the linear region ([S] ≪ Km) of the Michaelis-Menten plot. The ∼10-fold lower kcat/Km value of the T238A mutant relative to wild-type PYCR1 with l-P5C is consistent with the proposed role of Thr-238 hydrogen bonding to the substrate.
Discussion
We described the first high-resolution structures of a human PYCR isozyme. Notably, the complex of PYCR1 with NADPH and the P5C/Pro analog THFA is the first structure of a relevant ternary complex for any P5CR. This structure provides new information about the mechanism of the enzyme. Considering it to represent the E-NADPH-P5C Michaelis complex, the structure implies that P5C binds with its ring approximately parallel to the nicotinamide ring with C2 of P5C close to the C4 of NADPH, as expected for a hydride-transfer mechanism. The donor-acceptor distance inferred from the structure is 3.7 Å, whereas the expected distance in the transition state for hydride-transfer reactions is ∼2.7 Å (26). It is possible that fluctuations of the enzyme are needed to achieve the transition state from the configuration represented in the crystal structure. Alternatively, our structure represents the product complex (E-NADP+-Pro) rather than the substrate complex.
The structure also immediately suggests the stereochemistry of hydride transfer. Because the B-side of the nicotinamide contacts the implied substrate, PYCR1 is predicted to catalyze the transfer of the pro-4S hydrogen to P5C. This assignment agrees with a previous prediction based on superimposing separate structures of P5CR-NADP+ and P5CR-Pro complexes (14). We are not aware of any reports of the determination of the stereochemistry of P5CR by kinetic isotope effect measurements.
It has been suggested that a conserved Thr or Ser in the active site functions as a general acid that donates a proton to the imine nitrogen of P5C (14). Ser and Thr are not usually considered to be general acid/base catalysts due to their high pKa values. It is possible for the protein environment to lower the pKa of an active site residue so it can function as a general acid, as occurs for Lys-199 of the NAD-malic enzyme (27) and Arg-69 of Sin resolvase (28). However, the active site of PYCR1 does not have a preponderance of positively charged residues that could depress the pKa of the proposed residue. We mutated the residue in question of PYCR1 (Thr-238), which forms a hydrogen bond to the proline amino group in our structure (Fig. 6). The mutation to Ala did not substantially diminish kcat, suggesting that Thr-238 is not essential for catalysis, whereas kcat/Km with varied P5C was 10-fold lower relative to wild type, indicating that the loss of Thr impacts P5C binding. For reference, we note that mutation of Lys-69 to Ala in the NAD-malic enzyme decreases Vmax by 105 (27). Thus, it is concluded that Thr-238 does not function as the general acid in PYCR1 but appears to have important interactions with the imine group of P5C, as corroborated by the structural and kinetic data.
As detailed under “Results,” the noncanonical dinucleotide-binding site proposed by Meng et al. (17) is not supported by the experimental data. The original cofactor ligand density was poor, the difference density was uniformly and strongly negative, and the PDB_REDO pipeline essentially removed the contribution of the cofactor and Glu ligands by elevating their temperature factors to non-physical values (supplemental Table S2). The cofactor also had poor geometry and formed several bad contacts, all of which are inconsistent with a well-defined ligand-binding site.
This is in sharp contrast to the highly conserved nature of dinucleotide-binding sites in other Rossmann fold proteins. First reported in the 1970s (18, 29), the Rossmann fold consists of repeated β/α motifs and comprises six (typically) parallel β-strands with α-helices packed on either side of the sheet (30). The dinucleotide binds with an extended conformation in the cleft found at the C termini of the strands, with the pyrophosphate interacting with the N terminus of αA and the Gly-rich β1-αA loop. Each base interacts with the opposing faces of the sheet, forming a network of polar and non-polar interactions. Our previous analysis of 102 Rossmann fold protein structures representing 43 enzymes and 40 species demonstrated a remarkably high consistency of the binding site structure and cofactor conformation (21). In fact, the binding site is so consistent that a conserved water molecule bridging the Gly-rich loop and the pyrophosphate could effectively be considered part of the dinucleotide-binding motif (21). Indeed, this water molecule is present in our structures (Fig. 5). In this context, it is surprising that a completely novel dinucleotide-binding mode for PYCR1 was proposed with almost no discussion of the important way in which the site deviated from conventional wisdom.
Strong evidence from other structural studies also points to the mistaken nature of the unconventional binding site of Meng et al. (17). The core structural elements of human PYCR1 are superimposable with those of available bacterial P5CR structures, and the dinucleotide ligands in the bacterial enzyme structures conform to the classic paradigm for Rossmann fold proteins (14). Furthermore, the PDB contains an unpublished human PYCR1 structure (PDB code 2IZZ) that contains NAD+ bound in the same location as our structure and bacterial P5CR structures. All of the above structural work also supports the conclusion that the binding site described by Meng et al. (17) is incorrect.
Incorrect ligand-binding site notwithstanding, there are positive aspects to the work of Meng et al. (17). They were the first to establish that the fold of PYCR1 is consistent with homologous enzymes. In addition, they documented the oligomeric state as a pentamer of intertwined dimers. These contributions to the literature remain valid. Nevertheless, it is essential that the record be corrected regarding the erroneous NAD(P)H-binding site, and we recommend that correction take place. We also recommend that the recent in silico analysis of cutis laxa PYCR1 mutants, which is based on the incorrect active site, be reevaluated with the correct PYCR1 structure (32).
Our results might explain why the canonical dinucleotide-binding site was unoccupied in the Meng et al. (17) structures. To obtain complexes, Meng et al. (17) soaked crystals in 0.3 mm NADH or NADPH. Our measurements with PYCR1 indicate a Km of 0.07 mm for NADH and 0.3 mm for NADPH (Table 2). Thus, the dinucleotide concentrations used by Meng et al. (17) may not have been high enough for saturation.
Many examples of incorrectly built structures or misplaced ligands have appeared in the crystallographic literature over the past few years (33–36). Some have arisen from updates in genomic databases that revealed older structures solved with incorrect sequences (33, 37, 38). Others are due to the very large number of new structures appearing each year coupled with the fact that many of them come from laboratories with little crystallographic experience (39). Further, there is pressure now to produce faster, more numerous, and more novel results in science (40), and ligand-binding density would be an important source of novelty. Finally, reviewers are often judging structures without access to coordinate files and electron density maps, making detailed verification of ligands difficult. Taken together, this leads to a risk of inappropriate ligand placement.
It has been suggested that the risk of errors in ligand placement could be mitigated by utilizing currently available structural tools (41, 42). These tools emphasize inspection of electron density, stereochemistry, and ligand environment (34, 41). We suggest that the historical context of the ligand site also be considered. In the case of a family like Rossmann dinucleotide-binding proteins, which contain a highly conserved fold, very strong proof should be required of the authors before proposing a completely new binding site. Such safeguards would probably have prevented the misidentification of the active site of PYCR1.
Experimental procedures
Expression and purification of PYCR1
DNA encoding human mitochondrial PYCR1 transcript variant 1 (NCBI RefSeq number NP_001269209.1) in pET-24b(+) with codons optimized for expression in Escherichia coli was synthesized by GenScript Biotech Corp. The construct encodes residues 1–300 of the full-length 319-residue protein plus an N-terminal hexahistidine tag and tobacco etch virus protease cleavage site. This construct was designed for enhanced crystallization based on unpublished PDB entry 2IZZ.
The PYCR1 construct was transformed into BL21(DE3)-competent E. coli cells and plated onto LB agar containing 50 μg/ml kanamycin. A starter culture of Luria broth medium, shaken overnight at 37 °C and 250 rpm, was used to inoculate two 1-liter cultures. After 2 h of shaking at 37 °C and 250 rpm, the larger cultures were induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside and grown with continued shaking at 18 °C overnight. Centrifugation at 3,000 × g for 20 min at 4 °C was used to collect the cells, which were then resuspended in 50 mm HEPES, pH 7.8, 300 mm NaCl, 10 mm imidazole, and 5% (w/v) glycerol (buffer A). The resuspended pellet was stored at −80 °C until ready for purification.
A mixture of DNase I, lysozyme, and GoldBio ProBlock protease inhibitor mixture was added to thawed cells and stirred at 4 °C for 20 min. After lysing cells via sonication, cell debris was removed by centrifugation at 16,000 rpm for 1 h at 4 °C. The resulting supernatant was passed through a 0.45-μm Millex-HV syringe filter and loaded onto a 5-ml HisTrap Ni2+-nitrilotriacetic acid column (GE Healthcare) pre-equilibrated in buffer A using an AKTA Pure chromatography instrument. The column was washed with buffer A supplemented with 30 mm imidazole and eluted with 300 mm imidazole. The fractions containing PYCR1 as determined by SDS-PAGE were collected and dialyzed at 4 °C overnight into 300 mm NaCl, 2% (w/v) glycerol, and 50 mm HEPES at pH 7.5. Following dialysis, the protein was concentrated to 5 ml using a centrifugal concentrator (Amicon Ultra-15) and then further purified on a HiLoad 16/600 Superdex 200 size-exclusion column (GE Healthcare) using a column buffer with the same composition as the dialysis buffer. The His tag was not removed.
Crystallization of PYCR1
Initial crystallization conditions were identified using Hampton Research Index and Crystal Screen I and II reagents in sitting drops (CrystalEX microplates) with 150-μl reservoir volumes. All screening trials were performed at 22 °C using drops formed by mixing 1 μl of the protein stock solution with 1 μl of reservoir solution. Cryschem M sitting drop plates were used for optimizations, with 500-μl reservoir volumes and drops containing 1.5 μl of protein and 1.5 μl of the reservoir solution. Microcrystals from initial hit conditions were used for streak seeding to obtain diffraction-quality crystals.
Crystals in the space group P21212 were grown using reservoir solutions containing 300 mm Na2SO4, 16–18% (w/v) polyethylene glycol (PEG) 3350, and 0.1 m HEPES at pH 7.5. The unit cell dimensions are a = 162 Å, b = 88 Å, and c = 116 Å. The asymmetric unit contains five PYCR1 molecules, which corresponds to Vm of 2.43 Å3/Da and 49% solvent (43). The best P21212 crystals diffracted to 1.85 Å resolution (Table 1).
A second crystal form in space group C2 was grown from reservoir solutions containing 3 m NaCl and 0.1 m HEPES at pH 7.5–8.0. The unit cell dimensions are a = 184 Å, b = 121 Å, c = 88 Å, and β = 109°. The asymmetric unit contains five PYCR1 molecules, which corresponds to Vm of 2.71 Å3/Da and 55% solvent (43). The C2 crystals also diffracted to 1.85 Å resolution (Table 1). We note that this crystal form is different from the PYCR1 C2 forms of Meng et al. (17) and unpublished PDB entry 2IZZ.
Crystal soaking
The orthorhombic form was the better of the two crystal forms for ligand soaking. Ligand-free P21212 crystals were prepared for low temperature data collection by in situ serial transfer into a solution containing the reservoir supplemented with 20% (v/v) PEG 200, followed by rapid plunging into liquid nitrogen. The complex of PYCR1 with NADPH bound was obtained by soaking ligand-free crystals in the 20% (v/v) PEG 200 cryobuffer supplemented with 100 mm NADPH. The complex with proline bound was obtained by soaking a crystal in the reservoir supplemented with 1.8 m proline. We note that this level of proline provides cryoprotection (22). A ternary complex with NADPH and the P5C/proline analog THFA was formed by first soaking with the 20% PEG 200 cryobuffer supplemented with 100 mm NADPH, followed by another short soak in the cryobuffer supplemented with 50 mm THFA. The ligand soaking times in all cases were <10 min.
The C2 crystal form was less amenable to ligand soaking but nevertheless afforded a high-resolution structure of the ligand-free enzyme. The structure of the ligand-free enzyme in space group C2 was solved from a crystal soaked in the reservoir solution supplemented with 30% (w/v) glycerol for cryoprotection and 10 mm NADP+. Density for NADP+ was not strong enough to allow modeling of the ligand, so this structure is considered to be “ligand-free.”
X-ray diffraction data collection, phasing, and refinement
X-ray diffraction data were collected on Advanced Light Source beamline 4.2.2 at Lawrence Berkeley National Laboratory using a Taurus-1 detector in shutterless mode. Data were integrated and scaled with XDS (44). Intensities were merged and converted to amplitudes with Aimless (45). Data processing statistics are shown in Table 1.
Initial phases were calculated with molecular replacement using PHASER (46). The search model was derived from an unpublished 1.95 Å resolution structure of PYCR1 (PDB code 2IZZ). Coot (47) and phenix.refine (48) were used for model building and refinement. The B-factor model consisted of one TLS group per protein chain and an isotropic B-factor for each non-hydrogen atom. MolProbity (49) was used for structure validation. Structure refinement statistics are shown in Table 1.
Analytical ultracentrifugation
Sedimentation-velocity experiments were conducted with a Beckman XL-I analytical ultracentrifuge using an An50Ti rotor and a two-sector cell. Sample sedimentation was continuously monitored at 20 °C using Rayleigh interference optics at 35,000 rpm for a total of 300 scans spaced at 2-min intervals. To observe concentration dependence of oligomerization, sedimentation-velocity experiments were performed at two different protein concentrations as determined by the Pierce BCA protein assay.
Sedimentation-velocity data were processed using Sedfit (50), assuming a partial specific volume of 0.73 ml/g. Analysis of the data in Sedfit allowed for determination of apparent sedimentation coefficient (c(S)) and molecular mass (c(M)) distributions. The approximate frictional ratio for decameric PYCR1 was determined by allowing the value to vary in the c(S) calculation of the 6 mg/ml PYCR1 sample. The frictional ratio refined to a value of 1.94. This value was applied to the analysis of the 0.8 mg/ml sample for consistency in decamer peak identification. Analyzed data were normalized and plotted using Origin 2016.
Kinetic measurements
All PYCR1 kinetic data were collected in triplicate using a Varian Cary BIO 50 UV-visible spectrophotometer following procedures adapted from De Ingeniis et al. (8). PYCR enzyme activity assays were performed by measuring the P5C-dependent oxidation of NADPH (Sigma-Aldrich) or NADH (Sigma-Aldrich) at 340 nm (ϵ340 = 6,200 m−1 cm−1) and 380 nm (1,314 m−1 cm−1) as described previously (8). dl-P5C was synthesized according to a previous protocol (51) involving periodation of dl-5-hydroxylysine and stored in 1 m HCl at 4 °C. dl-P5C was quantified using o-aminobenzaldehyde and neutralized to pH 7.5 with 1 m Tris-HCl (pH 9.0) immediately before assays (51–53). NADH and NADPH stock concentrations were quantified at 340 nm. Assays (600-μl total volume) were performed at 37 °C in 0.1 m Tris-HCl (pH 7.5), 0.01% Brij-35 detergent (Santa Cruz Biotechnology), 1 mm EDTA disodium salt (Fisher), using 0.006 and 0.06 μm PYCR1 wild-type and mutant T238A enzymes. The Km and kcat for NADH and NADPH were determined by varying NADH (0–650 μm) and NADPH (0–2000 μm) while holding the dl-P5C concentration fixed at 3.5 mm. The kinetic parameters for P5C were determined by varying dl-P5C (0–3.5 mm) and holding NADPH fixed at 500 μm. The ionic strength in assays in which dl-P5C varied was kept constant at ∼450 mm, using 1 m Tris+Cl− (pH 7.5) as a balancing buffer (52). Reaction traces were followed for 2.5 min, and data were fit to the Michaelis-Menten equation with SigmaPlot version 12.0 to determine the kinetic parameters (31, 54). For estimating the Km for l-P5C, the concentration of l-P5C was considered to be half the total dl-P5C concentration.
Author contributions
E. M. C. generated protein and performed crystallography experiments and structure refinements. A. C. C. performed crystallization experiments. S. M. P. and E. M. C. performed kinetics experiments. D. A. K. performed analytical ultracentrifugation experiments and data analysis. J. J. T. collected some of the X-ray data and performed structure refinements. All authors analyzed and interpreted data. E. M. C., D. A. K., K. L. K., D. F. B., and J. J. T. wrote the paper.
Supplementary Material
Acknowledgments
We thank Dr. Jay Nix for help with X-ray diffraction data collection at beamline 4.2.2 of the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02-05CH11231.
This work was supported by NIGMS, National Institutes of Health, Grant R01GM065546. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5UAT, 5UAU, 5UAV, 5UAW, and 5UAX) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains supplemental Tables S1 and S2 and Figs. S1–S9.
- P5C
- Δ1-pyrroline-5-carboxylate
- P5CR
- Δ1-pyrroline-5-carboxylate reductase
- PYCR
- human Δ1-pyrroline-5-carboxylate reductase
- THFA
- l-tetrahydro-2-furoic acid
- PDB
- Protein Data Bank.
References
- 1. Phang J. M. (1985) The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr. Top. Cell. Regul. 25, 91–132 [DOI] [PubMed] [Google Scholar]
- 2. Adams E., and Frank L. (1980) Metabolism of proline and the hydroxyprolines. Annu. Rev. Biochem. 49, 1005–1061 [DOI] [PubMed] [Google Scholar]
- 3. Tanner J. J. (2008) Structural biology of proline catabolism. Amino Acids 35, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Ingeniis J., Ratnikov B., Richardson A. D., Scott D. A., Aza-Blanc P., De S. K., Kazanov M., Pellecchia M., Ronai Z., Osterman A. L., and Smith J. W. (2012) Functional specialization in proline biosynthesis of melanoma. PLoS One 7, e45190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reversade B., Escande-Beillard N., Dimopoulou A., Fischer B., Chng S. C., Li Y., Shboul M., Tham P. Y., Kayserili H., Al-Gazali L., Shahwan M., Brancati F., Lee H., O'Connor B. D., Schmidt-von Kegler M., et al. (2009) Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 41, 1016–1021 [DOI] [PubMed] [Google Scholar]
- 6. Phang J. M., Liu W., and Zabirnyk O. (2010) Proline metabolism and microenvironmental stress. Annu. Rev. Nutr. 30, 441–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nilsson R., Jain M., Madhusudhan N., Sheppard N. G., Strittmatter L., Kampf C., Huang J., Asplund A., and Mootha V. K. (2014) Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 5, 3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Ingeniis J., Ratnikov B., Richardson A. D., Scott D. A., Aza-Blanc P., De S. K., Kazanov M., Pellecchia M., Ronai Z., Osterman A. L., and Smith J. W. (2012) Functional specialization in proline biosynthesis of melanoma. PLoS One 7, e45190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ou R., Zhang X., Cai J., Shao X., Lv M., Qiu W., Xuan X., Liu J., Li Z., and Xu Y. (2016) Downregulation of pyrroline-5-carboxylate reductase-2 induces the autophagy of melanoma cells via AMPK/mTOR pathway. Tumour Biol. 37, 6485–6491 [DOI] [PubMed] [Google Scholar]
- 10. Loayza-Puch F., Rooijers K., Buil L. C., Zijlstra J., Oude Vrielink J. F., Lopes R., Ugalde A. P., van Breugel P., Hofland I., Wesseling J., van Tellingen O., Bex A., and Agami R. (2016) Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530, 490–494 [DOI] [PubMed] [Google Scholar]
- 11. Phang J. M., Liu W., Hancock C., and Christian K. J. (2012) The proline regulatory axis and cancer. Front. Oncol. 2, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zeng T., Zhu L., Liao M., Zhuo W., Yang S., Wu W., and Wang D. (2017) Knockdown of PYCR1 inhibits cell proliferation and colony formation via cell cycle arrest and apoptosis in prostate cancer. Med. Oncol. 34, 27. [DOI] [PubMed] [Google Scholar]
- 13. Nakayama T., Al-Maawali A., El-Quessny M., Rajab A., Khalil S., Stoler J. M., Tan W. H., Nasir R., Schmitz-Abe K., Hill R. S., Partlow J. N., Al-Saffar M., Servattalab S., LaCoursiere C. M., Tambunan D. E., et al. (2015) Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am. J. Hum. Genet. 96, 709–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nocek B., Chang C., Li H., Lezondra L., Holzle D., Collart F., and Joachimiak A. (2005) Crystal structures of Δ1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitides and Streptococcus pyogenes. J. Mol. Biol. 354, 91–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruszkowski M., Nocek B., Forlani G., and Dauter Z. (2015) The structure of Medicago truncatula δ1-pyrroline-5-carboxylate reductase provides new insights into regulation of proline biosynthesis in plants. Front. Plant Sci. 6, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Franklin M. C., Cheung J., Rudolph M. J., Burshteyn F., Cassidy M., Gary E., Hillerich B., Yao Z. K., Carlier P. R., Totrov M., and Love J. D. (2015) Structural genomics for drug design against the pathogen Coxiella burnetii. Proteins 83, 2124–2136 [DOI] [PubMed] [Google Scholar]
- 17. Meng Z., Lou Z., Liu Z., Li M., Zhao X., Bartlam M., and Rao Z. (2006) Crystal structure of human pyrroline-5-carboxylate reductase. J. Mol. Biol. 359, 1364–1377 [DOI] [PubMed] [Google Scholar]
- 18. Adams M. J., Ford G. C., Koekoek R., Lentz P. J., McPherson A. Jr, Rossmann M. G., Smiley I. E., Schevitz R. W., and Wonacott A. J. (1970) Structure of lactate dehydrogenase at 2–8 Å resolution. Nature 227, 1098–1103 [DOI] [PubMed] [Google Scholar]
- 19. Rossmann M. G., Moras D., and Olsen K. W. (1974) Chemical and biological evolution of nucleotide-binding protein. Nature 250, 194–199 [DOI] [PubMed] [Google Scholar]
- 20. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 21. Bottoms C. A., Smith P. E., and Tanner J. J. (2002) A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 11, 2125–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pemberton T. A., Still B. R., Christensen E. M., Singh H., Srivastava D., and Tanner J. J. (2012) Proline: Mother Nature's cryoprotectant applied to protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 68, 1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Touw W. G., Joosten R. P., and Vriend G. (2016) New biological insights from better structure models. J. Mol. Biol. 428, 1375–1393 [DOI] [PubMed] [Google Scholar]
- 24. Kleywegt G. J., Harris M. R., Zou J. Y., Taylor T. C., Wählby A., and Jones T. A. (2004) The Uppsala Electron-Density Server. Acta Crystallogr. D Biol. Crystallogr. 60, 2240–2249 [DOI] [PubMed] [Google Scholar]
- 25. DeLano W. L. (2015) The PyMOL Molecular Graphics System, version 1.8, Schroedinger, LLC, New York [Google Scholar]
- 26. Hammes-Schiffer S. (2002) Comparison of hydride, hydrogen atom, and proton-coupled electron transfer reactions. Chemphyschem 3, 33–42 [DOI] [PubMed] [Google Scholar]
- 27. Liu D., Karsten W. E., and Cook P. F. (2000) Lysine 199 is the general acid in the NAD-malic enzyme reaction. Biochemistry 39, 11955–11960 [DOI] [PubMed] [Google Scholar]
- 28. Keenholtz R. A., Mouw K. W., Boocock M. R., Li N. S., Piccirilli J. A., and Rice P. A. (2013) Arginine as a general acid catalyst in serine recombinase-mediated DNA cleavage. J. Biol. Chem. 288, 29206–29214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buehner M., Ford G. C., Moras D., Olsen K. W., and Rossman M. G. (1974) Three-dimensional structure of d-glyceraldehyde-3-phosphate dehydrogenase. J. Mol. Biol. 90, 25–49 [DOI] [PubMed] [Google Scholar]
- 30. Brändén C., and Tooze J. (1991) Introduction to Protein Structure, Chapter 10, Garland Publishing, Inc., New York [Google Scholar]
- 31. Michaelis L., Menten M. L., Johnson K. A., and Goody R. S. (2011) The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry 50, 8264–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sang P., Hu W., Ye Y. J., Li L. H., Zhang C., Xie Y. H., and Meng Z. H. (2016) In silico screening, molecular docking, and molecular dynamics studies of SNP-derived human P5CR mutants. J. Biomol. Struct. Dyn. 10.1080/07391102.2016.1222967 [DOI] [PubMed] [Google Scholar]
- 33. Stec B. (2013) Time passes yet errors remain: comments on the structure of N10-formyltetrahydrofolate synthetase. Protein Sci. 22, 671–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weichenberger C. X., Pozharski E., and Rupp B. (2013) Visualizing ligand molecules in Twilight electron density. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weiss M. S., Diederichs K., Read R. J., Panjikar S., Van Duyne G. D., Gregory Matera A., Fischer U., and Grimm C. (2016) A critical examination of the recently reported crystal structures of the human SMN protein. Hum. Mol. Genet. 10.1093/hmg/ddw298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muller Y. A. (2013) Unexpected features in the Protein Data Bank entries 3qd1 and 4i8e: the structural description of the binding of the serine-rich repeat adhesin GspB to host cell carbohydrate receptor is not a solved issue. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 1071–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Celeste L., Lovelace L., and Lebioda L. (2013) Response to Boguslaw Stec's letter to the editor of Protein Science. Protein Sci. 22, 675–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Celeste L. R., Chai G., Bielak M., Minor W., Lovelace L. L., and Lebioda L. (2012) Mechanism of N10-formyltetrahydrofolate synthetase derived from complexes with intermediates and inhibitors. Protein Sci. 21, 219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wlodawer A., Minor W., Dauter Z., and Jaskolski M. (2013) Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 280, 5705–5736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edwards M. A., and Roy S. (2017) Academic research in the 21st century: maintaining scientific integrity in a climate of perverse incentives and hypercompetition. Environ. Eng. Sci. 34, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deller M. C., and Rupp B. (2015) Models of protein-ligand crystal structures: trust, but verify. J. Comput. Aided Mol. Des. 29, 817–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pozharski E., Weichenberger C. X., and Rupp B. (2013) Techniques, tools and best practices for ligand electron-density analysis and results from their application to deposited crystal structures. Acta Crystallogr. D Biol. Crystallogr. 69, 150–167 [DOI] [PubMed] [Google Scholar]
- 43. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 44. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Williams I., and Frank L. (1975) Improved chemical synthesis and enzymatic assay of δ-1-pyrroline-5-carboxylic acid. Anal. Biochem. 64, 85–97 [DOI] [PubMed] [Google Scholar]
- 52. Moxley M. A., Sanyal N., Krishnan N., Tanner J. J., and Becker D. F. (2014) Evidence for hysteretic substrate channeling in the proline dehydrogenase and δ1-pyrroline-5-carboxylate dehydrogenase coupled reaction of proline utilization A (PutA). J. Biol. Chem. 289, 3639–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singh R. K., Larson J. D., Zhu W., Rambo R. P., Hura G. L., Becker D. F., and Tanner J. J. (2011) Small-angle X-ray scattering studies of the oligomeric state and quaternary structure of the trifunctional proline utilization A (PutA) flavoprotein from Escherichia coli. J. Biol. Chem. 286, 43144–43153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Michaelis L., and Menten M. L. (1913) Die kinetik der invertinwirkung. Biochem. Z. 49, 333–369 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.