

Abstract
Porcine reproductive and respiratory syndrome, a highly infectious disease caused by porcine reproductive and respiratory syndrome virus (PRRSV), has developed various strategies to evade the host innate immune response, including the suppression of type I IFN activation. The mitochondrial antiviral signalling protein (MAVS) is an important bridging adaptor of retinoic acid-inducible gene I/melanoma differentiation-associated protein 5 signalling pathways. Here, we demonstrated that the 3C-like protease (3CLSP) of PRRSV prevented the induction of IFN-β by cleaving MAVS in a proteasome- and caspase-independent manner. Moreover, this cleavage ability was dependent on the protease activity of 3CLSP. Mutations specifically disrupting the cysteine protease activity of 3CLSP eliminated MAVS cleavage and the inhibition of IFN induction. Subsequently, we determined that 3CLSP cleaved MAVS at Glu268. Remarkably, a MAVS point mutation at Glu268 rendered MAVS resistant to 3CLSP cleavage. These results reveal a novel PRRSV mechanism to escape host immunity by directly cleaving MAVS.
Introduction
Porcine reproductive and respiratory syndrome (PRRS) leads to enormous economic losses in swine production worldwide (Neumann et al., 2005). The prevalent clinical manifestations of PRRS are severe reproductive failure in sows and gilts and respiratory problems in pigs of all ages (Albina, 1997; Halbur et al., 1996). Its causative agent, PRRSV, which is classified in the order Nidovirales, family Arteriviridae, genus Arterivirus, is an enveloped, positive-sense ssRNA virus (Music and Gagnon, 2010). The PRRSV genome contains ORF1a, ORF1b, ORF2a, ORF2b, ORF3–7 and ORF5a, which was most recently identified. ORF1a and ORF1b encode two long non-structural polyproteins, pp1a and pp1ab. Non-structural protein 4 (nsp4) is a 3C-like serine protease (3CLSP), which processes pp1a/pp1ab of the virus into 10 mature non-structural proteins (nsp3–nsp12) (Tian et al., 2009; Fang & Snijder, 2010). Structural analysis of 3CLSP has revealed two chymotrypsin-like β-barrel domains and an extra C-terminal α/β-domain with a canonical catalytic triad located in the open cleft between the two β-barrel domains, which comprises Ser118, His39 and Asp64.
Type I IFNs, which comprise 13 subtypes for IFN-α and a single subtype for IFN-β, IFN-ω, IFN-κ and IFN-ε in humans, are the main antiviral cytokines in the innate immune response (Bowie and Unterholzner, 2008; Sadler and Williams, 2008). The innate immune response recognizes viral infections through host pattern recognition receptors (PRRs), which trigger antiviral immune responses (Albina, 1997). Pathogen-associated molecular patterns (PAMPs) trigger type I IFN expression through the activation of retinoic acid-inducible gene I (RIG-I)-like receptors and Toll-like receptors (Kawai and Akira, 2006). RIG-I and melanoma differentiation-associated protein 5 (MDA5) form a known receptor for intracellular viral dsRNA. However, RIG-I can also recognize viral ssRNA lacking a 5′ cap or with three phosphates at the 5′ end. On engaging viral dsRNA or ssRNA, RIG-I and MDA5 become activated and interact with the caspase activation and recruitment domain (CARD) of the mitochondrial antiviral signalling protein (MAVS), which is anchored to the mitochondrial outer-membrane through its C-terminal transmembrane (TM) domain (Hou et al., 2011). MAVS forms prion-like aggregates that interact with cytosolic signalling proteins, such as TNF receptor-associated factors, leading to the activation of IκB kinase and serine/threonine protein kinase. Subsequently, the transcription factors NF-κB and IFN regulatory factor 3 (IRF3) are activated through phosphorylation-dependent mechanisms, migrate into the nucleus and activate promoters of type I IFNs (Albina, 1997; Goodbourn et al., 2000). MAVS plays an essential role in the RIG-I and MDA5 signalling pathways, which is a shared pathway that activates both NF-κB and IRF3.
PRRSV infection leads to delayed production of neutralizing antibodies and suppression of innate immune responses. PRRSV-infected pigs do not invoke a type I IFN response (Sun et al., 2012). Many strategies in which PRRSV regulates type I IFN responses have been identified in several reports describing at least four non-structural proteins (nsp1, nsp2, nsp4 and nsp11) that play significant roles in IFN suppression pathways (Kim et al., 2010; Sun et al., 2012). A recent study reported that PRRSV nsp4 has the ability to inhibit IFN-β production by targeting the E349/S350 site of NF-κB essential modulator (NEMO) (Huang et al., 2014). PRRSV 3CLSP is multifunctional and can suppress host antiviral signalling pathways through a variety of mechanisms. In the present study, we found that PRRSV 3CLSP inhibits IFN-β production stimulated by Sendai virus (SeV) through mediation of MAVS cleavage. Furthermore, we showed that 3CLSP cleaves MAVS at E268/G269, and that the cleavage products lose the ability to individually activate type I IFN responses.
Results
PRRSV 3CLSP inhibits SeV-induced type I IFN expression
The results of our previous study demonstrated that PRRSV suppresses IFN production by interfering with RIG-I signalling pathways (Luo et al., 2008). To determine whether PRRSV 3CLSP inhibits IFN-β induction, we transfected expression vectors encoding 3CLSP into HEK-293T cells together with a luciferase reporter plasmid under the control of the porcine IFN-β promoter (IFN-β–Luc) and an internal control reporter plasmid (pRL-TK). At 24 h post-transfection, cells were treated with SeV, which can activate IFN expression, for 8 h and then measured luciferase activity. Overexpression of 3CLSP strongly suppressed SeV-stimulated IFN-β promoter activity, compared with cells transfected with an empty vector and 3CLSP inhibited IFN-β activation in a dose-dependent manner (Fig. 1a). These results showed that 3CLSP blocked SeV-induced IFN-β activation.
Fig. 1.

3CLSP inhibits the promoter activation of IFN-β, IRF3 and NF-κB, and the protease activity of 3CLSP is required to block SeV-mediated IFN-β induction. (a) PRRSV 3CLSP inhibits the promoter activation of IFN, IRF-3 and NF-κB. IFN-β–Luc, IRF3–Luc and NF-κB–Luc were co-transfected into HEK-293T cells, together with the pRL-TK plasmid and increasing quantities (0, 0.1, 0.2, 0.4 or 0.8 μg) of plasmids encoding 3CLSP. After 24 h transfection, the cells were treated with SeV for 8 h and then luciferase assays were performed. (b) 3CLSP blocks RIG-I-mediated IFN-β induction. HEK-293T cells were co-transfected with IFN-β–Luc, the pRL-TK plasmid and pCAGGS-HA-nsp4 together with constructs expressing RIG-I, its constitutively active mutant RIG-IN, MDA-5 or MAVS. Luciferase assays were performed 32 h after transfection. **P < 0.01 (3CLSP vs vector). Results are shown as means ± sd.
3CLSP disrupts RIG-I/MDA5 signalling for activation of both IRFs and NF-κB
To further determine whether PRRSV 3CLSP impairs the activation of IRF3 and/or NF-κB, which are essential IFN-β transcription factors, HEK-293T cells were co-transfected with expression vectors encoding 3CLSP and pRL-TK together with IRF3–Luc and NF-κB–Luc reporter plasmids. The results showed that 3CLSP overexpression inhibited the SeV-induced promoter activities of IRF3 and NF-κB in a dose-dependent manner (Fig. 1a). These data also revealed that 3CLSP efficiently antagonizes IFN-β production by impairing the activation of IRF3 and NF-κB signalling.
To further investigate whether 3CLSP blocks RIG-I-mediated IFN-β induction, HEK-293T cells were co-transfected with 3CLSP expression plasmids and expression constructs encoding RIG-I, its constitutively active mutant RIG-IN, MDA-5 or MAVS, together with IFN-β–Luc and pRL-TK. Luciferase activities were examined 32 h after co-transfection. As shown in Fig. 1(b), 3CLSP overexpression inhibited RIG-I/MDA5-mediated signalling to antagonize IFN-β expression.
3CLSP activity is indispensable for nsp4 to block IFN-β production
3CLSP, which contains a canonical catalytic triad of His39-Asp64-Ser118, is the main proteolytic enzyme of the viral polyprotein, and the activity of 3CLSP is abolished by any mutation to the amino acids in the catalytic triad (Tian et al., 2009). To determine whether serine protease catalytic activity is 3CLSP mediated in IFN-β antagonism, we constructed a series of catalytically deficient 3CLSP mutants, H39A, D64A and S118A. Each of the mutant vectors or a 3CLSP expression plasmid was then co-transfected into HEK-293T cells along with the IFN-β–Luc, IRF3–Luc or NF-κB–Luc vectors and pRL-TK. Luciferase activities were measured after SeV treatment for 8 h. Mock cells were induced to IFN-β expression by SeV treatment. In contrast to WT 3CLSP, 3CLSP mutants did not significantly inhibit SeV-induced activities of the IFN-β promoter and IRF3-responsive promoter (Fig. 2). These results showed that 3CLSP activity is essential for nsp4 inhibition of IFN-β production.
Fig. 2.

3CLSP suppression of IFN-β induction is dependent on protease activity. HEK-293T cells were co-transfected with IFN-β–Luc, IRF3–Luc or NF-κB–Luc along with pRL-TK plasmid and the 3CLSP mutants/3CLSP expression plasmids (0.8 μg). An empty vector (pCAGGS-HA) was used as a control. At 24 h post-transfection, cells were treated with SeV for 8 h and then luciferase activity was measured. *P < 0.05 (WT 3CLSP vs 3CLSP mutants). WT 3CLSP significantly inhibited IFN-β expression compared with the mutants and mock cells. Results are shown as means ± sd;ns, not significant.
PRRSV 3CLSP blocks IFN-β induction by cleaving MAVS
MAVS is a crucial adaptor protein in the antiviral signalling pathway, which has been shown to induce activation of both IRF3 and NF-κB and thus type I IFN production (Seth et al., 2005). MAVS acts as a shared bridge protein between IRF3 and NF-κB signalling. RIG-I/MDA5 signalling to IRFs and NF-κB diverges at MAVS in human and mouse cells. The results of this experiment showed that PRRSV 3CLSP disrupted RIG-I/MDA5 signalling for activation of both IRFs and NF-κB. The results of our previous study indicated that MAVS may be the target through which PRRSV interference suppresses IFN-β production (Luo et al., 2008). PRRSV 3CLSP-mediated inhibition of IFN activation may be caused by 3CLSP targeting of MAVS. To determine whether MAVS is the proteolytic target of PRRSV 3CLSP, HEK-293T cells were co-transfected with nsp4 and Flag–MAVS expression plasmids. At 32 h after co-transfection, Western blotting was performed to assess the interactions between 3CLSP and MAVS. Compared with an empty vector, co-expression of 3CLSP led to a reduction in MAVS abundance, concomitant with the generation of a faster-migrating MAVS fragment (∼37 kDa), presumably a MAVS cleavage product (Fig. 3a). Importantly, the degree of MAVS cleavage was related to 3CLSP expression in a dose-dependent manner (Fig. 3b).
Fig. 3.
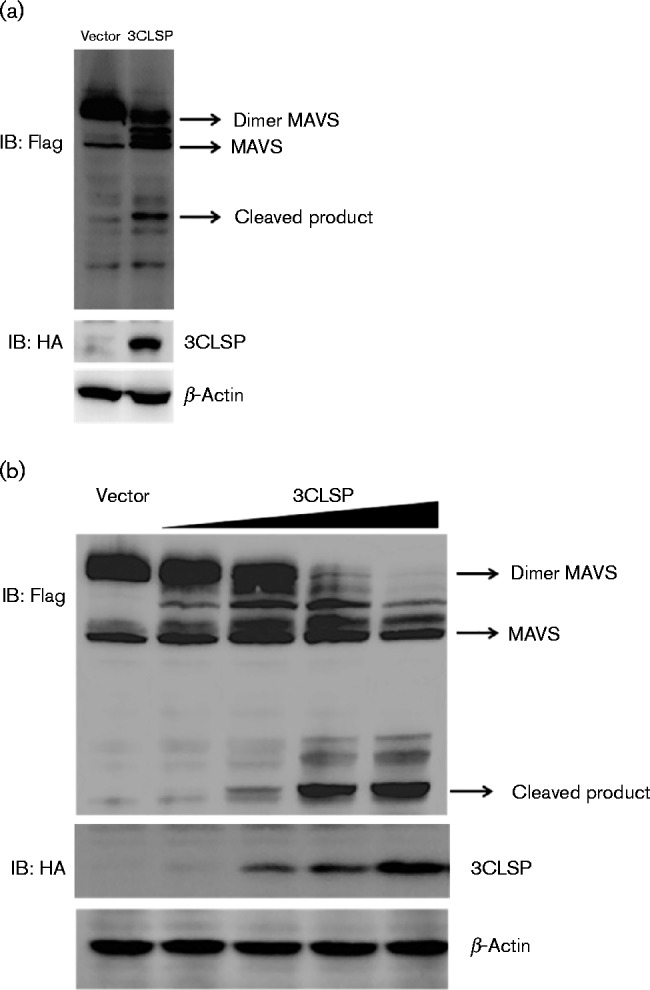
PRRSV 3CLSP cleaves MAVS. (a) HEK-293T cells were transfected with Flag-tagged MAVS expression plasmids (3 μg) along with 3CLSP expression plasmids (0.5 μg). At 32 h after co-transfection, the cells were collected and analysed by Western blotting. (b) HEK-293T cells were transfected with Flag-tagged MAVS expression plasmids (3 μg) along with increasing quantities (0, 0.1, 0.2, 0.4 or 0.8 μg) of plasmid encoding 3CLSP. The cells were collected and analysed by Western blotting at 32 h post-transfection.
We found that PRRSV 3CLSP cleaved MAVS in a dose-dependent manner. Subsequently, we assessed whether expression of PRRSV 3CLSP was sufficient to cleave endogenous MAVS in infected cells. Therefore, Marc-145 cells were infected with various doses of PRRSV or UV-inactivated PRRSV (UV-PRRSV), and endogenous MAVS protein was detected using endogenous MAVS antibodies. Although no cleavage of MAVS was observed, MAVS expression was significantly reduced in PRRSV-infected cells compared with non-infected cells (Fig. 4). These results further indicated that endogenous MAVS closely interacts with PRRSV in host cells.
Fig. 4.
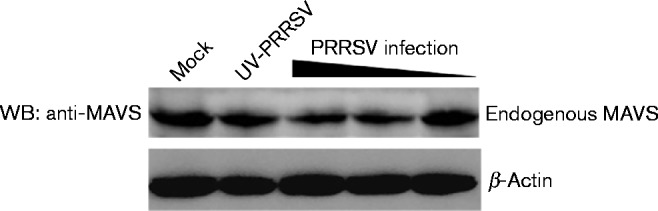
Marc-145 cells were infected with different doses (m.o.i. of 0.01, 0.1 and 1) of PRRSV or UV-PRRSV as a control. The cells were lysed 36 h p.i. and analysed by Western blotting (WB) using rabbit anti-MAVS antibody.
3CLSP activity is responsible for MAVS cleavage
We showed that 3CLSP activity is indispensable to nsp4 to block IFN-β production. Therefore, we next investigated whether 3CLSP cleaves MAVS via its protease activity. Compared with WT 3CLSP, the mutant vectors of 3CLSP (H39A, D64A and S118A) lost the ability to cleave MAVS (Fig. 5a). Hence, MAVS cleavage by PRRSV 3CLSP appears to be dependent on 3CLSP activity.
Fig. 5.
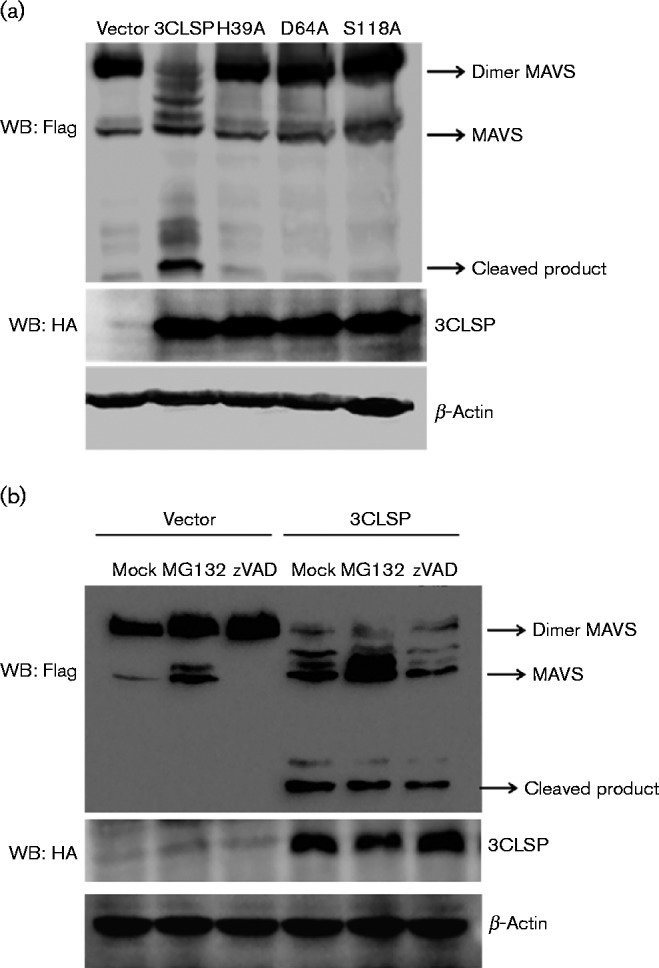
3CLSP activity is indispensable to MAVS cleavage. (a) HEK-293T cells were transfected with Flag-tagged MAVS expression plasmids (3 μg) along with the 3CLSP or 3CLSP mutant plasmids (0.5 μg). The cells were lysed at 32 h post-transfection and analysed by Western blotting (WB). (b) HEK-293T cells were transfected with Flag-tagged MAVS expression plasmids (3 μg) along with the 3CLSP expression plasmid or an empty vector (0.5 μg). At 24 h post-transfection, the cells were treated with MG132 (20 μM) or zVAD-FMK (20 μM) for 8 h. The cells were lysed and analysed by Western blotting.
Induction of cell death reportedly leads to rapid MAVS cleavage independent of the apoptosis and proteasome pathways (Scott and Norris, 2008). To investigate whether 3CLSP cleavage of MAVS was also through the apoptosis and/or proteasome pathways, MAVS expression plasmids along with plasmids encoding 3CLSP were transfected into HEK-293T cells. At 24 h post-transfection, the cells were treated with the caspase inhibitor z-VAD-FMK or the proteasome inhibitor MG132 for 8 h. The results showed that neither z-VAD-FMK nor MG132 affected MAVS cleavage by 3CLSP (Fig. 5b). Therefore, we concluded that MAVS cleavage is specifically attributable to the protease activity of 3CLSP and is independent of the apoptosis and proteasome pathways.
PRRSV 3CLSP cleaves MAVS at Glu268
Previous studies have indicated that 3CLSP preferentially cleaves glutamic acid/glycine (E/G), glutamic acid/alanine (E/A) and/or glutamic acid/serine (E/S) bonds of viral polyproteins and cellular targets (Tian et al., 2009; Huang et al., 2014). Therefore, we next examined the residues of MAVS for potential 3CLSP cleavage sites. The cleavage product of N-terminal, Flag-tagged MAVS is predicted to be a ∼37 kDa product, while intact MAVS is predicted to be about a 55 kDa product encoding 540 aa. In consideration of the ubiquitination of the N-terminal of MAVS, we predicted that potential 3CLSP cleavage sites existed between aa 250 and 320. Hence, we selected three sites (E268/G269, E274/S275 and E288/A289) to construct MAVS mutants (Fig. 6a), in which the invariant glutamine, which is a possible targeted cleavage site, was mutated to alanine. We co-transfected WT MAVS or MAVS mutants (E268A, E274A and E288A), along with 3CLSP or an empty vector into HEK-293T cells. Compared with WT MAVS, only E268A was resistant to 3CLSP-mediated cleavage in HEK293 cells (Fig. 6a). These results demonstrated that Glu268 is the probable cleavage site of 3CLSP.
Fig. 6.


3CLSP-mediated MAVS cleavage is involved in the inhibition of type I IFN induction. (a) Schematic representation of WT MAVS and its derivatives. HEK-293T cells were co-transfected with Flag-tagged WT MAVS or MAVS mutants (E268A, E274A and E288A) along with 3CLSP or an empty vector. These cells were lysed at 32 h post-transfection and analysed by Western blotting (WB). (b) HEK-293T cells were co-transfected with IFN-β–Luc, the pRL-TK plasmid and a plasmid encoding 3CLSP (0.3 μg) together with the empty vector, MAVS or MAVS (E268A) expression vector (0.3 μg). Luciferase activity was examined at 32 h post-transfection. The MAVS E268A mutant was significantly different from WT MAVS (*P < 0.01). (c) MAVS or MAVS truncated mutants are shown. Complete MAVS contained the N-terminal CARD domain, proline-rich domain and C-terminal TM domain. HEK-293T cells were co-transfected with the indicated reporter plasmid (0.1 μg), pRL-TK (0.1 μg) or the Flag-tagged MAVS expression plasmid (0.5 μg) along with a plasmid encoding Flag-tagged WT MAVS, 3CLSP-induced cleavage fragments of MAVS or an empty vector. **P < 0.01. Results are shown as means ± sd.
Interestingly, the E268A mutation of MAVS preserved its IFN-β promoter activity, while the E268A mutation of MAVS remarkably overcame the 3CLSP blockade on activation of the IFN-β promoter, compared with WT MAVS (Fig. 6b). Together, these results further confirmed that the MAVS cleavage site of PRRSV 3CLSP is E268/G269.
CLSP-induced MAVS cleavage fragments exhibit impaired type I IFN signalling
For MAVS, the N-terminal CARD domain, proline-rich (PRO) domain and C-terminal TM domain are required for induction of type I IFN in both the TLR and RLR signalling pathways (Seth et al., 2005; Moresco et al., 2011). Our results showed that PRRSV 3CLSP cleaved MAVS at E268, which is located between the PRO domain and TM domain. To determine whether either of the 3CLSP-mediated cleavage fragments of MAVS remained active, two MAVS cleavage mutants, the N-terminal fragment (aa 1–268) and C-terminal fragment (aa 269–540) of MAVS were constructed. We then investigated the ability of these two truncated mutants to activate IFN-β and IRF3/NF-κB-dependent promoters. As shown in Fig. 6(c), compared with the full-length MAVS, neither fragment (aa 1–268 and aa 269–540) was capable of efficiently activating any of the three promoters. These results further indicated that nsp4 antagonizes IFN-β expression through 3CLSP-mediated MAVS cleavage and that the cleavage site is at E268.
Discussion
Previous publications have reported that many viruses, including PRRSV, have evolved various strategies to antagonize innate immune responses by blocking the production or action of IFNs (Murtaugh et al., 2002; Seth et al., 2005; Luo et al., 2008; Beura et al., 2010). Although IFNs can efficiently inhibit PRRSV replication (Albina et al., 1998), PRRSV infection triggers poor type I IFN responses and inhibits the production of type I IFNs induced by poly(I : C) or transmissible gastroenteritis virus (Albina et al., 1998; Buddaert et al., 1998; Van Reeth et al., 1999; Miller et al., 2004). Previous studies have revealed that PRRSV nsp4 plays an important role in the inhibition of type I IFN production. It has been reported that IFN-β promoter activation by IRF3 was suppressed to various degrees by PRRSV nsp1, nsp2, nsp4 and nsp11 (Beura et al., 2010), indicating that PRRSV nsp4 may interfere with signalling pathways to type I IFN activation. A very recent study reported that PRRSVnsp4 had the ability to inhibit dsRNA-induced type I IFN expression by PRRSV 3CLSP-mediated cleavage of NEMO (Huang et al., 2014). In the present study, we showed that PRRSV 3CLSP acts as a multifunctional protein that antagonizes type I IFN activation. Besides the cleavage of NEMO, MAVS cleavage by PRRSV 3CLSP presents an alternative mechanism to inhibit IFN-β expression.
IFNs are secreted cytokines and have multifunctional roles in antiviral defence and in shaping adaptive immunity (Goodbourn et al., 2000). Type I IFNs are the main cytokines in the innate immune response against viral infections via induction and activation of a large number of IFN-stimulated genes that code for effector proteins with well-defined functions to limit virus replication (Krebs and Hilton, 2001; Sadler and Williams, 2008). During viral infection, host-cell PRRs recognize PAMPs, such as the lipid envelope, nucleic acids and glycoproteins, as well as dsRNA, the by-product of the replication cycle, and then initiate activation of the IFN pathway. The cytoplasmic RIG-I-like receptors and membrane-associated Toll-like receptors represent two major classes of PRRs, which detect viral infection and activate signalling cascades culminating in the production of type I IFNs. MAVS, which plays an important role in the RLR signalling pathway, contains an N-terminal CARD domain, a PRO region and a C-terminal hydrophobic TM region, which are all requisite for MAVS function. An increasing number of publications have reported that cleavage of the adaptor protein MAVS is important to mediate host innate immune activity. Some viruses can specifically disrupt the signal transduction of MAVS to evade host innate immune responses. For example, enterovirus 71 protease 2Apro, hepatitis C virus protease NS3/4A and hepatitis A virus 3ABC, a precursor of the sole hepatitis A virus protease, can cleave MAVS at different sites to disrupt RIG-I/MDA5 signalling (Li et al., 2005; Yang et al., 2007; Wang et al., 2013). Canine hepacivirus NS3 serine protease and coxsackievirus B 3Cpro can cleave the adaptor proteins MAVS and TRIF (Mukherjee et al., 2011; Parera et al., 2012). MAVS is cleaved during human rhinovirus 1a infection (Drahos and Racaniello, 2009). In our study, 3CLSP-mediated cleavage of MAVS was identified as an alternative molecular mechanism of type I IFN evasion utilized by PRRSV. Our results revealed that PRRSV 3CLSP is able to cleave specific host innate immune molecules via different mechanisms. In addition, the 3C proteases of other viruses have been reported to have similar characteristics to cleave MAVS. These viruses include foot-and-mouth disease virus (Wang et al., 2011), coronavirus (Clementz et al., 2010), bovine viral diarrhea virus (Chen et al., 2007), enterovirus 71 (Lei et al., 2011), coxsackievirus B (Mukherjee et al., 2011) and hepatitis A virus (Qu et al., 2011). Therefore, we speculate that 3CLSP of other arteriviruses, such as equine artervirus, may cleave MAVS or other immune molecules. In future studies, we plan to further investigate this hypothesis.
Western blots demonstrated remarkable reductions of MAVS expression in PRRSV-infected cells compared with mock cells, although endogenous MAVS cleavage was not detected in infected cells. The cause may be as follows: (i) virus–host interaction is more complicated in infected cells – it is possible that the MAVS cleavage fragment was further degraded to undetected cleavage; it only shows the difference of MAVS expression; or (ii) the amount of endogenous MAVS cleavage may be relatively less, and therefore the cleavage cannot be detected in the infected cells as the detecting method may not be sensitive enough.
The PRRSV 3CLSP cleavage site of MAVS is at E268/G269, and this cleavage renders MAVS incapable of activating the IFN-β promoter. MAVS is composed of an N-terminal CARD domain, PRO domain and C-terminal TM domain. Each domain is required for induction of type I IFN activation in both the TLR and RLR signalling pathways (Seth et al., 2005; Moresco et al., 2011). The cleavage of MAVS may interfere or block the IFN signalling pathway initiated by binding of dsRNA to RIG-I or MDA-5. Moreover, MAVS cleavage was specifically attributable to the protease activity of 3CLSP, rather than the apoptosis or proteasome pathways. Previously, Huang et al. (2014) reported that PRRSV nsp4 can antagonize IFN-β expression by targeting NEMO. Here, we determined that nsp4 blocked the IFN signalling pathway by cleaving MAVS. This finding indicates that PRRSV nsp4 plays an important role in PRRSV pathogenesis and immunological responses. MAVS is an upstream signal to NEMO in IFN signalling cascades. MAVS plays an essential role in the RIG-I and MDA5 signalling pathways, which is a shared pathway that activates both NF-κB and IRF3. If the MAVS signal is disrupted, both the NF-κB and IRF3 signalling cascade of IFN-β expression can be inhibited. Nevertheless, NEMO cleavage may only inhibit the NF-κB signalling cascade. In contrast, MAVS cleavage by 3C protease of PRSRV may be more efficient.
In summary, our data indicated that PRRSV 3CLSP can inhibit IFN-β expression by impairing the activation of both IRF3 and NF-κB. Moreover, 3CLSP-mediated proteolytic cleavage of MAVS was directly involved in the inhibition of type I IFN activation and cleavage of MAVS at Glu268. Our future studies will aim to determine which bond is cleaved by the virus protease. These findings should further contribute to our understanding of the molecular mechanisms of innate immunity evasion strategies utilized by PRRSV.
Methods
Viruses, cells and chemicals
SeV was obtained from the Centre of Virus Resource and Information, Wuhan Institute of Virology, Chinese Academy of Sciences (Wuhan, China). HEK-293T cells were cultured in RPMI 1640 (Invitrogen) supplemented with 10 % FBS at 37 °C in a humidified 5 % CO2 incubator. The broad caspase inhibitor z-VAD-FMK and the proteasome inhibitor MG132 were obtained from the Beyotime Institute of Biotechnology. Mouse mAbs against Flag and haemagglutinin (HA) were purchased from Medical and Biological Laboratories. mAbs against β-actin were obtained from Beyotime Institute of Biotechnology. Rabbit polyclonal antibodies directed against MAVS were obtained from Proteintech.
Plasmids
Construction of the 4 × PRDIII/I–Luc (referred to as IRF3–Luc) and 4 × PRDII–Luc (referred to as NF-κB–Luc) luciferase reporter plasmids has been described elsewhere (Wang et al., 2011). The expression plasmids for WT RIG-I (pEF-Flag-RIG-I), its constitutively active mutant (pEF-Flag-RIG-IN), which only contains the active CARD domain of the RIG-I N terminus and has the negative regulating domain of the RIG-I C terminus deleted, and p125–Luc (IFN-β–Luc) were gifts from T. Fujita (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). The cDNA expression constructs encoding porcine MAVS and MDA5 have been described previously (Wang et al., 2011). The empty vector control was designated ‘mock’. cDNA encoding nsp4 of PRRSV strain WUH3 was amplified by standard reverse transcription PCR from total RNA extracted from Marc-145 cells infected with PRRSV strain WUH3 and cloned into vector pCAGGS-HA. Mutagenesis of MAVS (E268A, E274A and E288A) and PRRSV 3CLSP (H39A, D64A and S1188A) constructs was carried out by overlap-extension PCR using PrimeSTAR HS DNA polymerase (TaKaRa Bio).
Luciferase reporter gene assay
HEK-293T cells grown in 24-well plates were co-transfected with 0.1 μg reporter plasmid per well, 0.1 μg plasmid pRL-TK (Promega) per well as an internal control for normalization of transfection efficiency, and the indicated expression plasmids or an empty control plasmid. Where indicated, the cells were infected with SeV (10 haemagglutinating activity units per well) 24 h after co-transfection. The cells were lysed after 12 h and firefly luciferase and Renilla luciferase activities were determined using a Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. The data are presented as relative firefly luciferase activities normalized to Renilla luciferase activities (mean ± sd) and are representative of three independent experiments.
Immunoblot analysis
Briefly, HEK-293T cells grown in 60 mm dishes were transfected with the appropriate plasmids. After 36 h, the cells were harvested with lysis buffer, and protein concentrations in whole-cell extracts were measured. Equal sample amounts were then subjected to SDS-PAGE and analysed for the expression of WT and mutant MAVS proteins by Western blotting using an anti-Flag antibody (Medical & Biological Laboratories). To confirm the expression levels of HA-tagged WT and mutant PRRSV 3CLSP proteins, an anti-HA antibody (Medical & Biological Laboratories) was used for immunoblotting. β-Actin expression was detected with an anti-β-actin mouse mAb (Beyotime Institute of Biotechnology) to demonstrate equal protein sample loading. The experiments were repeated three times. The data are representative of three independent experiments.
Statistical analysis
The bioinformatics program GraphPad Prism was used for statistical analysis. ANOVA and Student's t-test were used to evaluate statistical differences of luciferase induction. The results were considered to be statistically significant for P < 0.05.
Acknowledgements
We would like to thank Dr L. Rui and Dr W. Dang for technical assistance and helpful discussions. This work was supported by grants from the Special Fund for Agro-scientific Research in the Public Interest of China (grant no. 201203039), the National Key Basic Research Program of China (973 Program) (grant no. 2014CB542703) and the Natural Science Foundation of China (grant no. 30800046).
References
- Albina E. (1997). Epidemiology of porcine reproductive and respiratory syndrome (PRRS): an overview Vet Microbiol 55309–316 10.1016/S0378-1135(96)01322-3. [DOI] [PubMed] [Google Scholar]
- Albina E., Piriou L., Hutet E., Cariolet R., L'Hospitalier R. (1998). Immune responses in pigs infected with porcine reproductive and respiratory syndrome virus (PRRSV) Vet Immunol Immunopathol 6149–66 10.1016/S0165-2427(97)00134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura L.K., Sarkar S.N., Kwon B., Subramaniam S., Jones C., Pattnaik A.K., Osorio F.A. (2010). Porcine reproductive and respiratory syndrome virus nonstructural protein 1β modulates host innate immune response by antagonizing IRF3 activation J Virol 841574–1584 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie A.G., Unterholzner L. (2008). Viral evasion and subversion of pattern-recognition receptor signalling Nat Rev Immunol 8911–922 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddaert W., Van Reeth K., Pensaert M. (1998). In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV) Adv Exp Med Biol 440461–467 10.1007/978-1-4615-5331-1_59. [DOI] [PubMed] [Google Scholar]
- Chen Z., Rijnbrand R., Jangra R.K., Devaraj S.G., Qu L., Ma Y., Lemon S.M., Li K. (2007). Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus Virology 366277–292 10.1016/j.virol.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., other authors (2010). Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases J Virol 844619–4629 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drahos J., Racaniello V.R. (2009). Cleavage of IPS-1 in cells infected with human rhinovirus J Virol 8311581–11587 10.1128/JVI.01490-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Snijder E.J. (2010). The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins Virus Res 15461–76 10.1016/j.virusres.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodbourn S., Didcock L., Randall R.E. (2000). Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures J Gen Virol 812341–2364. [DOI] [PubMed] [Google Scholar]
- Halbur P.G., Paul P.S., Meng X.J., Lum M.A., Andrews J.J., Rathje J.A. (1996). Comparative pathogenicity of nine US porcine reproductive and respiratory syndrome virus (PRRSV) isolates in a five-week-old cesarean-derived, colostrum-deprived pig model J Vet Diagn Invest 811–20 10.1177/104063879600800103. [DOI] [PubMed] [Google Scholar]
- Hou F., Sun L., Zheng H., Skaug B., Jiang Q.X., Chen Z.J. (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response Cell 146448–461 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Zhang Q., Guo X.K., Yu Z.B., Xu A.T., Tang J., Feng W.H. (2014). Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-κB essential modulator J Virol 8810934–10945 10.1128/JVI.01396-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Akira S. (2006). Innate immune recognition of viral infection Nat Immunol 7131–137 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kim O., Sun Y., Lai F.W., Song C., Yoo D. (2010). Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells Virology 402315–326 10.1016/j.virol.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs D.L., Hilton D.J. (2001). SOCS proteins: negative regulators of cytokine signaling Stem Cells 19378–387 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- Lei X., Sun Z., Liu X., Jin Q., He B., Wang J. (2011). Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3 J Virol 858811–8818 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.D., Sun L., Seth R.B., Pineda G., Chen Z.J. (2005). Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity Proc Natl Acad Sci U S A 10217717–17722 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R., Xiao S., Jiang Y., Jin H., Wang D., Liu M., Chen H., Fang L. (2008). Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-β-production by interfering with the RIG-I signaling pathway Mol Immunol 452839–2846 10.1016/j.molimm.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L.C., Laegreid W.W., Bono J.L., Chitko-McKown C.G., Fox J.M. (2004). Interferon type I response in porcine reproductive and respiratory syndrome virus-infected MARC-145 cells Arch Virol 1492453–2463 10.1007/s00705-004-0377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresco E.M., Vine D.L., Beutler B. (2011). Prion-like behavior of MAVS in RIG-I signaling Cell Res 211643–1645 10.1038/cr.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A., Morosky S.A., Delorme-Axford E., Dybdahl-Sissoko N., Oberste M.S., Wang T., Coyne C.B. (2011). The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling PLoS Pathog 7e1001311. 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh M.P., Xiao Z., Zuckermann F. (2002). Immunological responses of swine to porcine reproductive and respiratory syndrome virus infection Viral Immunol 15533–547 10.1089/088282402320914485. [DOI] [PubMed] [Google Scholar]
- Music N., Gagnon C.A. (2010). The role of porcine reproductive and respiratory syndrome (PRRS) virus structural and non-structural proteins in virus pathogenesis Anim Health Res Rev 11135–163 10.1017/S1466252310000034. [DOI] [PubMed] [Google Scholar]
- Neumann E.J., Kliebenstein J.B., Johnson C.D., Mabry J.W., Bush E.J., Seitzinger A.H., Green A.L., Zimmerman J.J. (2005). Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States J Am Vet Med Assoc 227385–392 10.2460/javma.2005.227.385. [DOI] [PubMed] [Google Scholar]
- Parera M., Martrus G., Franco S., Clotet B., Martinez M.A. (2012). Canine hepacivirus NS3 serine protease can cleave the human adaptor proteins MAVS and TRIF PLoS One 7e42481. 10.1371/journal.pone.0042481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L., Feng Z., Yamane D., Liang Y., Lanford R.E., Li K., Lemon S.M. (2011). Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD PLoS Pathog 7e1002169. 10.1371/journal.ppat.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler A.J., Williams B.R. (2008). Interferon-inducible antiviral effectors Nat Rev Immunol 8559–568 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott I., Norris K.L. (2008). The mitochondrial antiviral signaling protein, MAVS, is cleaved during apoptosis Biochem Biophys Res Commun 375101–106 10.1016/j.bbrc.2008.07.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R.B., Sun L., Ea C.K., Chen Z.J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3 Cell 122669–682 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Sun Y., Han M., Kim C., Calvert J.G., Yoo D. (2012). Interplay between interferon-mediated innate immunity and porcine reproductive and respiratory syndrome virus Viruses 4424–446 10.3390/v4040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X., Lu G., Gao F., Peng H., Feng Y., Ma G., Bartlam M., Tian K., Yan J., other authors (2009). Structure and cleavage specificity of the chymotrypsin-like serine protease (3CLSP/nsp4) of porcine reproductive and respiratory syndrome virus (PRRSV) J Mol Biol 392977–993 10.1016/j.jmb.2009.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Reeth K., Labarque G., Nauwynck H., Pensaert M. (1999). Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: correlations with pathogenicity Res Vet Sci 6747–52 10.1053/rvsc.1998.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Fang L., Li P., Sun L., Fan J., Zhang Q., Luo R., Liu X., Li K., other authors (2011). The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase J Virol 853758–3766 10.1128/JVI.02589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Xi X., Lei X., Zhang X., Cui S., Wang J., Jin Q., Zhao Z. (2013). Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses PLoS Pathog 9e1003231. 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Liang Y., Qu L., Chen Z., Yi M., Li K., Lemon S.M. (2007). Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor Proc Natl Acad Sci U S A 1047253–7258 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]