

Abstract
Background
Deletion of the recurrent ~600 kb BP4-BP5 chromosomal region 16p11.2 has been associated with a wide range of neurodevelopmental outcomes.
Method
To clarify the phenotype of 16p11.2 deletion, we examined psychiatric and developmental presentation of predominantly clinically referred individuals, with a particular emphasis on broader autism phenotype characteristics in individuals with recurrent ~600 kb chromosome 16p11.2 deletions. 85 Individuals with the 16p11.2 deletion, and 153 familial controls, were evaluated for symptom presentation and clinical diagnosis using an extensive standardized assessment battery across 3 clinical sites.
Results
Individuals with the 16p11.2 deletion presented with a high frequency of psychiatric and developmental disorders (>90%). The most commonly diagnosed conditions were developmental coordination disorder, phonological processing disorder, expressive and receptive language disorders (71% of individuals over 3 years of age with a speech and language related disorder), and autism spectrum disorder (ASD). Individuals with the 16p11.2 deletion not meeting diagnostic criteria for ASD had significantly higher prevalence of autism-related characteristics compared to the familial non-carrier control group. Individuals with the 16p11.2 deletion had a range of intellectual ability, but IQ scores were 26 points lower than non-carrier family members on average.
Conclusion
Clinically referred individuals with the 16p11.2 deletion have high rates of psychiatric and developmental disorders and provide a genetically well-defined group to study the emergence of developmental difficulties, particularly those associated with the broader autism phenotype.
Keywords: 16p11.2 deletion, genetics, autism, developmental disability, psychiatric diagnosis
INTRODUCTION
Deletion of the recurrent ~600 kb BP4-BP5 region on 16p11.2 (chr 16: 29,649,996–30,199,855 in hg19) has been associated with a wide range of neurodevelopmental outcomes with a prevalence of approximately 0.6% (ranging from 0.3 to 1% across studies) of all patients ascertained for autism spectrum disorder (ASD), and 0.4% (ranging from 0.3 to 0.7%) in large series of patients with intellectual disability (ID) or birth defects (1–3). These rates are significantly higher than the estimated background population prevalence of .04–.05% (4, 5)
Previous research taking a “genetics first” approach with 16p11.2 copy number variants (CNVs) has reported significant heterogeneity in the phenotype of individuals with the 16p11.2 deletion (1–3, 6–9), but with consistent findings of increased frequency of ASD, intellectual and learning disabilities, and possible increased frequency of psychiatric disorders. Shinawi et al. (2010) found that 14 of 16 individuals had speech delays, 3 met criteria for ASD, and 6 had other behavioral difficulties. Even when excluding all psychiatric cases, findings of a cognitive deficit, particularly in verbal ability, persisted in the Icelandic population study by Stefansson et al (2013). Hanson et al. (2010) identified 21 individuals with 16p11.2 deletion who presented with a variety of neurodevelopmental disorders, including developmental delay, variable cognitive presentation, features of ASD, high incidence of language impairment, and a wide range of behavioral and psychiatric conditions. Zufferey et al. (2012) ascertained a total of 285 16p11.2 deletion carriers through several cohorts, including data on 56 probands from a European consortium gathered from questionnaires completed by referring clinicians, 45 probands from the Simons Variation in Individuals Project (Simons VIP), and 117 through literature review (which included participants ascertained for developmental/intellectual disabilities, obesity, and from the general population). Results revealed that full-scale intelligence quotients (FSIQ) were two standard deviations lower in carriers relative to familial controls, with verbal IQ being generally lower than nonverbal IQ. Further, 15% of carriers were classified as having ASD, many required speech therapy, and more than 70% were found to have comorbid psychiatric diagnoses. This heterogeneity in phenotypic presentation is also found in other chromosomal CNVs (e.g., 1q21.1) and gene mutations (e.g., NLGN4, NRXN1, SHANK2) associated with ASD and psychiatric disorders (10–15). However, limited phenotypic assessment has been completed in many of these rare genetic disorders, limiting the assessment of the true phenotypic heterogeneity of these disorders.
In these prior studies characterizing 16p11.2 deletion carriers, diagnostic characterization was established through multiple methods, including clinical assessment, questionnaires, and, in some cases, medical-history reviews; often this process lacked a standardization of the clinical assessment. Finally, several of these studies included multiple modes of ascertainment. These limitations stress the need for large sample sizes ascertained uniformly for the presence of the 16p11.2 deletion, and assessed with a standardized neuropsychological battery to assess the diversity of difficulties previously found to be common in the disorder, as well as standardized assessments of non-deletion relatives to serve as familial controls.
Familial comparisons offer a valid design (16–18), as well as an efficient way to overcome potential confounding problems inherent to unrelated case control designs, including differing genetic backgrounds and socioeconomic statuses (19, 20).
In order to deeply characterize the psychiatric and developmental problems such as ASD in a genetically well-defined CNV, we performed detailed cognitive, adaptive, language, psychiatric, behavioral and diagnostic testing, including standardized ASD assessment, on a large number of individuals with 16p11.2 deletions and their non-carrier siblings and parents. Because of the high likelihood of ASD in this population, we also explored whether differences between the carriers and familial controls were associated with the deficits inherent to this diagnosis or if these deficits were seen even when controlling for ASD related difficulties.
METHODS AND MATERIALS
Subjects
Subjects included individuals with the same recurrent 600 kb BP4-BP5 16p11.2 deletion without other pathogenic CNVs or known genetic diagnoses, the biological siblings of the individual with the deletion, and the biological parents of the individual with the deletion (see Table 1). Siblings were selected for participation based on closeness in age to the carrier. One half sibling was included. Adoptive parents were not used as control subjects but were interviewed for information about their carrier child. The majority of individuals with the 16p11.2 deletion were clinically identified, but cascade genetic testing within the families (see below) did identify some additional carriers.
Table 1.
Measures employed, number of individuals, mean score and standard deviation, and mean age and standard deviation of individuals tested in years.
| 16p11.2 Carrier age±SD score±SD |
Non-Carrier age±SD score±SD |
Carrier v Non Carrier p-value not controlling for any other factors | Carrier v Non Carrier p-value controlling for age, sex, NVIQ | Carrier v Non Carrier p-value controlling for age, sex, NVIQ, and ASD | |
|---|---|---|---|---|---|
| Vineland Adaptive Behavior Scales Second Edition (VABSII) | n=81 10.2 ± 7.3 |
n=46 11.2 ± 4.9 |
|||
| Composite | 80.0±18.2 | 104.2±10.8 | <.0001 | <.0001 | <.0001 |
| Social | 81.4±14.9 | 103.6±10.0 | <.0001 | <.0001 | <.0001 |
| Communication | 77.4±13.4 | 104.2±10.7 | <.0001 | <.0001 | <.0001 |
| Daily Living | 82.2±13.4 | 104.4±11.8 | <.0001 | <.0001 | <.0001 |
| Intelligence Quotient (IQ) | n=85 | n=153 | |||
| 11.1 ±8.9 | 30.5 ± 14.2 | ||||
| Verbal IQ1 | 79.0 ± 18.0 | 106.6 ± 11.9 | <.0001 | <.0001 | <.0001 |
| Nonverbal IQ1 | 86.8± 15.1 | 110.3 ± 13.2 | <.0001 | <.0001 | <.0001 |
| Full Scale IQ1 | 82.7 ± 15.0 | 109.5 ± 12.3 | <.0001 | <.0001 | <.0001 |
| Comprehensive Assessment of Spoken Language (CASL; 3–12) | n=68 9.6 ±3.8 76.8± 15.5 |
n=43 10.6 ± 4.2 105.9± 10.9 |
<.0001 |
<.0001 |
<.0001 |
| Edinburgh Handedness Inventory (EHI) | n=75 8.8 ± 4.0 |
n=38 12.5 ± 8.4 |
.0002 |
.03 |
.02 |
| Child Behavior Checklist (CBCL) 6–18 | n=56 10.5 ±2.9 |
n=36 11.4± 3.5 |
|||
| Total T | 61.2±10.8 | 47.0±10.7 | <.0001 | .01 | .01 |
| Internalizing T | 60.4±10.0 | 48.8±11.0 | <.0001 | .03 | .05 |
| Externalizing T | 54.3±11.1 | 47.8±8.8 | .006 | .54 | .76 |
| Conduct Problems T | 56.1±7.0 | 52.2±4.9 | .002 | .37 | .61 |
| ADHD Problems T | 62.3±8.1 | 53.4±4.8 | <.0001 | .0002 | .001 |
| Wechsler Individual Achievement Test (WIAT; 1st grade+) | n=57 13.9 ±9.5 |
||||
| Word Reading | |||||
| Reading | 75.5±15.6 | ||||
| Comprehension | 83.5±16.4 | ||||
| Sentence Composition | 79.4±16.5 | ||||
| Numerical Operations | 79.9±17.6 | ||||
| Comprehensive Test of Phonological Processing (CTOPP) Non-Word | n=63 10.1 ±3.5 |
n=44 12.7 ± 7.6 |
|||
| Repetition (5–24) | 5.4 ± 2.3 | 7.8 ± 2.4 | <.0001 | .01 | .02 |
| Behavior and Sensory Interests Questionnaire (BSIQ) | n=80 | n=39 | |||
| 8.9 ±4.0 | |||||
| 9.3 ± 7.1 | 10.5 ± 4.2 | ||||
| 2.5 ± 3.4 | <.0001 | .002 | .003 | ||
| Social Responsiveness Scale Parent Report (SRS; 4–18) | n=68 9.5 ±3.4 77.8 ± 32.5 |
n=41 10.5 ± 3.9 20.9 ± 17.0 |
<.0001 |
<.0001 |
<.0001 |
Not controlled for NVIQ
Biological or adoptive families which included an individual with the recurrent ~600 kb 16p11.2 BP4-BP5 deletion mediated by segmental duplications (chr16:29,652,999 to 30,199,351; hg19) identified through clinical diagnostic evaluations and who expressed interest in participating in research on the Simons VIP Connect website were invited to participate. All deletion carriers had the same recurrent deletion and no additional pathogenic CNVs or known monogenic disorders. Recruitment included directing traffic to the Simons VIP Connect website (SimonsVIPConnect.org) from Google Ads, links from patient advocacy websites, social media sites as well as through collaborations with clinical molecular cytogenetics laboratories that informed treating physicians of the study and through direct mailings to medical professionals. (Please see Simons VIP Consortium, 2012 (21) for more details on recruitment and inclusion/exclusion criteria.) Cascade genetic testing was conducted for all family members using a custom-designed oligonucleotide array containing genome-wide coverage at a resolution of ~400 kb and targeting known disease gene coverage at a resolution of ~50 kb (OGT 60K, Oxford Gene Technologies, Tarrytown, NY), according to previously published methods of analysis (22) to determine if the deletion was de novo or inherited and to identify other deletion carriers within the family.
Following screening, families participated in data collection at one of three Simons VIP phenotyping sites (Boston, Houston, and Seattle) for a comprehensive and standardized multi-day evaluation. The study was approved by the Institutional Review Board at each participating institution; all participants provided informed consent prior to data collection. All diagnostic interviewing and cognitive testing of children under 5 years old was videotaped for later review. Standardization of measurement across sites included mandatory formalized, standardized training on all measures through in-person training sessions and webinars for all clinicians, cross-site reliability and maintenance through monthly clinician conference calls and periodic videotape review, and validation and diagnostic confirmation through data review and observation of video recorded sessions by independent consultants.
The current analyses were limited to individuals age 3 and older because of lack of complete data sets in very young children and infants, to control for the possible instability of IQ measurement in very young children, particularly those with ASD and developmental disability (23–26) (27, 28), and to control for changes in presentation and rates of DSM diagnoses over time in very young children (29–31).
Phenotypic Assessment
Psychiatric diagnosis
Experienced, licensed clinicians gave best-estimate, clinical DSM-IV-TR (32) diagnoses using all information obtained during the research evaluation. Information was based on the standardized interview, questionnaire, and observation processes described below as well as results from standardized administration of the Diagnostic Interview Schedule for the Children (34), SCL-90 (35), and review of available medical records and prior testing. In order to capture the range of psychiatric presentation, exclusionary criteria for diagnoses were not considered (e.g., if a child met criteria for both ADHD and ASD, both diagnoses were considered). Autism-specific diagnostic measures included the Autism Diagnostic Observation Scale (36) and the Autism Diagnostic Interview – Revised (37). ASD symptom severity was calculated using the Calibrated Severity Score (CSS; (38)).
Autism related symptom measures
The Social Responsiveness Scale (SRS) was completed by parents about their children with the 16p11.2 deletion and the designated siblings (39). Total raw scores from the SRS were calculated and used within analyses for children between 4 and 18 years of age. Raw scores were used to provide greater differentiation of scores at the lower and higher end of the scales. The SRS-Adult version was completed for each parent by a spouse or partner (40). The Behavior and Sensory Interests Questionnaire (BSIQ) was also completed by parents about their children with the deletion and designated siblings. The BSIQ is an 87-item interview assessing repetitive and stereotyped interests/behaviors.
Cognitive and Behavioral Measures
Participants were administered a developmentally appropriate cognitive measure: Mullen Scales of Early Learning (41), Differential Abilities Scale, Second Edition (DAS-II; (42)), or Wechsler Abbreviated Scales of Intelligence (WASI; (43)). Standard scores or ratio intelligence quotient (IQ) scores were used to calculate Full Scale IQ (FSIQ), Verbal IQ (VIQ), and Nonverbal IQ (NVIQ), when possible. Other measures included: Vineland Adaptive Behavior Scales, Second Edition (VABS-II; parent interview version (44)); Wechsler Individual Achievement Test (WIAT, (45)); Comprehensive Assessment of Spoken Language (CASL, (46)) ; Comprehensive Test of Phonological Processing (CTOPP), Non-Word Repetition (47), Child Behavior Checklist 6–18 (CBCL) (48–50); Edinburgh Handedness Inventory (51). Only data from individuals who met age and basal criteria stated in the test manuals were used for analysis.
Data Analysis
We examined differences in psychiatric diagnosis, autism related symptoms, cognitive and adaptive skills, social and language abilities, and behavioral symptoms between 16p11.2 deletion carriers and non-carrier familial controls. We used random intercept linear mixed models (LMMs) for continuous outcomes and generalized estimating equations (GEEs) with a compound symmetric correlation structure for categorical and count outcomes. Deletion carriers were divided into three groups: de novo carriers, inherited carriers, and carriers with unknown inheritance status. In all the LMMs and GEEs, we estimated the differences in the outcome measures between de novo cases and inherited cases and between each carriers group and non-carrier familial controls, while accounting for correlated measures within family. We also constructed a contrast to estimate the differences between all carriers and non-carrier familial controls. Group differences were compared, unless otherwise noted, after controlling for age, sex, and non-verbal IQ (NVIQ). Additional LMM and GEE analyses also were conducted controlling for ASD diagnosis, to examine the effect of ASD on the behavioral presentation of individuals with 16p11.2 deletion. Due to the limited differences found between de novo and inherited carriers or between any inheritance group and carriers with unknown inheritance status, as well as small number of inherited cases, we reported the combined differences between all carriers and non-carrier familial controls in the main text and showed the comparisons between de novo and inherited carriers in the supplementary tables. To account for multiple comparisons, Bonferroni Correction was used, yielding a corrected alpha value of .0018 for the 28 comparisons. All statistical analyses were conducted using SAS 9.3 (52), IBM SPSS v19, and R 3.0.2 (53).
RESULTS
Psychiatric Diagnoses
Individuals with the 16p11.2 deletion presented with multiple psychiatric comorbid disorders (supplemental Figures 1 and 2): ninety-three percent of carriers had at least 1 diagnosis, compared to only 21% of controls. Developmental coordination disorder, phonological processing disorder, language disorders and ASD were the most common psychiatric diagnoses observed in carrier participants. Overall, there was a profile of speech- and language-based disorders among the 16p11.2 deletion carriers, with 71% of the individuals having a speech and language related disorder (Table 2). GEE analysis indicated that individuals with the 16p11.2 deletion had a higher expected number of psychiatric diagnoses even when controlling for NVIQ, age and sex (p<.0001). The mean number of distinct diagnoses in non-carriers was 0.3, while individuals with the deletion had an average of 2.9 diagnoses (a nearly ten-fold increase in number; Table 2). In addition, these differences in expected number of diagnoses persisted after controlling for ASD diagnosis (p<.0001; Supplemental Table 3).
Table 2.
Frequency of DSM-IV Psychiatric Disorders
| Child Carrier (Age 3–17) |
Adult Carrier | Non Carrier Child (Age 3–17) |
Non Carrier Adult | Carrier v Non Carrier p-value not controlling for any other factors | Carrier v Non Carrier p-value controlling for age, sex, NVIQ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| n=78 | n=7 | n=40 | n=115 | |||||||
| Phonological Processing Disorder (315.39) | 44 (56%) | 0 | 3 (8%) | 0 | <.0001 | .0002 | ||||
| Developmental Coordination Disorder (315.4) | 45 (58%) | 0 | 0 | 0 | ||||||
| Language Disorders (315.31, 315.32) | 36 (46%) | 1 (14%) | 1 (3%) | 0 | <.0001 | .003 | ||||
| Enuresis (307.6) | 16 (21%) | 0 | 2 (5%) | 0 | .0002 | .03 | ||||
| Autism Spectrum Disorders (299.00, 299.80) | 20 (26%) | 0 | 0 | 0 | ||||||
| ADHD Diagnosis (314.00, 314.01, 314.9) | 15 (19%) | 0 | 3 (8%) | 3 (3%) | .01 | .22 | ||||
| Borderline Intellectual Functioning (v62.89) | 10 (13%) | 2 (29%) | 0 | 3 (3%) | ||||||
| Intellectual Disability (317, 318, 319) | 8 (10%) | 0 | 0 | 0 | ||||||
| Behavior Disorders (312.9, 313.82) | 10 (13%) | 0 | 1 (3%) | 0 | ||||||
| Learning Disorders (315.0, 315.1, 315.2, 315.9) | 10 (13%) | NA | NA | NA | ||||||
| Anxiety Disorders (300.0, 300.02, 300.4, 300.9) | 5 (6%) | 0 | 3 (8%) | 10 (9%) | ||||||
| Tic Disorder (307.2, 307.22, 307.3) | 5 (6%) | 0 | 0 | 0 | ||||||
|
|
|
|
|
|||||||
| Number of Diagnoses | 3.1±1.6 | 0.6±0.5 | 0.4±0.8 | 0.3±0.6 | <.0001 | <.0001 | ||||
Cognitive Ability
As shown in Table 1, individuals with the 16p11.2 deletion demonstrated an average FSIQ score of 82.7, representing a 26.8 point (1.8 SD) shift downward compared to the FSIQ average of 109.5 in non-deletion controls. The same pattern was observed for Nonverbal and Verbal IQ. In line with their increased odds of a diagnosis related to language difficulties reported above, 27% of deletion participants demonstrated a VIQ< NVIQ discrepancy of 15 or more points, while only 7% showed a pattern of NVIQ<VIQ. This pattern is consistent across ages and includes adults.
Autism-related Symptoms
Social functioning
Children age 4–18 years with the 16p11.2 deletion had significantly higher SRS total scores compared to controls (Table 1), even when controlling for presence of ASD.
Restricted and repetitive behaviors
Almost all children with 16p11.2 deletion had some reported level of restricted and repetitive behavior patterns, with 88% of carriers versus 33% of controls reporting more than two of these types of behaviors. There was a significant difference in total BSIQ scores for restricted and repetitive behaviors in individuals with the 16p11.2 deletion compared to the non-carriers, which decreased slightly when controlling for age, sex, NVIQ and ASD (Table 1).
Adaptive skills
Individuals with 16p11.2 deletion exhibited a wide range of adaptive skill abilities on both composite and subdomain (Social, Communication, Daily Living Skills) scores. Poorer abilities in individuals with 16p11.2 deletion relative to non-carriers were found across all subdomains when controlling for age, sex and NVIQ (Table 1).
Academic Skills
On the WIAT, school age and adult individuals with the 16p11.2 deletion performed in the below-average to borderline range across reading comprehension, word reading, sentence composition, and numerical operations, on average (Table 1). Seventy-nine percent fell at least one standard deviation (SD=15) below the test mean (100) in basic word reading while 67% fell two or more standard deviations below the test mean. Sixty-five percent of carriers scored at least one standard deviation below the test mean in math (numerical operations subtest), and 32% of those individuals were lower by two standard deviations or more. Thirty one percent of individuals with the deletion showed a discrepancy of at least 1 SD between reading or math achievement scores and FSIQ.
Language Ability
Overall CASL scores for individuals with the 16p11.2 deletion were significantly lower than scores for non-carrier controls. On the non-word repetition task from the CTOPP, there was a significant difference between verbal individuals with 16p11.2 deletion and familial controls, which decreased slightly when controlling for age, NVIQ, sex, and ASD diagnosis (Table 1).
Behavioral difficulties
Analyses revealed that mean T-scores for children with 16p11.2 deletion ages 6–18 years on the CBCL were higher (more impaired) than those for control siblings on the Total CBCL score, internalizing domain score, ADHD and affective problems subscales. Importantly, 46% of the child deletion carriers showed problems in the clinically significant range on the total-problem scale, 23% on externalizing problems and 38% on internalizing problems. A significant difference in odds of ADHD diagnosis between carriers and controls persisted even after controlling for NVIQ, age, sex, and ASD (Table 1).
Handedness
Handedness information based on the Edinburgh Handedness Inventory was available for 75 deletion carriers (including all ages) and 38 non-carrier family members. 19% of carriers reported left hand dominance and 29% reported mixed dominance compared to 3% and 11% of non-carrier family members, respectively. GEE revealed significantly higher odds of left hand or mixed dominance for deletion carriers (Table 1).
Site Effects
Across all quantitative dimensional measures used, after controlling for multiple comparisons, there were no significant differences between sites. Across diagnostic categories, there were differences across the sites with regard to the rate at which developmental coordination disorder (p<.0002), language disorder (p=0.003) and enuresis disorder (p=0.02) were diagnosed. However, the same pattern of commonly occurring diagnoses was observed.
As noted in our methods, analyses of the differences between the de novo and inherited cases were performed as part of the LMM and GEE analyses on each measure and there were no significant differences (see Supplementary Tables 1–5).
DISCUSSION
We performed detailed diagnostic, cognitive, and behavioral testing, including standardized ASD assessment, on individuals who were ascertained after clinical identification of the 16p11.2 deletion and family member cascade testing and compared them to their familial controls. Our protocol addressed challenges in prior studies by way of standardization and comprehensive phenotyping. Our analyses clearly indicate that individuals with the deletion have a high frequency and range of psychiatric and developmental disorders compared to non-carrier controls. The most commonly observed diagnoses were developmental coordination disorder, phonological processing disorder, language disorders and ASD. One or more speech and language diagnoses were present in 71% of all individuals with 16p11.2 deletion, highlighting the specific contribution of this CNV to language development. Importantly, there was significant psychiatric comorbidity; many individuals met criteria for multiple diagnoses (Figure S2). This diagnostic overlap and clustering provides avenues for further understanding of the phenotype of the 16p11.2 deletion.
Further, while 24% of all individuals with the 16p11.2 deletion had a diagnosis of ASD, the majority of individuals with the deletion had significantly higher rates of autism-related characteristics, such as social and behavioral difficulties as reported on the SRS and repetitive and stereotyped behaviors as indexed by the BSIQ, when compared to non-deletion family members. Children who did not meet criteria for any psychiatric diagnosis still had sub-threshold challenges in the social-communication and behavioral traits related to ASD. This finding highlights the quantitative effect of the 16p11.2 deletion on autism related traits, even when full ASD-diagnostic criteria are not met.
A novel finding we report in this sample is that of increased odds of left hand or mixed hand dominance in individuals with 16p11.2 deletion. This underscores potential differences in brain development and cerebral asymmetry and provides insight into the deficits observed.
Finally, individuals with the 16p11.2 deletion demonstrated varying levels of intellectual ability, and the average IQ was approximately one standard deviation below the population mean. However, relative to non-deletion family members, participants with the deletion showed a 1.8 standard deviation decrement in IQ. Together with the results of social and behavioral deficits, these IQ findings suggest that the 16p11.2 deletion, regardless of psychiatric diagnosis, broadly affects several aspects of brain development and function, including language, cognition, and social cognition.
Previous studies of the 16p11.2 deletion with smaller sample sizes have reported wide ranging phenotypes, including cognitive impairments, language deficits, ASD, and behavioral problems (3, 6). The findings in this large series, using a standardized assessment battery and experienced clinicians trained to reliability, confirm and expand upon these previous findings compared to familial controls. We report similar findings of commonly observed language impairments, presence of a subgroup with ASD, and consistent cognitive impairments. Additionally these results highlight the presence of articulation challenges, language disorders, and motor impairments in a significant minority of participants with a deletion as well as left hand and mixed hand dominance.
Additionally, when these findings are compared against other clinically ascertained individuals with other deleterious CNVs, the uniqueness of the articulation, language, and motor impairments in the16p11.2 deletion is apparent. That is, many similarly ascertained clinical patients with recurrent CNVs also share an effect on IQ and phenotypic variability, but lack the specificity of language and motor based impairments in the 16p11.2 deletion. For example, about 25% of clinical patients with 22q11.2 deletions (DiGeorge or velocardiofacial syndrome) have a psychiatric disturbance, such as ASD, ADHD or schizophrenia (54). Clinically ascertained patients with 1q21.1 deletions have similar psychiatric phenotypic variability including intellectual disability, ASD, and schizophrenia (55). However, neither shares the constellation of articulation, language and motor impairments. Language, articulation, and motor challenges are common in ASD (56–61), underscoring the relevance of this locus to ASD specifically.
The composition of our sample provides insight into the 16p11.2 deletion phenotype. Despite efforts to broadly include all individuals with 16p11.2 deletion, our sample contains only 7 adults with the deletion. It is essential to consider the ascertainment approach and study design employed in this study in light of these findings. Participants were recruited through the Simons VIP Connect website to which they were referred via medical genetics clinics, genetic counselors, and internet searches and had been clinically diagnosed with a 16p11.2 deletion due to clinical concerns that led to ordering a chromosome microarray. Participants completed a screening process and traveled (often a great distance) to the clinical testing site. All expenses were paid to remove financial barriers. Once a patient was identified in a family, cascade genetic testing identified other family members with the deletion. This design led to self-selection of parents who could navigate the research recruitment and screening process, and navigate travel with family members with a deletion. The small proportion of adults in the sample is likely a result of the challenges associated with study participation that preclude participation of adults with the deletion unless another adult (often a spouse) could navigate this system and coordinate participation. Interestingly, several participants in the sample with inherited deletions were adopted, with records reporting behavioral challenges in the biological parent with the deletion that would have precluded participation in the study or caring for a child. In addition there is an ascertainment bias against clinically asymptomatic individuals since they would not have known they carried the deletion since most asymptomatic individuals have not had a chromosome microarray. Finally, the sample also contains far fewer individuals with inherited compared to de novo events, suggesting the possibility that the 16p11.2 deletion affects reproductive fitness, thus reducing the likelihood that this CNV will be transmitted.
Whereas our conclusions are potentially constrained by possible ascertainment biases as described above, recent work from a large study in Iceland (21) suggests that our findings may be broadly representative of most carriers of the CNV. As part of a larger study on CNVs, the researchers identified all carriers of the 16p11.2 deletion (n=43) in a population sample of 101,655 Icelanders, representing roughly one third of the entire population of Iceland. The investigators administered an abbreviated cognitive battery to seven carriers of the CNV who did not have a psychiatric diagnosis. These carriers showed marked impairments in verbal and performance IQ, as well as measures of verbal fluency and other cognitive domains, suggesting that our related observations in the Simons VIP sample are not an artifact of clinical ascertainment. The Icelandic sample also sheds light on the small number of individuals with inherited deletions, as fecundity in carriers was sharply reduced relative to other CNV carriers or the population at large; this likely explains the reduced likelihood that the CNV would be transmitted.
One study limitation is the potential increased noise resulting from the concatenation of data from multiple sites. The use of three sites employing experienced clinicians provided the ability to work with large numbers of individuals in a short amount of time. Examination of site influence on assessments revealed no differences between sites relative to the quantitative measures, suggesting that participants across site were similar in cognitive, adaptive, social, language, behavioral functioning. However, differences in diagnostic assignment across the sites in three psychiatric diagnoses were noted, suggesting the possibility of differences in diagnostic practices. Given similarity of sample sizes at each site and the overall rates for these three diagnoses, while introducing additional noise, our findings indicate the robustness of the diagnostic picture of 16p11.2 deletion and provide a reasonable estimate of the frequency with which each diagnosis is observed in clinical practice in which clinicians may apply different diagnostic labels.
These results have clinical implications. First, the consistent finding of a spoken-language deficit encompassing both receptive and expressive language, as well as articulation, highlights the importance of identifying and addressing communication challenges early in development. Second, given the high frequency of ASD and presence of the broader autism phenotype, careful consideration of autism-related symptoms is essential in any diagnostic characterization, as well as to treatment planning. Sub-diagnostic challenges in ASD-related domains can potentially moderate treatment outcome and adherence. Finally, motor coordination problems were found in over 50% of our deletion carriers. Increased clumsiness, motor delays, and fine and gross motor coordination have not consistently been noted in the literature. However, given the high prevalence of motor impairment in our sample, this should be carefully assessed and appropriate physical and/or occupational therapy initiated.
In summary, our analyses using data from a large, well characterized series of individuals with the 16p11.2 deletion revealed a consistent, quantitative detrimental effect on cognition, language ability, motor coordination, increased rates of ASD and the broader ASD phenotype (social difficulties, communication difficulties, stereotyped and repetitive behaviors and interests) as well as psychiatric difficulties compared to familial and population norms.
Supplementary Material
Figure 1.
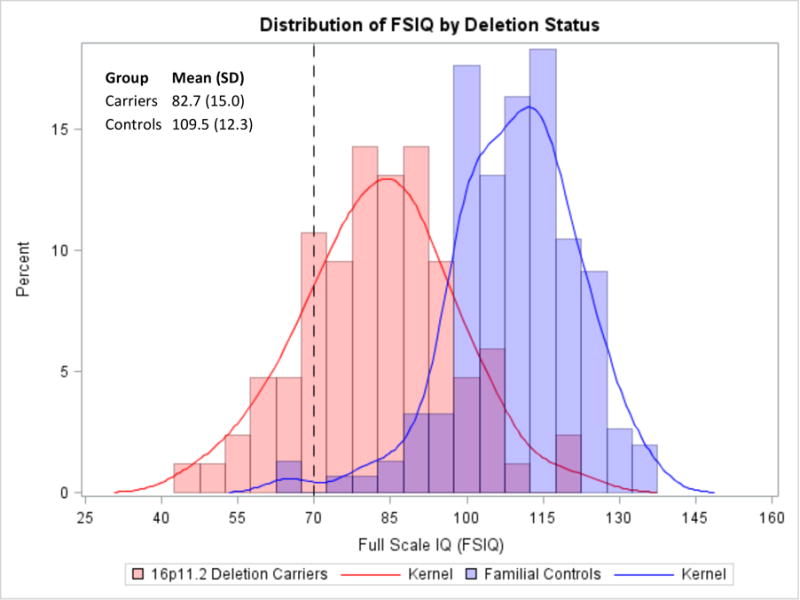
Distribution of FSIQ scores by deletion status highlights a 1.8 SD decrement in scores in 16p11.2 deletion cases relative to non-carrier family members. Dotted vertical line represents cutoff value for intellectual disability.
Acknowledgments
This work was supported by two grants from the Simons Foundation (SFARI award #198677 to EH, RB, RGK, & WKC and SFARI award #312100 to WAF, CLM, DHL) and a grant from the National Institutes of Health (MH074090 to DHL & CLM).
We are grateful to all of the families at the participating Simons Variation in Individuals Project (Simons VIP) sites, as well as the Simons VIP working group (Simons VIP consortium, Neuron, 73(6):1063–1067, 2012). We are also grateful for the guidance and support from Cathy Lord and Helen Tager-Flusberg on the development and implementation of this project.
Footnotes
We appreciate obtaining access to phenotypic data on SFARI Base.
Approved researchers can obtain the Simons VIP population dataset described in this study by applying at https://base.sfari.org.
FINANCIAL DISCLOSURES
None
References
- 1.Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. European journal of medical genetics. 2009;52(2–3):77–87. doi: 10.1016/j.ejmg.2009.03.006. Epub 2009/03/25. [DOI] [PubMed] [Google Scholar]
- 2.Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE, Shaffer LG, et al. Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. Journal of neurodevelopmental disorders. 2010;2(1):26–38. doi: 10.1007/s11689-009-9037-4. Epub 2010/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. Journal of medical genetics. 2010;47(5):332–41. doi: 10.1136/jmg.2009.073015. Epub 2009/11/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478(7367):97–102. doi: 10.1038/nature10406. Epub 2011/09/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505(7483):361–6. doi: 10.1038/nature12818. Epub 2013/12/20. [DOI] [PubMed] [Google Scholar]
- 6.Hanson E, Nasir RH, Fong A, Lian A, Hundley R, Shen Y, et al. Cognitive and behavioral characterization of 16p11.2 deletion syndrome. Journal of developmental and behavioral pediatrics: JDBP. 2010;31(8):649–57. doi: 10.1097/DBP.0b013e3181ea50ed. Epub 2010/07/09. [DOI] [PubMed] [Google Scholar]
- 7.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. The New England journal of medicine. 2008;358(7):667–75. doi: 10.1056/NEJMoa075974. Epub 2008/01/11. [DOI] [PubMed] [Google Scholar]
- 8.Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. Journal of medical genetics. 2012;49(10):660–8. doi: 10.1136/jmedgenet-2012-101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller DT, Nasir R, Sobeih MM, Shen Y, Wu BL, Hanson E. In: 16p11.2 Microdeletion. Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews; Seattle (WA): 2009. [Google Scholar]
- 10.Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. The New England journal of medicine. 2008;359(16):1685–99. doi: 10.1056/NEJMoa0805384. Epub 2008/09/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature genetics. 2003;34(1):27–9. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–55. doi: 10.1038/nrg2346. Epub 2008/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wisniowiecka-Kowalnik B, Nesteruk M, Peters SU, Xia Z, Cooper ML, Savage S, et al. Intragenic rearrangements in NRXN1 in three families with autism spectrum disorder, developmental delay, and speech delay. American journal of medical genetics Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2010;153B(5):983–93. doi: 10.1002/ajmg.b.31064. [DOI] [PubMed] [Google Scholar]
- 14.Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nature genetics. 2010;42(6):489–91. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- 15.Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet neurology. 2013;12(4):406–14. doi: 10.1016/S1474-4422(13)70011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milne RL, John EM, Knight JA, Dite GS, Southey MC, Giles GG, et al. The potential value of sibling controls compared with population controls for association studies of lifestyle-related risk factors: an example from the Breast Cancer Family Registry. International journal of epidemiology. 2011;40(5):1342–54. doi: 10.1093/ije/dyr110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donovan SJ, Susser E. Commentary: Advent of sibling designs. International journal of epidemiology. 2011;40(2):345–9. doi: 10.1093/ije/dyr057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Boehnke M, Abecasis GR. Efficient study designs for test of genetic association using sibship data and unrelated cases and controls. American journal of human genetics. 2006;78(5):778–92. doi: 10.1086/503711. Epub 2006/04/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinberg CR, Umbach DM. Choosing a retrospective design to assess joint genetic and environmental contributions to risk. American journal of epidemiology. 2000;152(3):197–203. doi: 10.1093/aje/152.3.197. [DOI] [PubMed] [Google Scholar]
- 20.Gauderman WJ, Witte JS, Thomas DC. Family-based association studies. Journal of the National Cancer Institute Monographs. 1999;(26):31–7. doi: 10.1093/oxfordjournals.jncimonographs.a024223. [DOI] [PubMed] [Google Scholar]
- 21.Simons Vip Consortium. Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron. 2012;73(6):1063–7. doi: 10.1016/j.neuron.2012.02.014. Epub 2012/03/27. [DOI] [PubMed] [Google Scholar]
- 22.Baldwin EL, Lee JY, Blake DM, Bunke BP, Alexander CR, Kogan AL, et al. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genetics in medicine: official journal of the American College of Medical Genetics. 2008;10(6):415–29. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- 23.Wishart JG, Duffy L. Instability of performance on cognitive tests in infants and young children with Down’s syndrome. Br J Educ Psychol. 1990;60(Pt 1):10–22. doi: 10.1111/j.2044-8279.1990.tb00918.x. Epub 1990/02/01. [DOI] [PubMed] [Google Scholar]
- 24.Pianta RC, Egeland B. Relation between depressive symptoms and stressful life events in a sample of disadvantaged mothers. Journal of consulting and clinical psychology. 1994;62(6):1229–34. doi: 10.1037//0022-006x.62.6.1229. [DOI] [PubMed] [Google Scholar]
- 25.Fulker DW, DeFries JC, Plomin R. Genetic influence on general mental ability increases between infancy and middle childhood. Nature. 1988;336(6201):767–9. doi: 10.1038/336767a0. [DOI] [PubMed] [Google Scholar]
- 26.Sattler J. Assessment of Children: Cognitive Applications. 4th. La Mesa, CA: Jerome Sattler Publisher Inc.; 2001. [Google Scholar]
- 27.Lord C, Schopler E. Stability of assessment results of autistic and non-autistic language-impaired children from preschool years to early school age. Journal of child psychology and psychiatry, and allied disciplines. 1989;30(4):575–90. doi: 10.1111/j.1469-7610.1989.tb00269.x. Epub 1989/07/01. [DOI] [PubMed] [Google Scholar]
- 28.Lord C, Schopler E. The role of age at assessment, developmental level, and test in the stability of intelligence scores in young autistic children. Journal of autism and developmental disorders. 1989;19(4):483–99. doi: 10.1007/BF02212853. Epub 1989/12/01. [DOI] [PubMed] [Google Scholar]
- 29.Dyson L, Edgar E, Crnic K. Psychological predictors of adjustment by siblings of developmentally disabled children. American Journal Of Mental Retardation: AJMR. 1989;94(3):292–302. [PubMed] [Google Scholar]
- 30.Postert C, Averbeck-Holocher M, Beyer T, Muller J, Furniss T. Five systems of psychiatric classification for preschool children: do differences in validity, usefulness and reliability make for competitive or complimentary constellations? Child psychiatry and human development. 2009;40(1):25–41. doi: 10.1007/s10578-008-0113-x. [DOI] [PubMed] [Google Scholar]
- 31.Luby JL, Si X, Belden AC, Tandon M, Spitznagel E. Preschool depression: homotypic continuity and course over 24 months. Archives of general psychiatry. 2009;66(8):897–905. doi: 10.1001/archgenpsychiatry.2009.97. Epub 2009/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Association AP. Diagnostic and Statistical Manual of Mental Disorders. 4th. Washington, DC: American Psychiatric Association; 2000. Text Revision ed. [Google Scholar]
- 33.Bishop SL, Guthrie W, Coffing M, Lord C. Convergent validity of the Mullen Scales of Early Learning and the differential ability scales in children with autism spectrum disorders. Am J Intellect Dev Disabil. 2011;116(5):331–43. doi: 10.1352/1944-7558-116.5.331. Epub 2011/09/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaffer D, Fisher P, Lucas C, Dulcan MK, Schwab-Stone M. NIMH Diagnostic Interview Schedule for Children, Version IV (NIMH DISC-IV): Description, differences from previous versions and reliability of some common diagnoses. J Am Acad Child Adoles Psychiatry. 2000;39(1) doi: 10.1097/00004583-200001000-00014. [DOI] [PubMed] [Google Scholar]
- 35.Derogatis L. Symptom Checklist- 90-R. Minneapolis, MN: Pearson Assessments; 1994. [Google Scholar]
- 36.Lord C, Risi S, Lambrecht L, Cook EH, Jr, Leventhal BL, DiLavore PC, et al. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. Journal of autism and developmental disorders. 2000;30(3):205–23. Epub 2000/10/31. [PubMed] [Google Scholar]
- 37.Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of autism and developmental disorders. 1994;24(5):659–85. doi: 10.1007/BF02172145. Epub 1994/10/01. [DOI] [PubMed] [Google Scholar]
- 38.Gotham K, Pickles A, Lord C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. Journal of autism and developmental disorders. 2009;39(5):693–705. doi: 10.1007/s10803-008-0674-3. Epub 2008/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Constantino JN, Gruber CP. The Social Responsiveness Scale Manual. Los Angeles, CA: Western Psychological Services; 2005. [Google Scholar]
- 40.Constantino JN, Gruber CP. The Social Responsiveness Scale- Adult Reseach Version. Los Angeles, CA: Western Psychological Services; 2005. [Google Scholar]
- 41.Mullen EM. In: Scales of Early Learning. AGS, editor. Circle Pines, MN: Pearson Assessments; 1995. [Google Scholar]
- 42.Elliott CD. Differential Ability Scales. 2nd. San Antonio, TX: Harcourt Assessment; 2007. [Google Scholar]
- 43.Weschsler D. Wechsler Abbreviated Scale of Intelligence. San Antonio, TX: The Psychological Corporation; 1999. [Google Scholar]
- 44.Sparrow SS, Chicchetti DV, Balla DA. Vineland Adaptive Behavior Scales. 2nd. Circle Pines, MN: America; 2005. [Google Scholar]
- 45.Weschsler D. Wechsler Individual Achievement Test. San Antonio, TX: Psychological Corporation; 2009. [Google Scholar]
- 46.Carrow-Woolfolk E. Comprehensive Assessment of Spoken Language. Torrance, CA: Western Psychological Services; 1999. [Google Scholar]
- 47.Wagner RK, Torgesen JK, Rashotte CA. Comprehensive Test of Phonological Processing. Austin, TX: Pro: Ed; 1999. [Google Scholar]
- 48.Achenbach TM, Dumenci L. Advances in empirically based assessment: revised cross-informant syndromes and new DSM-oriented scales for the CBCL, YSR, and TRF: comment on Lengua, Sadowksi, Friedrich, and Fischer (2001) Journal of Consulting and Clinical Psychology. 2001;69(4):699–702. [PubMed] [Google Scholar]
- 49.Achenbach TM, Rescorla LA. Manual for the ASEBA Adult Forms & Profiles. Burlington, VT: University of Vermont, Research Center for Children, Youth, & Families; 2003. [Google Scholar]
- 50.Achenbach TM, Ruffle TM. The Child Behavior Checklist and related forms for assessing behavioral/emotional problems and competencies. American Academy of Pediatrics. 2000;21(8):265–71. doi: 10.1542/pir.21-8-265. [DOI] [PubMed] [Google Scholar]
- 51.Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia. 1971;9(1):97–113. doi: 10.1016/0028-3932(71)90067-4. [DOI] [PubMed] [Google Scholar]
- 52.SAS Version 9.2. Cary, NC, USA: SAS Institute Inc.; 2002–2005. [Google Scholar]
- 53.Team RC. R: A language and environment for statistical computing. Viennam Austria: R Foundation for Statistical Computing; 2013. Available from: http://www.R-project.org/ [Google Scholar]
- 54.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455(7215):919–23. doi: 10.1038/nature07458. Epub 2008/10/17. [DOI] [PubMed] [Google Scholar]
- 55.Mefford HC, Cooper GM, Zerr T, Smith JD, Baker C, Shafer N, et al. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome research. 2009;19(9):1579–85. doi: 10.1101/gr.094987.109. Epub 2009/06/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boucher J. Research review: structural language in autistic spectrum disorder - characteristics and causes. Journal of child psychology and psychiatry, and allied disciplines. 2012;53(3):219–33. doi: 10.1111/j.1469-7610.2011.02508.x. Epub 2011/12/23. [DOI] [PubMed] [Google Scholar]
- 57.Kjelgaard MM, Tager-Flusberg H. An Investigation of Language Impairment in Autism: Implications for Genetic Subgroups. Lang Cogn Process. 2001;16(2–3):287–308. doi: 10.1080/01690960042000058. Epub 2006/05/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maski KP, Jeste SS, Spence SJ. Common neurological co-morbidities in autism spectrum disorders. Curr Opin Pediatr. 2011;23(6):609–15. doi: 10.1097/MOP.0b013e32834c9282. Epub 2011/10/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ming X, Brimacombe M, Wagner GC. Prevalence of motor impairment in autism spectrum disorders. Brain Dev. 2007;29(9):565–70. doi: 10.1016/j.braindev.2007.03.002. Epub 2007/05/01. [DOI] [PubMed] [Google Scholar]
- 60.Shriberg LD, Paul R, McSweeny JL, Klin AM, Cohen DJ, Volkmar FR. Speech and prosody characteristics of adolescents and adults with high-functioning autism and Asperger syndrome. J Speech Lang Hear Res. 2001;44(5):1097–115. doi: 10.1044/1092-4388(2001/087). Epub 2001/11/16. [DOI] [PubMed] [Google Scholar]
- 61.Tager-Flusberg H, Joseph RM. Identifying neurocognitive phenotypes in autism. Philos Trans R Soc Lond B Biol Sci. 2003;358(1430):303–14. doi: 10.1098/rstb.2002.1198. Epub 2003/03/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.