

Abstract
Mesenchymal stem cells (MSCs) are a type of adult stem cell that have been exploited for the treatment of a variety of diseases, including cancer. In particular, MSCs have been studied extensively for their ability to treat glioblastoma (GBM), the most common and deadly form of brain cancer in adults. MSCs are attractive therapeutics because they can be obtained relatively easily from patients, are capable of being expanded in vitro, can be easily engineered, and because they inherently are capable of homing to tumors, making them ideal vehicles for delivering biological anti-tumoral agents. Oncolytic viruses are promising biological therapeutic agents that have been employed in the treatment of GBMs, and MSCs are currently being explored as means of delivering these viruses. Here we review the role of MSCs in the treatment of GBMs, focusing on the intersection of MSCs and oncolytic viruses.
Keywords: Mesenchymal Stem Cell, Gliomas, Adenovirus
Introduction
Mesenchymal stem cells (MSCs) have gained increasing attention over the past several decades because of their potential application in the treatment of disease. The therapeutic prospect of MSCs lies primarily in their inherent capacity to home to injured or inflamed tissue, their ability to secrete anti-inflammatory, tissue-rejuvenating factors, and the ease with which they can be modified or engineered to serve as delivery vehicles of exogenous biological agents. Unmodified MSCs have been used in the treatment of degenerative diseases [1, 2], myocardial infarction [3], stroke [4], and trauma [5]. Engineered MSCs have been used as cellular carries of anti-tumoral agents in various cancers, including glioblastoma (GBM), the most common and deadly malignant brain tumor in adults [6–11]. Multiple studies have shown that MSCs avidly home to solid tumors, including GBMs, presumably because the microenvironment or stromal milieu of cancer, particularly brain tumors, is similar to that of non-healing wounds. Of the various anti-cancer cargoes that have been loaded into MSCs, oncolytic viruses are amongst the most promising in the treatment of brain tumors, and MSCs loaded with oncolytic viruses will soon be tested in clinical trials in patients with GBM. Oncolytic viruses are replication competent viruses that have been genetically modified to selectively infect and replicate in tumor cells compared with normal cells. This review focuses on the recent advances in the use of MSCs in the treatment of brain tumors, emphasizing the role of MSCs as delivery vehicles for oncolytic viruses.
Therapeutic Challenges of Glioblastoma
GBM (World Health Organization [WHO] Grade IV astrocytoma) is the most common and deadly primary adult brain tumor. Despite aggressive microsurgery followed by concurrent radiation/chemotherapy and adjuvant chemotherapy, patients with GBM survive on average less than 15 months following diagnosis [12]. Recent clinical trials have shown that altering the dose or schedule of standard cytotoxic chemotherapy or inhibiting angiogenesis has little impact on patient survival [13–15]. Likewise, targeted therapies that have been effective in other cancers have not been effective against brain tumors. This poor outcome is due to the complex molecular and cellular biology of GBMs. GBMs are highly infiltrative as tumor cells migrate into normal brain parenchyma, which narrows the therapeutic window of most therapies. Furthermore, GBMs contain glioma stem-like cells (GSCs) that render GBMs resistant to most therapies. Finally, GBMs are very heterogeneous, containing many different cellular clones that results in outgrowth of therapeutic resistant subclones. The poor outcome is equally due to the inability to deliver therapeutic agents to the tumor because of the blood brain/tumor barrier (BBB/BTB) which functionally excludes most drugs from entering the tumor. Given these problems there has been an urgent need both to develop novel therapies for GBM and to develop innovative ways to deliver these therapies.
Stem Cells as Delivery Vehicles in GBM
Recent evidence suggests that stem cells may be effective delivery vehicles for the treatment of cancer including brain tumors. Originally, stem cells, particularly hematopoietic stem cells, were utilized in cancer therapy to replace bone marrow containing residual tumor cells after “conditioning” with aggressive chemotherapy as part of autologous, allogeneic, or syngeneic bone marrow transplant strategies. Since then, the application of stem cells in cancer therapy has expanded to their use as biological vehicles for delivering novel anti-tumor therapies to solid tumors[6], especially brain tumors [7].
Neural Stem Cells (NSCs) were the first type of stem cells to be investigated as potential cellular vehicles to deliver therapeutic agents to brain tumors. NSCs are found in specific periventricular regions of the central nervous system (CNS) and are destined to become the cells comprising the brain, including neurons and glial cells (astrocytes, oligodendrocytes, and ependymal cells). Because NSCs possess an intrinsic capacity for extensive migration within the brain [16], early research investigated whether these migratory properties could allow NSCs to track down infiltrating tumor cells that reside outside of the main tumor mass. The seminal publication in 2000 by Aboody et al. first described the use of NSCs in the treatment of gliomas [17]. They showed that NSCs (genetically immortalized by transfection with MYC) could distribute themselves throughout the tumor and could migrate to infiltrative tumor cells that extended out of the main tumor mass and dispersed into normal brain. Equally important, they showed that these immortalized NSCs could be engineered to carry the therapeutic transgene cytosine deaminase (CD), an enzyme that converts the prodrug 5-fluorocytosine (5-FC) to 5-fluorouracil (5-FU). This publication set the field of cell-based therapies for GBM into motion [17] and since then multiple publications have reported the use of NSCs to deliver a variety of anti-glioma agents, including interleukin-4 (IL-4) [18], IL-12 [19], IL-23 [20], soluble variant of tumor necrosis factor-related apoptosis inducing ligand [19, 21–23], prodrug converting enzymes cytosine deaminase [17, 24, 25], antiangiogenic protein thrombospondin [26], and oncolytic viruses [27, 28].
Because the acquisition of NSCs for clinical use requires isolation of tissue from the brains of fetuses or from the periventricular zone of adult brains during surgery, alternative sources of stem cells were sought. One alternative source has been the adult human bone marrow, which is a rich reservoir of harvestable stem cells. Compared with NSCs, bone marrow stem cells are attractive because: 1) they are easily acquired from patients via aspiration of the iliac crest or sternum, 2) patients can act as their own donors making autologous transplant possible and obviating immune-mediated rejection, and 3) no ethical issues surround their use. Of the various stem cells within the bone marrow, mesenchymal stem cells (MSCs) are particularly attractive for clinical applications because the methods for acquiring MSCs are well established, in vitro culture is straightforward, and the techniques for engineering and manipulating MSCs are known [29]. MSCs also express low level of major histocompatibility complex (MHC) class I molecules and do not express MHC class II on the cell surface, rendering allogeneic transplant feasible [30]. In fact, MSCs generated from adult human healthy donors have been approved for the treatment of acute graft versus host disease in Canada, New Zealand[31] and Japan [32].
The prospect of using MSCs for the treatment of solid tumors was revealed with the seminal publication by Studeny et al. in 2002 [6]. Soon thereafter, the use of MSCs for the treatment of various other types of cancers was reported, including in the treatment of, lung [33], colon [8], ovarian [34], pancreatic [35], renal [11], breast cancers [36] and sarcoma [9, 10]. There are several clinical trials underway evaluating MSCs as delivery vehicles. For example, in a phase I trial, patients with advanced head and neck cancer received intratumoral injection of MSCs transduced with IL-12, called GX-051 (NCT 02079324). In another phase I trial, eligible ovarian cancer patients will undergo intraperitoneal infusion of MSCs loaded with interferon-β (INF-β) (NCT 02530047). In another trial, prostate cancer patients who are scheduled to undergo prostatectomies will receive allogeneic MSCs intravenously. After surgery the relative amount of donor MSC DNA to recipient DNA in prostate specimens will be quantified to determine if systemically infused MSCs home to prostate cancer (NCT 01983709).
The first report of the use of MSCs for the treatment of GBM was in 2004 by Nakamura et al [7]. The investigators showed that rat MSCs could migrate toward syngeneic rat brain tumors after intracranial injection of the MSCs into the contralateral hemisphere. They further showed that these MSCs were able to deliver the anti-tumor cargo IL-2. Shortly thereafter, Nakamizo et al. demonstrated for the first time that human bone marrow-derived MSCs were capable of homing to human GBMs after intravascular injection. Specifically, after intracarotid injection into glioma-bearing mice, fluorescently labeled MSCs were visualized exclusively within the tumor, but were absent from the normal brain parenchyma. They showed human MSCs engineered to deliver IFN-β increased survival of tumor-bearing mice compared with control treatment mice. Subsequent studies by Shinojima et al. showed that when delivered into the carotid artery human MSCs also home to intracranial patient-derived GSC xenografts, which are the current gold-standard models of human gliomas [37]. Sasportas et al. and Menon et al. also demonstrated that bone marrow-derived MSCs can be used to deliver pro-apoptotic proteins to malignant glioma cells. In these independent studies, MSCs were transduced with a lentivirus expressing secretable tumor necrosis factor related apoptosis-inducing ligand (S-TRAIL), and were injected into glioma xenografts resulting in inhibition of tumor growth by inducing apoptosis [38, 39]. Since these studies, MSCs have been used to deliver other therapeutic agents including CD [40, 41], herpes simplex virus type I thymidine kinase (HSV-TK) combined with valproic acid (VPA) [42], single-chain antibody (scFv) against EGFRvIII [43], and nanoparticle encapsulated doxorubicin [44].
Although delivery by direct intratumoral injection or by intravascular delivery into the carotid artery have been shown to be effective, the efficacy of intravenous delivery of MSCs has been more controversial. On the one hand, Yang et al. demonstrated that human bone marrow MSCs migrated to brainstem gliomas after intravenous (tail vein) injection in nude mice [45]. On the other hand, Bexell and colleagues could not demonstrate efficient MSCs homing in rat syngeneic glioma models after intravenous injection [46]. However, they also reported rat MSCs did not migrate toward rat gliomas after extratumoral implantation into the ipsilateral or contralateral hemisphere [47]. This lack of homing seen by these investigators might be due to different species (i.e., human MSCs versus rat MSCs) or differences in the factors produced by the tumor that mediate MSC homing, e.g. TGF-β[37], platelet-derived growth factor-B [48], and vascular endothelial growth factor A [49]. Nevertheless, others have corroborated that intravenous injection of MSCs is inefficient due to the sequestering of MSCs in the lungs [50]. Alternatively, others have shown that MSCs can be delivered by intranasal injection, or by encapsulating MSCs in a hydrogel prior to transplantation. This technique significantly improved survival in several glioma models (reviewed in [51]).
Although originally and most commonly isolated from bone marrow, MSCs have now been identified in most organs of the body allowing more options for isolating MSCs. Isolation of MSCs from different sources has been demonstrated and applied for GBM treatment, such as umbilical cord blood [52], adipose tissue [53], and amniotic fluid [54]. Adipose tissue is an attractive source for MSCs because of the easy and repeatable access to subcutaneous adipose tissue, and because adipose tissue is a high yield source of MSCs [55]. Umbilical cord blood also may be a useful source of MSCs, because umbilical cord blood is routinely discarded at parturition [56].
It is now well accepted that MSCs can migrate toward brain tumors after intracranial injection, can home to brain tumors after intravascular injection, and can deliver a variety of anti-glioma cargoes as described in Table 1. In addition to their ability to deliver secretable biological molecules, MSCs have also been used to deliver live oncolytic viruses. Given the growing enthusiasm for these viruses to treat cancer, particularly GBM, it is worthwhile to review in more detail the intersection of MSCs and oncolytic viral therapies.
Table 1.
MSCs as therapeutic delivery vectors for brain tumor
| Therapeutic | Transgene/modification | MSC source | Route of admin. | Glioma (source) | Ref. |
|---|---|---|---|---|---|
| Cytokine | IL-2 | rat | i.c. / i.t. | 9L (rat) | [7] |
| IL-12 | human | i.t. | GL26 (mouse) | [52] | |
| IFN-β | human | i.t. / i.c.r. | U87 (human) | [126] | |
| sTRAIL | human | i.t. | Gli36 (human) | [38] | |
| human | i.c. | U87 (human) | [39] | ||
| Prodrug converting enzymes | CD | rat | i.t. | C6 (rat) | [40] |
| rat | i.t. | 9L (rat) | [41] | ||
| HSV-tk and VPA | human | i.t. | U87 (human) | [42] | |
| rCE | human | i.t. | F98 (rat) | [53] | |
| Endostatin | human | i.t. | U87 (human) | [54] | |
| Oncolytic viruses | CRAd | human | i.c.r. | U87 (human) | [50] |
| Antibodies | scFv anti-EGFRv III | human | i.t. | U87 (human) | [43] |
| Nanoparticles | Silica nanorattle-DOX | human | i.t. | U87 (human) | [44] |
Abbreviations: CD, cytosine deaminase; CRAd, conditionally-replicating adenovirus; HSV-tk, Herpes simplex virus type 1 thymidine kinase; DOX, doxorubicin; i.c., intracerebral; i.t., intratumoral; i.v., intravenous; i.c.r., intracarotid; IFN, interferon; IL, interleukin; LNCs, lipid nanocapsules; MSC, mesenchymal stem cell; PLA-NPs, poly-lactic acid nanoparticles; rCE, rabbit carboxylesterase enzyme; scFv, single-chain antibody fragment; sTRAIL, soluble variant of tumor necrosis factor-related apoptosis inducing ligand; VPA, valproic acid.
Virotherapy for the treatment of GBM: history and challenges
Oncolytic viruses are live, replication competent viruses that selectively replicate within cancer cells. Viral infection causes the cancer cells to lyse, which releases more viral particles into the surrounding tissue. These new viral progeny can subsequently infect neighboring cancer cells and with each round of infection, replication, lysis and release, the virus can propagate and spread throughout the tumor, potentially eradicating the entire tumor mass.
Virotherapy actually began in the 1950s when early pioneers of virotherapy, such as Moore [57], Southam [58], and others [59, 60], sought to identify cancer-killing viruses that were not toxic to normal tissues, based on the notion of “viral evolution,” i.e., that viruses could be adapted and selected for propagation in tumors [61]. Unfortunately, these initial attempts elicited unpromising results because of the unwanted toxic side effects of the viruses on normal cells and tissues [57, 62]. It was not until the development of recombinant DNA technology in the 1990s that the potential of viral genome manipulation allowed for the development of viruses that were tumor selective, i.e., they killed tumor cells but not normal cells, and the concept of oncolytic virotherapy really took hold [61]. The first genetically modified oncolytic virus was a modified herpes simplex virus (HSV), called dlsptk, developed in the 1990s by Martuza and colleagues, for the treatment of GBM. This virus contained a deletion in the thymidine kinase (TK) gene [63], which is one of the 70 genes encoded by HSV that is essential for viral replication in non-dividing cells, but not in dividing cells [64, 65]. Because of the deletion in TK, the dlsptk virus was able to replicate only in dividing GBM cells, but not in post-mitotic cells, such as neurons [66]. Since this initial report, a variety of viruses have been used for the treatment of cancer [67]. For brain tumors, studies with HSV spurred the development of viral therapy using adenovirus [68], measles virus [69], poliovirus [70], reovirus [71], retrovirus [72], parvovirus [73], etc. Table 2 lists several virus candidates that have been used in clinical trials for the treatment of GBMs.
Table 2.
Clinical trials to investigate the safety and efficacy of oncolytic viruses in glioma patients
| Virus type | Virus name | Modifications | Phase | Tumor | Route | Status | Combination | ID | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Herpes simplex virus | HSV-1716 | γ34.5(−) | 1 | Newly diagnosed and recurrent HGG | IT | Completed | - | [127–129] | |
| G207 | γ34.5(−), ICP6(−) | 1/2 | Recurrent HGG | IT | Completed | - | NCT00028158 | [130, 131] | |
| 1 | Recurrent HGG | IT | Completed | RT | NCT 00157703 | [97] | |||
| 1 | Recurrent supratentorial brain tumor | IT | Recruiting | RT | NCT 02457845 | ||||
| M032 | γ34.5(−), IL-12 | 1 | Recurrent HGG | IT | Recruiting | - | NCT 02062827 | ||
| G47delta | γ34.5(−), ICP6(−),α47(−) | 1/2 | Recurrent GBM | IT | Ongoing | - | JPRN-UMIN000002661 | ||
| Adenovirus | Delta-24-RGD | Delta-24, RGD | 1/2 | Recurrent GBM | IT | Completed | - | NCT 01582516 | |
| 1 | Recurrent HGG | IT | Completed | - | NCT 00805376 | ||||
| 1 | Recurrent GBM | IT | Ongoing | TMZ | NCT 01956734 | ||||
| 1 | Recurrent GBM or gliosarcoma | IT | Recruiting | INF-γ | NCT 02197169 | ||||
| 2 | Recurrent GBM or gliosarcoma | IT | Recruiting | Pembrolizumab | NCT 02798406 | ||||
| 1/2 | Recurrent GBM | IT | Ongoing | - | EUCTR2007-001104-21 | ||||
| Onyx-015 | E1B-55k (−) | 1 | Recurrent HGG | IT | Completed | - | [100] | ||
| Newcastle disease virus | NDV-HUJ | - | 1/2 | Recurrent GBM | IV | Completed | - | [132] | |
| Reovirus | Reolysin | - | 1/2 | Recurrent HGG | IT | Completed | - | NCT 00528684 | [98] |
| 1 | Recurrent HGG | IV | Recruiting | Sargramostim | NCT 02444546 | ||||
| Poliovirus | PVS-RIPO | IRES | 1 | Recurrent GBM | IT | Recruiting | - | NCT 01491893 | |
| Parvovirus | H-1PV | - | 1/2 | Primary or Recurrent GBM | IT/IV | Completed | - | NCT 01301430 | [133] |
| Measles virus | MV-CEA | CEA | 1 | Recurrent GBM | IT | Recruiting | - | NCT 00390299 | |
| Reovirus | TOCA511 | CD | 1 | Recurrent HGG | IT/IV | Ongoing | 5-FC | NCT 01156584 | |
| 2/3 | Recurrent HGG | IT | Recruiting | 5-FC | NCT 02414165 | [134] | |||
| 1 | Recurrent HGG | IV/IT | Ongoing | 5-FC | NCT 01985256 | ||||
| 1 | Newly diagnosed HGG | IT | Not yet recruiting | 5-FC | NCT 02598011 | ||||
| 1 | Recurrent HGG | IT | Ongoing | 5-FC | NCT 01470794 |
Abbreviations: 5-FC, 5-fluorocytosine; CD, Cytosine deaminase; CEA, carcinogenic embryonic antigen; GBM, glioblastoma; HGG, high grade glioma; HSV, herpes simplex virus; ICP, infected cell protein; IL, interleukin; INF, interferon; IRES, internal ribosomal entry site; IT, intratumoral; IV, intravenous; MV, measles virus; NDV, New castle disease virus; PV, polio virus; RT, radiation therapy; TMZ, Temozolomide
A variety of genetic modifications have been used to enhance tumor selectivity [74–78]. One way to enhance tumor selectivity is to exploit viral genes that are critical to viral replication but are redundant in cancer cells. For example, the tumor selectivity of the oncolytic adenovirus Delta-24-RGD results from a 24-base pair deletion engineered into in the viral E1A gene. Because the main function of the protein product of viral E1A is to bind and inactivate cellular retinoblastoma (Rb), deleting a portion of E1A renders the virus unable to replicate in cells that have normal functioning Rb, i.e. normal cells [79–81] (Figure 1A) [82–84]. However, the virus is able to replicate in tumors cells because most, if not all tumor cells, including GBMs, have lost Rb, harbor a mutation of Rb, or have undergone inactivation of p16, the main upstream regulator of Rb (Figure 1B). Mutation in Rb or loss of p16 is common in most tumors [85], including GBMs [86]. Another oncolytic virus that was developed to take advantage of this approach is the ONYX-15 virus, which was engineered to contain a mutated E1B. The function of E1B is to inactivate the cellular p53 gene, thereby preventing viral infected cells from undergoing apoptosis and allowing the virus to replicate. When ONYX-015 infects cells, it is theoretically unable to replicate because the virus could not inactivate p53. However tumor cells commonly contain mutation sin p53 and therefore viral replication is permissive in tumor cells. Another adenovirus, HB101, contains deletions in E1B and E3 and also is permissive in tumor cells only (reviewed in [87]). In addition to these approaches, investigators modified viruses with the goal of enhancing tumor infectivity using strategies that modify the virus to recognize surface proteins that are highly abundant on the surface of cancer cells. For example, Delta-24-RGD also enhanced tumor infectivity because an RGD (Arginine-Glycine-Aspartic acid) peptide sequence has been engineered into the fiber knob, which allows Delta-24-RGD to enter tumor cells via integrins, which are highly expressed on tumor cells, and independent of the normal Cocksackie-Adenoreceptor, which is only poorly expressed on tumors. (Figure 1A). Another example of this approach is the echovirus type 1, which preferentially infects ovarian cancer cells due to overabundance of the I domain of integrin α2β1 [88].
Figure 1.
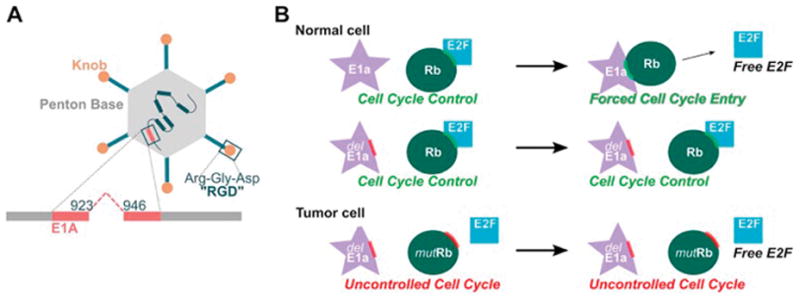
Delta-24-RGD harbors a 24-base pair deletion in the viral E1A region that is responsible for binding Retinoblastoma (Rb), and contains the inserted RGD sequence to enhance infectivity (Figure 1A). Rb protein normally prevents cells from entering S-phase. Because E1A is mutated, the virus is unable to replicate in cells that have functioning Rb, i.e. normal cells. However, the virus is able to replicate in cells that have lost or mutated Rb protein, i.e. tumor cells (Figure 1B).
Virotherapy and immune system involvement
Several preclinical and clinical studies have indicated that the efficacy of oncolytic viruses is due not only to direct oncolysis, but also to the ability of the virus to induce an anti-tumor cytotoxic (CD-8 mediated) adaptive immune response. For example, Andreansky et al. showed that intracerebral injection of HSV expressing IL-4 into GL-261 gliomas in C57BL/6 immunocompetent prolonged survival of the mice, whereas treatment with HSV expressing IL-4 or IL-10 resulted in survival rates similar to saline treated controls [89]. Todo et al showed that G207, a conditionally replicating HSV, injected intratumorally not only exhibited a prominent oncolytic antitumor effect in mice harboring subcutaneous N18 neuroblastoma cells, but also caused regression of remote, established tumors in the brain or in the periphery [90]. These results suggested that antitumor activity of oncolytic viruses may be mediated by or enhanced by induction of specific and systemic antitumor immunity. More recently, Jiang et al. demonstrated that Delta-24-RGD treatment elicited anti-glioma immunity in immunocompetent mice bearing GL-261 gliomas through infiltration of innate and adaptive immune cells. In these studies, Delta-24-RGD activated TH1 immunity at the tumor site which resulted in specific anti-glioma immunity, reduced tumor size, and prolonged animal survival. They also showed that Delta-24-RGD increased presentation of tumor-associated antigens to CD8+ T-cells, based on experiments using the ovalbumin modeling system as a surrogate for tumor antigens [68].
In clinical studies, treatment with oncolytic viruses has been reported to activate T cell, to trigger dendritic cells and to stimulate innate and adaptive antitumor immunity in several cancer types [91–93]. Talimogene laherparepvec is a genetically modified HSV type 1 virus that selectively replicates in tumors. Both copies of the viral gene coding for ICP34.5 were deleted and replaced with the gene coding for granulocyte macrophage colony-stimulating factor (GM-CSF), and the gene coding for ICP47 (which suppresses the immune response to the virus) was removed. Direct intratumoral injection of talimogene laherparepvec to patients with metastatic melanoma led to complete response in 8 of 50 treated patients, and more importantly led to regression of both injected and uninjected (including visceral) tumors [94], demonstrated that intratumoral administration of oncolytic viruses can intensify anticancer immunity and induce an adaptive endogenous vaccine effect. Talimogene laherparepvec was approved as the first oncolytic viral therapy by the U.S. Food and Drug Administration for the treatment of recurrent melanoma in October of 2015 [95]. The ability of oncolytic viruses to activate antiglioma immune responses has been shown in unpublished results of phase I trials of Delta-24-RGD [96].
Delivery Vehicles of Oncolytic Viruses
A major obstacle in the current brain tumor treatment paradigm using oncolytic viruses is the use of intratumoral injection as the primary mode of viral delivery. To date, in most clinical trials oncolytic viruses have been delivered directly into the MRI-defined enhancing portion of the tumor through a rigid biopsy needle or through a silicone catheter inserted via a small burr hole in the skull guided by stereotactic image-guided injection [97–99]. Oncolytic viruses have also been injected into the wall of the resection cavity after surgical removal of the main contrast-enhancing mass using a hand-held needle [100]. These methods have proven suboptimal as direct intratumoral injection is limited by backflow of the solution up the catheter or the needle [101]. This backflow results in loss of significant quantities of the injected solution, particularly for injections delivered close to the brain surface or to a traversed sulcus [102], and several clinical trials have recommended placing catheters ≥2–3cm from the brain surface [103]. Therefore, it is suspected that many patients do not receive the required viral dose when delivery relies on direct intratumoral injections. In addition, even if the virus is successfully deposited into the tumor mass, human gliomas are heterogeneous and contain multiple barriers to viral spread, including areas of necrosis, hemorrhage, cysts, and edema. These factors represent hurdles for the successful spread of the virus from the sites of injection to the edges of the tumor and thereby limit virus-mediated tumor eradication after intratumoral injection. Multiple injections into several sites may overcome these hurdles, but each injection increases the risk of intracranial bleeding and can be technically challenging. The use of Convection Enhanced Delivery (CED), in which a catheter is inserted into the tumor and viral solution is infused slowly under pressure over time creating a convective current, may improve intratumoral delivery, however, the capacity of CED to increase the spread of a virus through the tumor remains unknown and to date CED using other agents has shown minor success [101].
The “holy grail” of viral delivery in GBM is intravascular administration, either intravenously or intra-arterially. Systemic intravascular delivery is ideal as it should theoretically result in widespread initial viral distribution into the tumor, thereby overcoming many of the barriers to viral spread. This wide distribution would also increase the viral mediated presentation of tumor-associated antigens, given the known heterogeneity of brain tumors, and may enhance immune mediated anti-glioma effects. In addition, repeat dosing is possible and logistically feasible with intravascular injections, whereas repeating intratumoral injections is logistically difficult for brain tumors. Unfortunately, intravascular delivery of most “naked” viruses is prohibitive due to peripheral organ toxicity, particularly to the liver, and to immune-mediated inactivation of the virus.
To circumvent the problems associated with local delivery of oncolytic viruses, investigators sought to utilize cellular carriers to deliver these viruses. Although NSCs have been used in this application in GBM animal models [104, 105], MSCs are more commonly used because they are more easily obtained [106–110]. Interestingly, one study demonstrated that NSCs were more effective in supporting replication of the CRAd-S-pk7 oncolytic virus, compared with MSCs, and in prolonging survival in an intracranial glioma nude mouse model [111]. However, such results have not been reported by others and it is unclear if this result was specific to the particular virus being studied. Nevertheless, clinical application of NSCs is more difficult compared with MSCs due to logistical and ethical problems associated with harvesting NSCs. In clinical trials, MSCs have been utilized as carriers of oncolytic viruses in recurrent ovarian cancer via intraperitoneal injection (NCT02068794) and in metastatic solid tumors via intravenous injection (NCT01844661). Furthermore, Melen et al. investigated Celyvir, autologous bone marrow MSCs carrying oncolytic adenovirus ICOVIR-5, for treating children with advanced metastatic neuroblastoma, and found it was tolerated well with only mild viral-related toxicity after intravenous injection and it produced dramatic tumor reductions [39].
In addition to the ability of MSCs to home to and disperse therapeutics within tumors, MSCs are also capable of shielding the virus from the immune system (Trojan Horse approach) when traversing the bloodstream. Mader and colleagues demonstrated that human MSCs could protect recombinant oncolytic measles viruses from antiviral antibodies. In their study, athymic mice bearing intraperitoneal human ovarian tumor xenografts were passively immunized with measles-immune human serum. Survival of these mice was enhanced by treatment with measles virus-infected MSC via intraperitoneal injection, however naked virus treatment did not prolong animal survival [112]. A subsequent study by Ong et al. showed intravenously delivered measles virus-infected MSCs evaded the presence of anti-measles virus immunity in severe combined immunodeficiency (SCID) mice that were injected with human measles immune serum and harbored patient-derived human hepatocellular carcinomas [108]. Additionally, although oncolytic virotherapy has not drawn much attention for the treatment of hematological malignancies, Castleton et al. demonstrated that oncolytic measles virus-loaded MSCs could be used in a human passively immunized SCID mice model of acute lymphoblastic leukemia (ALL). The MSCs could deliver oncolytic measles virotherapy directly into ALL cells even in the presence of anti-measles virus immunity [109]. These results suggested that using MSCs could overcome the neutralizing effect of humoral antiviral antibodies.
With regards to brain tumors, In 2008 Lesniak and colleagues published the first report demonstrating that MSCs could be loaded with oncolytic viruses for the treatment of GBM [113]. The authors used a conditionally replicative adenovirus (CRAd) that specifically targeted the C-X-C chemokine receptor 4 (CXCR4), which is expressed in MSCs and whose promoter is active in glioma cells [114]. They found that the virus was able to first replicate within MSCs, and then could infect and replicate in glioma cells. The authors then showed that adenoviral antigens were detected in tumors seven days after MSC-CRAd-CXCR4 were injected into the brain 5mm anterior to the site where the glioma tumor cells were implanted, suggesting that MSCs harboring the oncolytic adenovirus retained their ability to migrate toward gliomas after intracranial injection in immunodeficient mice in vivo [113]. To better understand the immunosuppressive properties of MSCs, in 2010 the same group published a study utilizing a cotton rat model that was chosen because it is semipermissive to adenoviral infection (whereas mice are not permissive to adenoviruses). They found that adenovirus-loaded MSCs suppressed T-cell proliferation and the production of interferon-γ by activated T-cells. In addition, MSCs loaded with adenovirus enhanced the persistence of adenovirus compared with virus injected animals alone [115]. Whereas the majority of studies using oncolytic viruses for the treatment of GBM rely on intratumoral injection, Yong et al. reported the ability of MSCs loaded with the oncolytic virus Delta-24-RGD to deliver the virus into intracranial gliomas after intravascular injection into the carotid artery. They demonstrated that MSCs loaded with Delta-24-RGD retain their ability to selectively home to intracranial glioma xenografts after intracarotid injection. Once within the tumors, MSCs released Delta-24-RGD which subsequently infected glioma cells, resulting in enhanced animal survival. These data support the translation of this approach to patients with human gliomas and provides a clinical assay for assessing the extent to which MSCs are capable of delivering Delta-24-RGD into gliomas of patients. It is anticiaptied that clinial trials using MSCs to delvier Delta-24-RGD and to deliver other adenoviruses will be carried out in the near future.
One cause for concern regarding MSCs for the delivery of therapeutic compounds is the potential for MSCs to promote tumor growth. A number of animal studies have raised this concern. For example, in a breast cancer model, Karnoub et al. found that bone marrow MSCs accelerated tumor growth, and that subcutaneously implanting mixtures of cancer cells and MSCs resulted in a marked increase in the numbers of lung metastases compared with subcutaneously implanting tumors cells alone [116]. Similarly, Klopp et al. found that MSCs increased human mammary epithelial cell mammosphere formation and increased expression of N-cadherin, a phenomenon associated with breast cancer progression [117]. Furthermore, adipose stromal cells isolated from intra-abdominal omental adipose tissue were found to increase tumor vascularization and promoted endometrial tumor growth [118]. On the other hand, Qiao et al. showed that fetal MSCs derived from dermis inhibited the growth of breast cancer cells by interfering with Wnt signaling [119]. Whether MSCs promote or inhibit tumor growth may be dependent on their source. For example, in a GBM model, Sasportas et al. showed that bone marrow-derived MSCs had no significant influence on tumor progression in the brain [38]. On the other hand, Behnan et al. isolated MSCs from murine gliomas and showed these brain tumor-derived MSCs stimulated tumor proliferation in vitro and enhanced tumor growth in vivo [120]. Subsequently, Hossain et al. isolated MSCs from human glioma surgical specimens and showed that these glioma-associated human MSCs (GA-hMSCs) increase proliferation and self-renewal of glioma stem cells (GSCs) in vitro and enhance GSC tumorigenicity in vivo [121]. One strategy to ensure that exogenous MSCs do not promote tumor growth is to engineer them to contain an agent that serves both as a therapeutic for GBM, but that also destroys the MSC once the agent is released. Oncolytic viruses are one agent that meets this requirement.
Because of the ease to access and relative abundance of fat tissue, adipose tissue-derived MSCs have also been studied for the application of oncolytic virotherapy. For example, adipose tissue-derived MSCs infected with green fluorescent protein expressing myxoma virus (vMyxgfp) were permissive for myxoma virus replication, and when injected intracranially into an orthotopic GBM mouse model, resulted in increased survival [122]. ICOVIR17, a CRAd that expresses a soluble form of PH20 hyaluronidase to degrade hyaluronic acid, was tested for efficacy against established patient-derived GBMs. Compared with direct virus injection, adipose tissue derived MSCs loaded with ICOVIR17 improved survival in a mouse resection model [123].
Finally, several groups have focused on using hydrogels to deliver MCSs into the residual “cavity” after surgical resection of a tumor and these approaches have been specifically applied to MSCs loaded with oncolytic viruses. Based on the finding that encapsulation of MSCs in biodegradable synthetic extracellular matrices (sECM) enhanced the retention and therapeutic potential of the stem cells within the resection cavity [124], Duebgen and colleagues investigated this sECM technology for MSCs loaded with HSV. They showed that sECM-encapsulated MSC-oncolytic HSV or a proapoptotic variant of the virus significant improved the survival of tumor bearing mice compared with direct injection of oncolytic HSV in vivo [125]. A previous report indicated that encapsulation of MSCs allow for the retention of more MSCs in the tumor resection cavity compared with unencapsulated MSCs. This hydrogel technology expands the application of MSCs loaded with oncolytic viruses to the post resection clinical setting and overcomes the difficulties associated with directly injecting naked virus using hand-held injection needles.
Conclusions
Oncolytic viruses which selectively infect and destroy tumor cells, while sparing normal, healthy cells, and which induce an endogenous vaccine by activating an anti-tumoral immune response, have the potential to significantly alter the outcome of patients with brain tumors, particularly GBM. However, how these viruses will be delivered most efficiently to maximize viral distribution and to enhance anti-tumoral efficacy remains unclear. Conventional methods of viral delivery using local intratumoral injection of naked virus through needles results in insufficient viral delivery into only a small area of the tumor. However, using MSCs as cellular delivery vehicles of oncolytic viruses appears to overcome many of the current problems of viral delivery. Because MSCs home to tumors after intracranial injection, intratumoral instillation of MSCs may increase the initial distribution of the virus due to the ability of MSCs to migrate through the tumor. Of potentially greater clinical impact, is to deliver the viral-loaded MSCs intravascularly, thereby exploiting the unique property of circulating MSCs to home to tumors. Although intravenous delivery does not appear to be efficient, intra-arterial delivery appears to be highly effective. Importantly, with modern neuro-endovascular techniques, in which neuro-interventionists and neurosurgeons can access the cerebral circulation, including the feeding arteries of tumors, via transfemoral access, intra-arterial delivery of MSCs loaded with oncolytic viruses is now inherently feasible. This approach can be applied to unresectable newly diagnosed brain tumors, as well recurrent brain tumors. Intravascular delivery of MSCs may also be applicable after surgical resection and as part of current adjuvant therapies for brain tumors, including during concurrent radiochemotherapy, which is the current standard of care for the treatment of GBM. In addition, endovascular delivery of MSCs may be an ideal approach to treating patients with multiple intracranial tumors, such as multifocal GBM or even more impactful, in patients with multiple brain metastases. Clinical trials using MSCs to deliver oncolytic virus are currently under way and ultimately will determine the extent to which the promising preclinical results actually translate into meaningful clinical outcomes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dezawa M, et al. Bone marrow stromal cells generate muscle cells and repair muscle degeneration. Science. 2005;309(5732):314–7. doi: 10.1126/science.1110364. [DOI] [PubMed] [Google Scholar]
- 2.Li Y, et al. Intracerebral transplantation of bone marrow stromal cells in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neurosci Lett. 2001;316(2):67–70. doi: 10.1016/s0304-3940(01)02384-9. [DOI] [PubMed] [Google Scholar]
- 3.Grinnemo KH, et al. Xenoreactivity and engraftment of human mesenchymal stem cells transplanted into infarcted rat myocardium. J Thorac Cardiovasc Surg. 2004;127(5):1293–300. doi: 10.1016/j.jtcvs.2003.07.037. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, et al. Therapeutic benefit of intracerebral transplantation of bone marrow stromal cells after cerebral ischemia in rats. J Neurol Sci. 2001;189(1–2):49–57. doi: 10.1016/s0022-510x(01)00557-3. [DOI] [PubMed] [Google Scholar]
- 5.Lu D, et al. Adult bone marrow stromal cells administered intravenously to rats after traumatic brain injury migrate into brain and improve neurological outcome. Neuroreport. 2001;12(3):559–563. doi: 10.1097/00001756-200103050-00025. [DOI] [PubMed] [Google Scholar]
- 6.Studeny M, et al. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62(13):3603–8. [PubMed] [Google Scholar]
- 7.Nakamura K, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11(14):1155–64. doi: 10.1038/sj.gt.3302276. [DOI] [PubMed] [Google Scholar]
- 8.Hung SC, et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. 2005;11(21):7749–56. doi: 10.1158/1078-0432.CCR-05-0876. [DOI] [PubMed] [Google Scholar]
- 9.Duan X, et al. Murine bone marrow-derived mesenchymal stem cells as vehicles for interleukin-12 gene delivery into Ewing sarcoma tumors. Cancer. 2009;115(1):13–22. doi: 10.1002/cncr.24013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiang J, et al. Mesenchymal stem cells as a gene therapy carrier for treatment of fibrosarcoma. Cytotherapy. 2009;11(5):516–26. doi: 10.1080/14653240902960429. [DOI] [PubMed] [Google Scholar]
- 11.Gao P, et al. Therapeutic potential of human mesenchymal stem cells producing IL-12 in a mouse xenograft model of renal cell carcinoma. Cancer Lett. 2010;290(2):157–66. doi: 10.1016/j.canlet.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 12.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert MR, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilbert MR. Antiangiogenic Therapy for Glioblastoma: Complex Biology and Complicated Results. J Clin Oncol. 2016;34(14):1567–9. doi: 10.1200/JCO.2016.66.5364. [DOI] [PubMed] [Google Scholar]
- 15.Gilbert MR, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31(32):4085–91. doi: 10.1200/JCO.2013.49.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–39. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- 17.Aboody KS, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97(23):12846–51. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benedetti S, et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat Med. 2000;6(4):447–50. doi: 10.1038/74710. [DOI] [PubMed] [Google Scholar]
- 19.Ehtesham M, et al. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002;62(20):5657–63. [PubMed] [Google Scholar]
- 20.Yuan X, et al. Interleukin-23-expressing bone marrow-derived neural stem-like cells exhibit antitumor activity against intracranial glioma. Cancer Res. 2006;66(5):2630–8. doi: 10.1158/0008-5472.CAN-05-1682. [DOI] [PubMed] [Google Scholar]
- 21.Shah K, et al. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann Neurol. 2005;57(1):34–41. doi: 10.1002/ana.20306. [DOI] [PubMed] [Google Scholar]
- 22.Balyasnikova IV, et al. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 2011;310(2):148–59. doi: 10.1016/j.canlet.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bagci-Onder T, et al. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res. 2011;71(1):154–63. doi: 10.1158/0008-5472.CAN-10-1601. [DOI] [PubMed] [Google Scholar]
- 24.Lee SJ, et al. Combined treatment of tumor-tropic human neural stem cells containing the CD suicide gene effectively targets brain tumors provoking a mild immune response. Oncol Rep. 2011;25(1):63–8. [PubMed] [Google Scholar]
- 25.Kim JH, et al. Therapeutic effect of genetically modified human neural stem cells encoding cytosine deaminase on experimental glioma. Biochem Biophys Res Commun. 2012;417(1):534–40. doi: 10.1016/j.bbrc.2011.11.155. [DOI] [PubMed] [Google Scholar]
- 26.van Eekelen M, et al. Human stem cells expressing novel TSP-1 variant have anti-angiogenic effect on brain tumors. Oncogene. 2010;29(22):3185–95. doi: 10.1038/onc.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmed AU, et al. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol Ther. 2011;19(9):1714–26. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed AU, et al. A Preclinical Evaluation of Neural Stem CellBased Cell Carrier for Targeted Antiglioma Oncolytic Virotherapy. Jnci-Journal of the National Cancer Institute. 2013;105(13):968–977. doi: 10.1093/jnci/djt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pittenger MF, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 30.Le Blanc K, et al. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31(10):890–6. doi: 10.1016/s0301-472x(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 31.Introna M, Rambaldi A. Mesenchymal stromal cells for prevention and treatment of graft-versus-host disease: successes and hurdles. Curr Opin Organ Transplant. 2015;20(1):72–8. doi: 10.1097/MOT.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 32.Konishi A, et al. First Approval of Regenerative Medical Products under the PMD Act in Japan. Cell Stem Cell. 2016;18(4):434–5. doi: 10.1016/j.stem.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 33.Kanehira M, et al. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther. 2007;14(11):894–903. doi: 10.1038/sj.cgt.7701079. [DOI] [PubMed] [Google Scholar]
- 34.Komarova S, et al. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006;5(3):755–66. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- 35.Kallifatidis G, et al. Improved lentiviral transduction of human mesenchymal stem cells for therapeutic intervention in pancreatic cancer. Cancer Gene Ther. 2008;15(4):231–40. doi: 10.1038/sj.cgt.7701097. [DOI] [PubMed] [Google Scholar]
- 36.Hall SRRCZLWCML. Human mesenchymal stem cells support capillary-like structures in a 3D model of in vitro angiogenesis. The FASEB journal. 2007;21(5):A145. [Google Scholar]
- 37.Shinojima N, et al. TGF-beta mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2013;73(7):2333–44. doi: 10.1158/0008-5472.CAN-12-3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasportas LS, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106(12):4822–7. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menon LG, et al. Human bone marrow-derived mesenchymal stromal cells expressing S-TRAIL as a cellular delivery vehicle for human glioma therapy. Stem Cells. 2009;27(9):2320–30. doi: 10.1002/stem.136. [DOI] [PubMed] [Google Scholar]
- 40.Fei S, et al. The antitumor effect of mesenchymal stem cells transduced with a lentiviral vector expressing cytosine deaminase in a rat glioma model. J Cancer Res Clin Oncol. 2012;138(2):347–57. doi: 10.1007/s00432-011-1104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kosaka H, et al. Therapeutic effect of suicide gene-transferred mesenchymal stem cells in a rat model of glioma. Cancer Gene Ther. 2012;19(8):572–8. doi: 10.1038/cgt.2012.35. [DOI] [PubMed] [Google Scholar]
- 42.Ryu CH, et al. Valproic acid enhances anti-tumor effect of mesenchymal stem cell mediated HSV-TK gene therapy in intracranial glioma. Biochem Biophys Res Commun. 2012;421(3):585–90. doi: 10.1016/j.bbrc.2012.04.050. [DOI] [PubMed] [Google Scholar]
- 43.Balyasnikova IV, et al. Mesenchymal stem cells modified with a single-chain antibody against EGFRvIII successfully inhibit the growth of human xenograft malignant glioma. PLoS One. 2010;5(3):e9750. doi: 10.1371/journal.pone.0009750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano. 2011;5(9):7462–70. doi: 10.1021/nn202399w. [DOI] [PubMed] [Google Scholar]
- 45.Yang B, et al. Dual-targeted antitumor effects against brainstem glioma by intravenous delivery of tumor necrosis factor-related, apoptosis-inducing, ligand-engineered human mesenchymal stem cells. Neurosurgery. 2009;65(3):610–24. doi: 10.1227/01.NEU.0000350227.61132.A7. discussion 624. [DOI] [PubMed] [Google Scholar]
- 46.Bexell D, et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol Ther. 2009;17(1):183–90. doi: 10.1038/mt.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bexell D, et al. Rat multipotent mesenchymal stromal cells lack long-distance tropism to 3 different rat glioma models. Neurosurgery. 2012;70(3):731–9. doi: 10.1227/NEU.0b013e318232dedd. [DOI] [PubMed] [Google Scholar]
- 48.Hata N, et al. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery. 2010;66(1):144–56. doi: 10.1227/01.NEU.0000363149.58885.2E. discussion 156–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schichor C, et al. Vascular endothelial growth factor A contributes to glioma-induced migration of human marrow stromal cells (hMSC) Exp Neurol. 2006;199(2):301–10. doi: 10.1016/j.expneurol.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 50.Yong RL, et al. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Delta24-RGD to human gliomas. Cancer Res. 2009;69(23):8932–40. doi: 10.1158/0008-5472.CAN-08-3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah K. Encapsulated stem cells for cancer therapy. Biomatter. 2013;3(1) doi: 10.4161/biom.24278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryu CH, et al. Gene therapy of intracranial glioma using interleukin 12-secreting human umbilical cord blood-derived mesenchymal stem cells. Hum Gene Ther. 2011;22(6):733–43. doi: 10.1089/hum.2010.187. [DOI] [PubMed] [Google Scholar]
- 53.Choi SA, et al. Human adipose tissue-derived mesenchymal stem cells: characteristics and therapeutic potential as cellular vehicles for prodrug gene therapy against brainstem gliomas. Eur J Cancer. 2012;48(1):129–37. doi: 10.1016/j.ejca.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 54.Yin J, et al. hMSC-mediated concurrent delivery of endostatin and carboxylesterase to mouse xenografts suppresses glioma initiation and recurrence. Mol Ther. 2011;19(6):1161–9. doi: 10.1038/mt.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Utsunomiya T, et al. Human adipose-derived stem cells: potential clinical applications in surgery. Surg Today. 2011;41(1):18–23. doi: 10.1007/s00595-010-4415-9. [DOI] [PubMed] [Google Scholar]
- 56.Anzalone R, et al. New emerging potentials for human Wharton’s jelly mesenchymal stem cells: immunological features and hepatocyte-like differentiative capacity. Stem Cells Dev. 2010;19(4):423–38. doi: 10.1089/scd.2009.0299. [DOI] [PubMed] [Google Scholar]
- 57.Moore AE. Viruses with oncolytic properties and their adaptation to tumors. Ann N Y Acad Sci. 1952;54(6):945–52. doi: 10.1111/j.1749-6632.1952.tb39969.x. [DOI] [PubMed] [Google Scholar]
- 58.Southam CM, Moore AE. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. Cancer. 1952;5(5):1025–34. doi: 10.1002/1097-0142(195209)5:5<1025::aid-cncr2820050518>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 59.Georgiades J, et al. Research on the oncolytic effect of APC viruses in cancer of the cervix uteri; preliminary report. Biul Inst Med Morsk Gdansk. 1959;10:49–57. [PubMed] [Google Scholar]
- 60.Huebner RJ, et al. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer. 1956;9(6):1211–8. doi: 10.1002/1097-0142(195611/12)9:6<1211::aid-cncr2820090624>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 61.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–9. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 62.Hammon WM, et al. Oncolytic Potentials of Nonhuman Viruses for Human Cancer. I. Effects of Twenty-Four Viruses on Human Cancer Cell Lines. J Natl Cancer Inst. 1963;31:329–45. [PubMed] [Google Scholar]
- 63.Coen DM, et al. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A. 1989;86(12):4736–40. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dubbs DR, Kit S. Mutant Strains of Herpes Simplex Deficient in Thymidine Kinase-Inducing Activity. Virology. 1964;22:493–502. doi: 10.1016/0042-6822(64)90070-4. [DOI] [PubMed] [Google Scholar]
- 65.Jamieson AT, Gentry GA, Subak-Sharpe JH. Induction of both thymidine and deoxycytidine kinase activity by herpes viruses. J Gen Virol. 1974;24(3):465–80. doi: 10.1099/0022-1317-24-3-465. [DOI] [PubMed] [Google Scholar]
- 66.Martuza RL, et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–6. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- 67.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–70. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang H, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One. 2014;9(5):e97407. doi: 10.1371/journal.pone.0097407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Phuong LK, et al. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003;63(10):2462–9. [PubMed] [Google Scholar]
- 70.Ochiai H, et al. Targeted therapy for glioblastoma multiforme neoplastic meningitis with intrathecal delivery of an oncolytic recombinant poliovirus. Clin Cancer Res. 2006;12(4):1349–54. doi: 10.1158/1078-0432.CCR-05-1595. [DOI] [PubMed] [Google Scholar]
- 71.Wilcox ME, et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001;93(12):903–12. doi: 10.1093/jnci/93.12.903. [DOI] [PubMed] [Google Scholar]
- 72.Ostertag D, et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012;14(2):145–59. doi: 10.1093/neuonc/nor199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Di Piazza M, et al. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J Virol. 2007;81(8):4186–98. doi: 10.1128/JVI.02601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Veerapong J, et al. Systemic delivery of (gamma1)34.5-deleted herpes simplex virus-1 selectively targets and treats distant human xenograft tumors that express high MEK activity. Cancer Res. 2007;67(17):8301–6. doi: 10.1158/0008-5472.CAN-07-1499. [DOI] [PubMed] [Google Scholar]
- 75.Russell SJ, Peng KW. Measles virus for cancer therapy. Curr Top Microbiol Immunol. 2009;330:213–41. doi: 10.1007/978-3-540-70617-5_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shmulevitz M, Marcato P, Lee PW. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. 2005;24(52):7720–8. doi: 10.1038/sj.onc.1209041. [DOI] [PubMed] [Google Scholar]
- 77.Gromeier M, et al. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci U S A. 2000;97(12):6803–8. doi: 10.1073/pnas.97.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wrzesinski C, et al. Chimeric and pseudotyped parvoviruses minimize the contamination of recombinant stocks with replication-competent viruses and identify a DNA sequence that restricts parvovirus H-1 in mouse cells. J Virol. 2003;77(6):3851–8. doi: 10.1128/JVI.77.6.3851-3858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heise C, Kirn DH. Replication-selective adenoviruses as oncolytic agents. J Clin Invest. 2000;105(7):847–51. doi: 10.1172/JCI9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fueyo J, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19(1):2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 81.Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell. 1989;56(1):67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- 82.Aasen G, Fagertun H, Halse J. Regional fat mass by DXA: high leg fat mass attenuates the relative risk of insulin resistance and dyslipidaemia in obese but not in overweight postmenopausal women. Scand J Clin Lab Invest. 2008;68(3):204–11. doi: 10.1080/00365510701649524. [DOI] [PubMed] [Google Scholar]
- 83.Harlow E, et al. Association of adenovirus early-region 1A proteins with cellular polypeptides. Mol Cell Biol. 1986;6(5):1579–89. doi: 10.1128/mcb.6.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moran E. DNA tumor virus transforming proteins and the cell cycle. Curr Opin Genet Dev. 1993;3(1):63–70. doi: 10.1016/s0959-437x(05)80342-9. [DOI] [PubMed] [Google Scholar]
- 85.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9(11):785–97. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ko D, Hawkins L, Yu DC. Development of transcriptionally regulated oncolytic adenoviruses. Oncogene. 2005;24(52):7763–74. doi: 10.1038/sj.onc.1209048. [DOI] [PubMed] [Google Scholar]
- 88.Shafren DR, et al. Oncolysis of human ovarian cancers by echovirus type 1. Int J Cancer. 2005;115(2):320–8. doi: 10.1002/ijc.20866. [DOI] [PubMed] [Google Scholar]
- 89.Andreansky S, et al. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998;5(1):121–30. doi: 10.1038/sj.gt.3300550. [DOI] [PubMed] [Google Scholar]
- 90.Todo T, et al. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum Gene Ther. 1999;10(17):2741–55. doi: 10.1089/10430349950016483. [DOI] [PubMed] [Google Scholar]
- 91.Kaufman HL, et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718–30. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- 92.Koski A, et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther. 2010;18(10):1874–84. doi: 10.1038/mt.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liikanen I, et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther. 2013;21(6):1212–23. doi: 10.1038/mt.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Senzer NN, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27(34):5763–71. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 95.Greig SL. Talimogene Laherparepvec: First Global Approval. Drugs. 2016;76(1):147–54. doi: 10.1007/s40265-015-0522-7. [DOI] [PubMed] [Google Scholar]
- 96.Lang FF, et al. Phase I Clinical Trial of Oncolytic Virus Delta-24-Rgd (Dnx-2401) with Biological Endpoints: Implications for Viro-Immunotherapy. Neuro-Oncology. 2014:16. [Google Scholar]
- 97.Markert JM, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014;22(5):1048–55. doi: 10.1038/mt.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Forsyth P, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16(3):627–32. doi: 10.1038/sj.mt.6300403. [DOI] [PubMed] [Google Scholar]
- 99.Kicielinski KP, et al. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther. 2014;22(5):1056–62. doi: 10.1038/mt.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chiocca EA, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10(5):958–66. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 101.Chaichana KL, Pinheiro L, Brem H. Delivery of local therapeutics to the brain: working toward advancing treatment for malignant gliomas. Ther Deliv. 2015;6(3):353–69. doi: 10.4155/tde.14.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bauman MA, et al. Physical characterization of neurocatheter performance in a brain phantom gelatinwith nanoscale porosity: steady-state and oscillatory flows. Nanotechnology. 2004;15(1):92–97. [Google Scholar]
- 103.Kunwar S, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrent malignant glioma: a report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol. 2007;25(7):837–44. doi: 10.1200/JCO.2006.08.1117. [DOI] [PubMed] [Google Scholar]
- 104.Tyler MA, et al. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2009;16(2):262–78. doi: 10.1038/gt.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Morshed RA, et al. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther. 2015;22(1):55–61. doi: 10.1038/cgt.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xia X, et al. Mesenchymal stem cells as carriers and amplifiers in CRAd delivery to tumors. Mol Cancer. 2011;10:134. doi: 10.1186/1476-4598-10-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stoff-Khalili MA, et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat. 2007;105(2):157–67. doi: 10.1007/s10549-006-9449-8. [DOI] [PubMed] [Google Scholar]
- 108.Ong HT, et al. Systemically delivered measles virus-infected mesenchymal stem cells can evade host immunity to inhibit liver cancer growth. J Hepatol. 2013;59(5):999–1006. doi: 10.1016/j.jhep.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Castleton A, et al. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood. 2014;123(9):1327–35. doi: 10.1182/blood-2013-09-528851. [DOI] [PubMed] [Google Scholar]
- 110.Hakkarainen T, et al. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum Gene Ther. 2007;18(7):627–41. doi: 10.1089/hum.2007.034. [DOI] [PubMed] [Google Scholar]
- 111.Ahmed AU, et al. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol Pharm. 2011;8(5):1559–72. doi: 10.1021/mp200161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mader EK, et al. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res. 2009;15(23):7246–55. doi: 10.1158/1078-0432.CCR-09-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sonabend AM, et al. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells. 2008;26(3):831–41. doi: 10.1634/stemcells.2007-0758. [DOI] [PubMed] [Google Scholar]
- 114.Ulasov IV, et al. Comparative evaluation of survivin, midkine, and CXCR4 promoters for transcriptional targeting of glioma gene therapy. Cancer Biology & Therapy. 2007;6(5):679–685. doi: 10.4161/cbt.6.5.3957. [DOI] [PubMed] [Google Scholar]
- 115.Ahmed AU, et al. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus suppress the anti-adenoviral immune response in the cotton rat model. Mol Ther. 2010;18(10):1846–56. doi: 10.1038/mt.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Karnoub AE, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–63. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 117.Klopp AH, et al. Mesenchymal stem cells promote mammosphere formation and decrease E-cadherin in normal and malignant breast cells. PLoS One. 2010;5(8):e12180. doi: 10.1371/journal.pone.0012180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Klopp AH, et al. Omental adipose tissue-derived stromal cells promote vascularization and growth of endometrial tumors. Clin Cancer Res. 2012;18(3):771–82. doi: 10.1158/1078-0432.CCR-11-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Qiao L, et al. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008;269(1):67–77. doi: 10.1016/j.canlet.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 120.Behnan J, et al. Recruited brain tumor-derived mesenchymal stem cells contribute to brain tumor progression. Stem Cells. 2014;32(5):1110–23. doi: 10.1002/stem.1614. [DOI] [PubMed] [Google Scholar]
- 121.Hossain A, et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells. 2015;33(8):2400–15. doi: 10.1002/stem.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Josiah DT, et al. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol Ther. 2010;18(2):377–85. doi: 10.1038/mt.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Martinez-Quintanilla J, et al. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol Ther. 2015;23(1):108–18. doi: 10.1038/mt.2014.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kauer TM, et al. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat Neurosci. 2012;15(2):197–204. doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Duebgen M, et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst. 2014;106(6):dju090. doi: 10.1093/jnci/dju090. [DOI] [PubMed] [Google Scholar]
- 126.Nakamizo A, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 127.Harrow S, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004;11(22):1648–58. doi: 10.1038/sj.gt.3302289. [DOI] [PubMed] [Google Scholar]
- 128.Papanastassiou V, et al. The potential for efficacy of the modified (ICP 34.5(−)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9(6):398–406. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 129.Rampling R, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7(10):859–66. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 130.Markert JM, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867–74. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 131.Markert JM, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17(1):199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Freeman AI, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13(1):221–8. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 133.Geletneky K, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer. 2012;12:99. doi: 10.1186/1471-2407-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cloughesy TF, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8(341):341ra75. doi: 10.1126/scitranslmed.aad9784. [DOI] [PMC free article] [PubMed] [Google Scholar]