

Abstract
A 20-year-old Japanese woman had an attack of acute intermittent porphyria (AIP). Magnetic resonance imaging (MRI) revealed symmetrical lesions in the cerebrum and cerebellar hemisphere, corresponding to posterior reversible encephalopathy syndrome (PRES). Our administration of heme arginate gradually improved the clinical condition associated with AIP and the level of metabolite of nitric oxide (NO), which is a vascular dilator. Repeated MRI and magnetic resonance angiography revealed exacerbated PRES, part of which showed a small infarction, accompanied by progressive vasoconstriction. These findings suggest that the recovery of NO by heme replacement alone is insufficient for preventing brain damage during an AIP attack.
Keywords: acute intermittent porphyria, posterior reversible encephalopathy syndrome, vasoconstriction, ischemia, nitric oxide, nicardipine
Introduction
Acute intermittent porphyria (AIP) is an autosomal dominant disorder caused by a deficiency of hydroxymethylbilane synthase in heme biosynthesis (1,2). The majority of patients with AIP show acute attacks that manifest as a combination of abdominal pain, mild mental symptoms, and autonomic dysfunction (1). In addition, encephalopathy including mental symptoms may derive from the brain lesions in AIP, visualized as posterior reversible encephalopathy syndrome (PRES) (1).
Although the precise mechanism of encephalopathy in AIP remains unknown, an initial event might be that a severe heme deficiency during AIP acute attacks causes a lack of nitric oxide synthase (NOS), which is a heme protein (3). A decreased production of the major vascular dilator nitric oxide (NO) (4) due to the lack of NOS is thought to elicit vasoconstriction (5,6). The hypoperfusion associated with vasoconstriction might be involved in the incidence of vasogenic edema, which is a characteristic feature of PRES (7,8). In addition, prolonged vasoconstriction might cause ischemic necrosis (9).
However, the relationship between vasoconstriction and the level of NO has not been assessed during an acute attack of AIP. Under these circumstances, we measured the concentration of nitrate ion (NO3-), which is an oxidized metabolite of NO, in a patient with AIP presenting with prolonged vasoconstriction. To our knowledge, this is the first report showing that NO seems to have little involvement in prolonged vasoconstriction in AIP.
Case Report
A 20-year-old Japanese woman was admitted to a local general hospital in October 2014 for severe abdominal pain that occurred intermittently over a 3-day period. She also felt severe pain involving the entire body, especially in the lumbar area. On the 4th day after the onset, she was put on a mechanical ventilator because she experienced a general convulsion followed by emotional lability. She developed tachycardia and hypertension, which were intractable to treatment.
On the 5th day after the abdominal pain's onset, brain magnetic resonance imaging (MRI) showed symmetric high-intensity lesions in the frontal, parietal, and occipital lobes and cerebellar hemispheres on fluid-attenuated inversion recovery (FLAIR) imaging. These lesions showed slight hyperintensity on diffusion-weighted imaging (DWI) and hyperintensity on an apparent diffusion coefficient (ADC) map (Fig. 1). On the 7th day, magnetic resonance angiography (MRA) provided ambiguous visualization suggesting vasoconstriction in the bilateral posterior cerebral artery (PCA) (Fig. 2). The patient was otherwise well aside from a diagnosis of allergic rhinitis. She was on no other medication than dietary supplements, smoked approximately 10 cigarettes per day, and drank modestly. The date of her last menstrual period was 4 weeks prior to her admission.
Figure 1.

MRI findings obtained on the 5th day (A-C), 17th day (D-F), 31st day (G-I), and 51st day (J-L). The symmetric frontal, parietal, and occipital lesions observed on the 5th day were improved on the 17th day. These lesions showed high intensity on FLAIR images (A, D), slight high intensity on DWI (B, E), and high intensity on an ADC map (C, F) (arrowheads). Despite the recovery of the clinical condition, the brain lesions with the same characteristics as those observed earlier reappeared on the FLAIR image (G) and ADC map (I) on the 31st day (arrowheads). These areas were isointense on DWI (H). A part of the frontal lesions showed high intensity on DWI (H) and low intensity on an ADC map (I) (open arrowheads). These small lesions remained on FLAIR imaging (J) (open arrowheads) but were obscure on the DWI (J) (open arrowheads) and ADC map (L).
Figure 2.
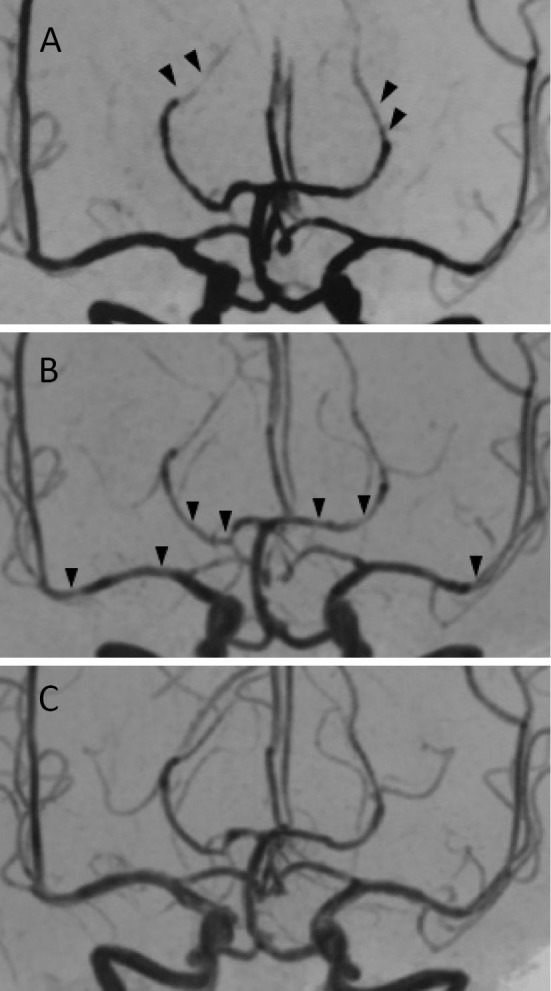
MRA findings obtained on the 7th day (A), 33rd day (B), and 51st day (C). The distal portion of the posterior cerebral artery (PCA) was visualized ambiguously on the 7th day. This abnormal finding spread to the proximal portion of the PCA and middle cerebral artery (MCA) on the 33rd day (arrowheads). As with the improvement in the MRI findings, the visualization of the MCA and PCA was nearly normalized on the 51st day.
She was transferred to our hospital for further evaluation. Her blood pressure was 176/93 mmHg, and her pulse was 113 beats/min with a regular rhythm, and this was refractory to the administration of nicardipine (0.86 μg/kg/min). Her blood test revealed increased levels as follows: white blood cells, 14,180 /μL; D-dimer, 6.5 μg/mL; C reactive protein, 1.26 mg/dL; total bilirubin, 4.7 mg/dL; direct bilirubin, 1.2 mg/dL; aspartate aminotransferase, 161 IU/L; alanine aminotransferase, 57 IU/L; creatine kinase, 17,009 IU/L; creatine kinase MB, 44 IU/L; and amylase, 183 IU/L. The patient had hyponatremia (123 mEq/L) due to inappropriate antidiuretic hormone secretion, which was later confirmed by elevated plasma arginine vasopressin (20.1 pg/mL). The results of a urinalysis were normal with the exception of discoloration of the urine. The findings on a cerebrospinal fluid examination was normal.
On the 8th day, she was weaned from ventilatory support. She had mild weakness of the iliopsoas muscle and a depressed deep tendon reflex. Nerve conduction studies showed normal findings except for a low incidence of F waves on the median, ulnar, and tibial nerves. Since the characteristic clinical course is a good predictor of AIP, we administered propranolol (0.01 mg/kg/h; the first choice for treating hypertension and tachycardia of AIP) as a substitute for nicardipine, the safety of which has not yet been established (10,11). A high-calorie and high-carbohydrate diet with glucose was also administered (10).
On the 19th day, the patient's levels of porphyrin precursors in a urine sample proved to be extremely high as follows: porphobilinogen (PBG), 196.1 mg/day (normal value: <2.0 mg/day) and δ-aminolevulinic acid (δ-ALA), 61.9 mg/L (normal value: <5.0 mg/L). Although a genetic diagnosis was not permitted by the patient's family, the characteristic symptoms and markedly increased level of porphyrin precursors confirmed the diagnosis of AIP. With respect to the patient's first-degree relatives, increased levels of urine δ-ALA and PBG were revealed in her elder sister but not in her mother. Her father refused the examination.
Following the daily intravenous administration of heme arginate (3 mg/kg) from the 20th day to the 23rd day, the patient's elevated liver enzymes, hypertension, and tachycardia moved gradually toward normal levels. The pain involving the entire body was also relieved. She has not suffered a relapse of general convulsions even after the withdrawal of anticonvulsant therapy. The deep tendon reflex returned to normal. On the 52nd day, the patient had recovered independent mobility and left the hospital.
Repeated MRI and MRA findings were as follows (Fig. 1, 2): On the 17th day, the high-intensity lesions had nearly disappeared with the exception of persistent partial lesions in the parietal and occipital lobes. On the 31st day (9 days after the end of the heme arginate administration), FLAIR imaging unexpectedly showed the reappearance of lesions in the frontal lobes as well as an increase in the size of the lesions in the parietal and occipital lobes. Some of the lesions in the bilateral frontal lobes located in the arterial ‘watershed’ and ‘borderzone’ regions showed hyperintensity on DWI and hypointensity on an ADC map. The lesions in the parietal to occipital lobes showed isointensity or slight hyperintensity on DWI and high intensity on the ADC map.
In addition, MRA revealed segmentally ambiguous visualization in the proximal portion of the middle cerebral artery (MCA) and PCA. Although the segmental stenosis of the MCA and PCA remained virtually unchanged on the 38th day, the relapsing lesions nearly disappeared except for small lesions in the frontal and parietal lobes. On the 51st day, the visualization of the MCA and PCA returned to nearly normal.
To clarify whether the ambiguous visualization of the MCA and PCA suggesting vasoconstriction was attributable to decreased NO during the patient's acute attack of AIP, we measured the level of serum nitrate ion (NO3-), which is an oxidized metabolite of NO, before and after administration of heme arginate. The level of NO3- measured by high-performance liquid chromatography (12) was 12 μmol/L on the 11th day, 21 μmol/L on the 17th day, and 35 μmol/L on the 46th day (normal value: 10-71 μmol/L).
Discussion
Our patient had exclusively neurovisceral phenomena, such as abdominal pain, autonomic dysfunction and mental symptoms. In addition, the DWI and ADC map findings suggested that the early cortical and subcortical lesions were vasogenic edema, consistent with the typical neuroimaging presentation of PRES (7,13). Although these findings prompted a differential diagnosis including hypertensive encephalopathy, infection, and autoimmune diseases (7), the markedly increased levels of urine PBG and δ-ALA indicated a diagnosis of AIP (2). The prompt administration of heme arginate (which replenishes the hepatic free heme pool (10)) resulted in a gradual improvement in the patient's hypertension, tachycardia, and generalized pain. The MRI lesions were improving but later showed exacerbation of PRES, which was likely an ischemic change, at least in part, accompanied by vasoconstriction expanded from the PCA to the MCA over time.
PRES accompanied by vasoconstriction is not frequently reported in AIP (14). Although the precise mechanism underlying such findings remains unknown, there are several hypotheses. Since hypertension during an acute attack of AIP might exceed the autoregulation limit (mean arterial pressure, 150-160 mmHg), hyperperfusion followed by breakdown of the blood-brain barrier could lead to vasogenic edema, which is part of the histopathology of PRES (7). In addition, severe heme deficiency during an acute attack of AIP causes a lack of the heme protein NOS (5). Since endothelial NOS (eNOS)-derived NO exerts a vasodilating effect for maintaining the cerebral blood flow (15), a lack of eNOS could be responsible for vasoconstriction. In fact, the level of NO3- before administration of heme arginate in our patient was at the low end of the normal range, suggesting a lack of eNOS.
However, these hypotheses are not likely to be applicable in our patient's case for several reasons. The hypertension observed in our patient did not reach the level of failed autoregulation. Even after the normalization of her hypertension, the findings of PRES worsened rather than improved. Although the infusion of heme arginate to replenish the hepatic free heme pool (10) from the 20th day resulted in further elevation of the level of NO3- on the 46th day, suggesting the recovery of eNOS, both the PRES and the vasoconstriction worsened. Alternatively, just as FK-506 and cyclosporine A can injure endothelial cells directly (16), excessive porphyrin precursors might cause endothelial cell damage followed by a breakdown of the blood-brain barrier and the release of potent vasoconstrictors, such as endothelin or thromboxane (6).
It has been reported that protracted vasoconstriction might be responsible for encephalopathy with a long clinical course of recovery, and that it can cause ischemic cytotoxic edema (9). Since our patient showed vasoconstriction that continued until at least the 38th day, the relapsing PRES, a part of which was the small ischemic lesions, might have been due to the prolonged vasoconstriction. With the exception that our patient did not show an acute onset of severe headache (i.e. a thunderclap headache), the clinical findings were similar to the clinical presentation of reversible cerebral vasoconstriction syndromes (RCVS) in the following aspects: (1) the reversibility of vasoconstriction within 12 weeks after onset; (2) evidence of brain infarction, particular in the arterial ‘watershed’ and ‘borderzone’ regions; and (3) the coexistence of changes consistent with PRES (17,18).
For the alleviation of symptoms and rapid reversibility of vascular abnormalities in RCVS, nicardipine with high lipophilicity seems to be considerably effective among the calcium-channel blockers (19,20). Indeed, our patient showed recurring exacerbation of PRES accompanied by the progression of vasoconstriction after the withdrawal of nicardipine. However, because nicardipine is metabolized by CYP3A4, which is a hemoprotein, a continuous nicardipine infusion for preventing vasoconstriction might cause exacerbation of an AIP attack due to a heme deficiency. In addition, excessive decreases in blood pressure with nicardipine might cause a watershed infarction in cerebral regions with hypoperfusion by a severely constricted cerebral artery (17). Therefore, nicardipine should be used cautiously under an adequate replacement of heme arginate and blood pressure monitoring.
In conclusion, PRES and vasoconstriction might be involved in the expression of encephalopathy in AIP. The time course of the present case as shown by MRI and MRA suggested the development of a small infarction that was probably due to prolonged vasoconstriction. Although the administration of heme arginate under supportive treatment increased our patient's level of NO3-, an oxidized metabolite of NO, the vasoconstriction worsened rather than improved. This finding suggests that the recovery of NO by heme replacement alone is insufficient for preventing brain damage. In addition, the discontinuation of nicardipine might have prolonged the vasoconstriction. These findings warrant further research to reveal the precise mechanism underlying the vasoconstriction and to establish the optimum nicardipine regimen during AIP attacks.
The authors state that they have no Conflict of Interest (COI).
References
- 1.Pischik E, Kauppinen R. Neurological manifestations of acute intermittent porphyria. Cell Mol Bio (Noisy-le-Grand) 55: 72-83, 2009. [PubMed] [Google Scholar]
- 2.Simon NG, Herkes GK. The neurologic manifestations of the acute porphyrias. J Clin Neurosci 18: 1147-1153, 2011. [DOI] [PubMed] [Google Scholar]
- 3.White KA, Marletta MA. Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry 31: 6627-6631, 1992. [DOI] [PubMed] [Google Scholar]
- 4.Lowenstein CJ, Snyder SH. Nitric oxide, a novel biologic messenger. Cell 70: 705-707, 1992. [DOI] [PubMed] [Google Scholar]
- 5.Kupferschmidt H, Bont A, Schnorf H, et al. Transient cortical blindness and bioccipital brain lesions in two patients with acute intermittent porphyria. Ann Intern Med 123: 598-600, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Utz N, Kinkel B, Hedde JP, Bewermeyer H. MR imaging of acute intermittent porphyria mimicking reversible posterior leukoencephalopathy syndrome. Neuroradiology 43: 1059-1062, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol 29: 1036-1042, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol 29: 1043-1049, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin JT, Wang SJ, Fuh JL, Hsiao LT, Lirng JF, Chen PM. Prolonged reversible vasospasm in cyclosporin A-induced encephalopathy. AJNR Am J Neuroradiol 24: 102-104, 2003. [PMC free article] [PubMed] [Google Scholar]
- 10.Gorchein A. Drug treatment in acute porphyria. Br J Clin Pharmacol 44: 427-434, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. The drug database for Acute Porphyria [Internet] [cited 2016 Apr 20] Available from: http://www.drugs-porphyria.org/
- 12.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126: 131-138, 1982. [DOI] [PubMed] [Google Scholar]
- 13.Ay H, Buonanno FS, Schaefer PW, et al. Posterior leukoencephalopathy without severe hypertension: utility of diffusion-weighted MRI. Neurology 51: 1369-1376, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Black KS, Mirsky P, Kalina P, et al. Angiographic demonstration of reversible cerebral vasospasm in porphyric encephalopathy. AJNR Am J Neuroradiol 16: 1650-1652, 1995. [PMC free article] [PubMed] [Google Scholar]
- 15.Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke 28: 1283-1288, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Zoja C, Furci L, Ghilardi F, Zilio P, Benigni A, Remuzzi G. Cyclosporin-induced endothelial cell injury. Lab Invest 55: 455-462, 1986. [PubMed] [Google Scholar]
- 17.Calabrese LH, Dodick DW, Schwedt TJ, Singhal AB. Narrative review: reversible cerebral vasoconstriction syndromes. Ann Intern Med 146: 34-44, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Ducros A. Reversible cerebral vasoconstriction syndrome. Lancet Neurol 11: 906-917, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Lu SR, Liao YC, Fuh JL, Lirng JF, Wang SJ. Nimodipine for treatment of primary thunderclap headache. Neurology 62: 1414-1416, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Kass-Hout T, Kass-Hout O, Sun CH, et al. A novel approach to diagnose reversible cerebral vasoconstriction syndrome: a case series. J Stroke Cerebrovasc Dis 24: e31-e37, 2015. [DOI] [PubMed] [Google Scholar]