

Abstract
l-Cysteine is a semi-essential amino acid and substrate for cystathionine-β-synthase (CBS) in the central nervous system. We previously reported that NaHS, an H2S donor, significantly alleviated brain damage after subarachnoid hemorrhage (SAH) in rats. However, the potential therapeutic value of l-cysteine and the molecular mechanism supporting these beneficial effects have not been determined. This study was designed to investigate whether l-cysteine could attenuate early brain injury following SAH and improve synaptic function by releasing endogenous H2S. Male Wistar rats were subjected to SAH induced by cisterna magna blood injection, and l-cysteine was intracerebroventricularly administered 30 min after SAH induction. Treatment with l-cysteine stimulated CBS activity in the prefrontal cortex (PFC) and H2S production. Moreover, l-cysteine treatment significantly ameliorated brain edema, improved neurobehavioral function, and attenuated neuronal cell death in the PFC; these effects were associated with a decrease in the Bax/Bcl-2 ratio and the suppression of caspase-3 activation 48 h after SAH. Furthermore, l-cysteine treatment activated the CREB–brain-derived neurotrophic factor (BDNF) pathway and intensified synaptic density by regulating synapse proteins 48 h after SAH. Importantly, all the beneficial effects of l-cysteine in SAH were abrogated by amino-oxyacetic acid, a CBS inhibitor. Based on these findings, l-cysteine may play a neuroprotective role in SAH by inhibiting cell apoptosis, upregulating CREB–BDNF expression, and promoting synaptic structure via the CBS/H2S pathway.
Keywords: l-cysteine, H2S, cystathionine-β-synthase, subarachnoid hemorrhage, early brain injury
Introduction
In patients with subarachnoid hemorrhage (SAH), early brain injury (EBI) is the primary cause of high mortality and morbidity (1). Multiple factors, including cell death, oxidative stress, abnormal inflammatory responses, and cerebral vasospasm, are involved in the mechanisms underlying EBI after SAH (2). Thus, identification of early neuroprotective strategies for potential clinical use is urgently needed.
l-Cysteine is a semi-essential amino acid and is important for regulating human metabolism (3). The three traditional endogenous sources of l-cysteine include absorption from the diet, the transsulfuration pathway, and protein degradation. Disruption of the extracellular l-cysteine/l-cystine ratio may be associated with oxidative stress (4, 5). Kimura et al. demonstrated that in the central nervous system (CNS), l-cysteine may be catalyzed by cystathionine-β-synthase (CBS), which is expressed in astrocytes, and may then produce endogenous hydrogen sulfide (H2S) (6, 7). Moreover, amino-oxyacetic acid (AOAA), a widely used selective CBS inhibitor, has been reported to block CBS-mediated H2S production in several organs (7, 8).
H2S plays multiple roles in the CNS under both physiological and pathological conditions (9). Interestingly, accumulating evidence has suggested that exogenous H2S can function as a powerful neuroprotective agent. Kimura and Kimura reported in 2004 that H2S protected primary rat cortical neurons from oxidative stress-induced injury (10). H2S also exerts a number of cytoprotective anti-apoptotic, antioxidant, and anti-inflammatory effects on the CNS (6, 11, 12). Our previous studies showed that H2S exhibited neuroprotective potential in an animal model of cerebral hypoxia injury (13, 14). Importantly, we observed that l-cysteine promoted the proliferation and neuronal differentiation of neural stem cells via the CBS/H2S system in vitro (15). Administering AOAA to animal models of cerebral hypoxia injury could inhibit H2S generation and induce physiological changes in blood pressure regulation or associative learning (7, 16).
Only limited information is available about the neuroprotective effects of H2S on SAH (17, 18). Furthermore, whether l-cysteine can safely exert protective effects on EBI after SAH by triggering CBS to produce H2S and the molecular mechanisms underlying these effects are still unknown. Thus, the aim of this study is to elucidate the potential therapeutic effect of l-cysteine on EBI after SAH and determine whether l-cysteine is associated with H2S function.
Animals and Methods
Animals
Male Wistar rats (280–350 g) were purchased from the Laboratory Animal Center, Shandong University. Upon arrival, the animals were housed under standard laboratory conditions (temperature 20 ± 2°C, 12 h:12 h light/dark cycle, lights on at 0800 h), provided free access to food and water and allowed to habituate to their new environment for 1 week.
SAH Model
Experimental SAH was induced in the rats using double blood injection according to our previous study (18). Briefly, esthesia was induced under 3.5% isoflurane and changed to continuous narcosis with 2.5% isoflurane during surgery. A catheter was inserted into the femoral artery under sterile conditions to withdraw blood and measure blood pressure. Two hundred microliters of autologous blood was withdrawn from the femoral artery and injected into the cisterna magna over a 3-min period.
Experimental Design
A total of 134 surgeries were conducted. The rats were randomly assigned to the following five groups: Sham (n = 22), Sham + l-cysteine (n = 22), SAH (n = 30), SAH + l-cysteine (n = 30), and SAH + l-cysteine + AOAA (n = 30). At 48 h after SAH, these rats were euthanized, and the prefrontal cortex (PFC) tissues were removed and prepared for analysis. The individual group mortality within 48 h after surgery was as follows: Sham 0% (0/22), Sham + l-cysteine 0% (0/22), SAH 23.3% (7/30), SAH + l-cysteine 10% (3/30), and SAH + l-cysteine + AOAA 16.7% (5/30).
Drug Administration
l-Cysteine (Sigma-Aldrich) was dissolved in vehicle (PBS) at a working concentration of 100 mM as determined by our previous research (15), and 30 µL of the l-cysteine solution was intracerebroventricularly administered 30 min after SAH. AOAA (Sigma-Aldrich) was dissolved in vehicle (PBS), and a 5 mg/kg dose was intraperitoneally administered with l-cysteine.
CBS Activity Assay and Measurement of H2S Production
The CBS activity of brain tissue was detected by a CBS assay kit (Genmed Scientifics Inc., China). This assay indirectly measures CBS activity by detecting CBS metabolites that interact with NADPH. Absorbance was measured at 340 nm using a microplate reader (Spectra Max 190, Molecular Devices, Sunnyvale, CA, USA).
To quantify H2S, we used the traditional methylene blue method. Briefly, the PFC tissue was homogenized and incubated with zinc acetate, which generates zinc sulfide that subsequently reacts with N,N-dimethyl-p-phenylenediamine sulfate (NNDPD). The absorbance value was determined at 670 nm, and the H2S level was calculated against an NaHS calibration curve.
Neurological Scores
At 48 h after SAH, neurological function was evaluated by two “blinded” investigators using a modified Garcia scoring system (19, 20). This system comprises the following seven subtests: spontaneous activity (0–3 points), reaction to side stroking (1–3 points) and to vibrissae touch (1–3 points), limb symmetry (0–3 points), forelimb outstretching (0–3 points), and climbing (0–3 points) and beam walking (0–4 points) abilities. The total score of these subtests reflected neurological function. High Garcia scores indicated better neurological function, and low scores indicated worse function, with the worst performances receiving 2 points (21).
Brain Water Content
Brain edema was determined according to the wet/dry method, where% brain water content = [(wet weight − dry weight)/wet weight] × 100%. Briefly, each brain sample (both cerebral hemispheres) was removed from the skull and weighed immediately. Then, the sample was dried at 100°C for 48 h and weighed to determine the dry weight.
Hematoxylin and Eosin Staining
Animals were perfused under deep anesthesia with 10% chloral hydrate followed by 4% paraformaldehyde. The brains were then removed and post-fixed in formalin. After fixation and dehydration in an ethanol gradient, the brain tissue was embedded in paraffin and sliced into 4-μm thick coronal sections using a section cutter (Leica, Germany). The sections (3 sections/rat) were stained with hematoxylin and eosin (H&E). In addition, four rats in each group were prepared for H&E staining. The morphology of the PFC (the cerebral cortex that covers the anterior portion of the frontal lobe) was observed under a light microscope (Olympus Corporation, Japan).
Transferase dUTP Nick End Labeling (TUNEL) Staining
Four samples from each group were prepared for terminal deoxynucleotidyl TUNEL staining. Apoptosis was detected using a TUNEL kit according to the manufacturer’s protocol (DeadEnd Fluorometric kit, Promega, WI, USA). Slides were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI), washed, coverslipped with a water-based mounting medium, and sealed with nail polish. Three microscope fields (20×) containing TUNEL-positive cells in the cortex were selected and imaged. The number of TUNEL/DAPI-positive cells was calculated as the mean of the numbers obtained from six images per rat. Counting was performed in a blinded manner.
Immunofluorescence Imaging
Slides (n = 4 samples per group) were fixed in 4% paraformaldehyde for 20 min and blocked with 10% goat serum in PBS. The slides were subsequently incubated overnight in a humidified chamber at 4°C with the following primary antibodies: NeuN (1:100, Abcam, Cambridge, MA, USA) and cleaved caspase-3 (1:100, Cell Signaling Tech., MA, USA). After primary antibody incubation, the samples were washed and incubated with an appropriate fluorescent-conjugated secondary antibody (1:500 dilution, Sigma-Aldrich) for 1 h. Images were captured using a Nikon TE2000U microscope. Three microscope fields (20×) containing cleaved caspase-3/NeuN double-positive cells in the cortex were chosen and imaged. The number of active caspase-3/NeuN double-positive cells was calculated as the mean of the numbers obtained from six images per rat. Counting was performed in a blinded manner.
Immunohistochemistry
The sections were deparaffinized using a standard procedure and washed with PBS as described previously. Briefly, after blocking for 30 min at room temperature, the sections were incubated with the following primary antibodies: CBS (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. After primary antibody incubation, the samples were washed and incubated with secondary antibodies for 2 h at room temperature. The sections were washed and then incubated with an avidin–biotinylated enzyme complex for 1 h at room temperature. The sections were visualized with diaminobenzidine. Nuclei were counterstained with hematoxylin. Finally, the sections were dehydrated in an alcohol gradient and cleared with xylene. Images were captured using a Nikon TE2000U microscope.
Sample Preparation for Transmission Electron Microscopy (TEM)
For TEM, we sacrificed three rats per group. PFC specimens, with an approximate volume of 1 mm3, were dissected quickly on ice and fixed in 2.5% glutaraldehyde for 2 h at 4°C. Following several washes in PBS, the specimens were fixed in 1% osmium tetroxide for 2 h and then dehydrated in a graded ethanol series. The tissues were subsequently infiltrated with 50/50 propylene oxide overnight and embedded. The tissues were prepared for sectioning on an Ultramicrotome (EM UC 7, Leica, Germany) and cut into 50-nm thick sections. After being stained with uranyl acetate, the sections were examined under a Hitachi H-7500 TEM.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from the PFC using a TRIzol reagent (Gibco, Invitrogen) according to the manufacturer’s instructions. The RNA concentration was determined using a spectrophotometer (Bio-Rad Labs) at 260 nm. Identical amounts of RNA (2 µg) were reverse transcribed into cDNA using a commercial RT-PCR kit (Fermentas, Vilnius, Lithuania) according to the manufacturer’s instructions. Then, the cDNA was subsequently amplified by PCR with specific primers (Table 1). The PCR products, which were separated on a 1.2% agarose/TAE gel, were visualized by staining with ethidium bromide. The densitometric values were normalized to those of β-actin. Band intensity was determined using Image-Pro Plus 6.0 software.
Table 1.
PCR primers used in this study.
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Bax | GGT TGC CCT CTT CTA CTT TGC | TCT TCC AGA TGG TGA GCG AG |
| Bcl-2 | GGA TGA CTT CTC TCG TCG CTA C | TGA CAT CTC CCT GTT GAC GCT |
| Brain-derived neurotrophic factor | AGC TGA GCG TGT GTG ACA GT | ACC CAT GGG ATT ACA CTT GG |
| Synaptophysin | CAAGAAATACCGCTACCAAGATG | CCCTCTGTTCCATTCACCTG |
| PSD95 | ATGGCACGTAATGGAGACTAC | TCTTGTGTAGTCGAACCATCTG |
| β-actin | CTA TTG GCA ACG AGC GGT TCC | CAG CAC TGT GTT GGC ATA GAG G |
Western Blot Analysis
Protein concentration in the PFC was determined using a BCA protein assay kit (Pierce Biotechnology, Inc.). A quantity of 30–50 µg of total proteins was loaded onto a 4–20% gradient polyacrylamide gel, electrophoretically transferred to a polyvinylidene difluoride membrane and probed with the following primary antibodies: Bax antibody (1:1,000, Santa Cruz Biotechnology, CA, USA), Bcl-2 antibody (1:1,000, Santa Cruz Biotechnology), cleaved caspase-3 (1:500, Cell Signaling Tech. MA, USA), caspase-3 (1:1,000, Cell Signaling), phospho-cAMP response element binding protein (p-CREB) (1:1,000, Cell Signaling Tech., MA, USA), CREB (1:1,000, Cell Signaling Tech., MA, USA), and brain-derived neurotrophic factor (BDNF) (1:1,000, Santa Cruz Biotechnology). β-actin (1:2,000; Sigma-Aldrich) was used as an internal control. The secondary antibody was horseradish peroxidase conjugated to goat/mouse anti-rabbit IgG (1:8,000, Sigma-Aldrich). The membranes were developed using an enhanced chemiluminescence detection system (Pierce, Rockford, IL, USA).
Statistical Analysis
SPSS 22.0 was used for statistical analysis. The neurological scores were analyzed by Kruskal–Wallis one-way analysis of variance (ANOVA) on ranks followed by Dunn’s post hoc test. Other data are presented as the mean ± SD; these data were analyzed by one-way ANOVA followed by Tukey’s post hoc analysis. Differences were considered significant at p < 0.05.
Results
The Effect of l-Cysteine on CBS Activity and H2S Production in SAH-Insulted Brain Tissue
Cystathionine-β-synthase has been reported to mainly localize to astrocytes in the CNS and catalyze l-cysteine to produce endogenous H2S (22, 23). Here, we investigated SAH-induced changes in CBS expression and CBS activity in response to l-cysteine treatment at 48 h after SAH. In agreement with previous findings, our immunohistochemical analysis revealed numerous CBS-positive cells in the PFC tissue of the Sham and Sham + l-cysteine groups, whereas CBS-positive cells were very rare in the SAH group (Figure 1A). Surprisingly, l-cysteine treatment significantly upregulated CBS expression in the SAH group (Figure 1A). Moreover, CBS expression in the PFC was evaluated at 48 h after SAH by Western blot and RT-PCR, and the results revealed that l-cysteine also increased the protein and mRNA expression levels of CBS (Figure 1B).
Figure 1.

Effects of l-cysteine on endogenous cystathionine-β-synthase (CBS) activity. (A) The expression of CBS in cells (red arrows indicated) was determined by immunohistochemistry at 48 h after subarachnoid hemorrhage (SAH). Scale bar = 50 μm (n = 4). (B) CBS was quantified by reverse transcription polymerase chain reaction and Western blotting at 48 h after SAH. Each value was normalized to β-actin. The bar graphs showing the quantification of mRNA and protein levels of CBS were generated by Image-Pro Plus 6.0 (n = 4). (C) CBS activity was assessed at 48 h after SAH (n = 6). (D) Production of endogenous H2S was evaluated by the methylene blue method at 48 h after SAH (n = 8). The values represent the means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 SAH vs Sham, #p < 0.05, ##p < 0.05, ###p < 0.001 SAH + l-Cys vs SAH, +p < 0.05, ++p < 0.01, +++p < 0.001 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
We further assessed CBS activity, which affects l-cysteine. Both the Sham + l-cysteine and SAH + l-cysteine groups exhibited dramatic upregulation of CBS activity, whereas the SAH group showed low CBS activity (Figure 1C). Next, we measured H2S production in the PFC of the different groups because H2S production is an indirect measure of CBS activity (24). The l-cysteine-treated groups (Sham + l-cysteine and SAH + l-cysteine) produced more H2S than the Sham or SAH groups. Exposure to SAH slightly decreased H2S levels in the PFC, but the levels in the SAH group were not significantly different from those in the Sham group (Figure 1D). Co-treatment with AOAA suppressed the effects of l-cysteine on SAH.
Administration of l-Cysteine Reduced Brain Edema and Improved Neurological Behavior at 48 h after SAH
Compared to the Sham group, the SAH groups had significantly lower neurological scores at 48 h (Figure 2A). l-Cysteine treatment improved neurological scores, but this treatment effect was reversed by AOAA (Figure 2A).
Figure 2.

l-Cysteine ameliorated subarachnoid hemorrhage (SAH)-induced brain injury. (A) Neurological scores were recorded at 48 h after SAH (n = 6). (B) Brain water content of the cerebral cortex was measured at 48 h after SAH (n = 6). (C) H&E staining was performed on brain tissues at 48 h after SAH. Pathological changes included focal edema in the prefrontal cortex (black arrows indicated) (n = 4). Scale bar = 100 μm. The values represent the mean ± SD. **p < 0.01, ***p < 0.001 SAH vs Sham, #p < 0.05, ##p < 0.01 SAH + l-Cys vs SAH, +p < 0.05 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
Brain edema (including both cerebral hemispheres) was evaluated immediately after the neurological assessment. The SAH-injured brain tissue had a significantly higher water content than that of the Sham and Sham + l-cysteine brain tissue. Post-SAH injection of l-cysteine reduced brain water content, but this outcome was reversed by AOAA administration (Figure 2B).
In the Sham and Sham + l-cysteine groups, the brain tissues had organized structural layers and cortical neurons with well-defined borders. However, in the SAH group, the cells were arranged sparsely, and the cell outline was fuzzy. Moreover, in the SAH group, we identified substantial edema in the PFC, which was pale in appearance, and shrunken neurons. l-Cysteine treatment ameliorated the edema and morphological damage induced by SAH (Figure 2C).
Additionally, a 5 mg/kg dose of AOAA did not induce further neuronal damage post-SAH (see Figure S1 in Supplementary Material).
l-Cysteine Attenuated SAH-Induced Brain Injury by Reducing Neuronal Apoptosis
In the PFC of rats in the Sham and Sham + l-cysteine groups, TUNEL-positive cells were rarely detected, while in the SAH group, many TUNEL-positive cells were identified. The apoptosis in response to SAH was significantly ameliorated by treatment with l-cysteine, but this effect was inhibited by AOAA administration (Figure 3).
Figure 3.

l-Cysteine attenuates subarachnoid hemorrhage (SAH)-induced apoptosis. (A) The detection of transferase dUTP nick end labeling (TUNEL)-positive cells in the prefrontal cortex was performed at 48 h after SAH. Scale bar = 50 μm. (B) Bar graphs showing the quantification of TUNEL-positive cells (n = 4). Scale bar = 50 μm. The values represent the means ± SD. ***p < 0.001 SAH vs Sham, ###p < 0.001 SAH + l-Cys vs SAH, ++p < 0.01 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
l-Cysteine Inhibited SAH-Induced Caspase-3 Activation
We used cleaved caspase-3/NeuN double staining to evaluate how l-cysteine treatment inhibited apoptosis after SAH. Few cleaved caspase-3-positive cells were detected in the Sham and Sham + l-cysteine groups, whereas numerous cleaved caspase-3/NeuN double-stained cells were seen in the SAH group (Figures 4A,B). l-Cysteine dramatically reduced cleaved caspase-3 expression levels, but this effect was blocked by AOAA. The effect of l-cysteine on SAH-induced caspase-3 activation was confirmed by Western blot (Figures 4C,D).
Figure 4.

The effect of l-cysteine on caspase-3 activation in subarachnoid hemorrhage (SAH). (A) Immunofluorescence staining revealed the colocalization of cleaved caspase-3 and NeuN in the prefrontal cortex at 48 h after SAH. Scale bar = 50 μm. (B) The bar graphs showing the quantification of cleaved caspase-3/NeuN-positive cells (n = 4). (C) The expression of cleaved caspase-3 was assessed using Western blot analysis. (D) Bar graphs showing the quantification of the protein levels of cleaved caspase-3 and caspase-3 were generated by Image-Pro Plus 6.0. The results are expressed as the cleaved caspase-3/caspase-3 ratio (n = 3). The values represent the mean ± SD. *p < 0.05, ***p < 0.001 SAH vs Sham, #p < 0.05, ###p < 0.001 SAH + l-Cys vs SAH, +p < 0.05, +++p < 0.001 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
l-Cysteine Restored Bcl-2 and Bax Expression Levels after SAH
Because Bcl-2 and Bax are key regulators of the mitochondrial apoptosis pathway in cells, we evaluated the expression levels of Bcl-2 and Bax at both the mRNA and protein levels. As shown in Figure 5, SAH markedly increased the Bax/Bcl-2 ratio at the mRNA and protein levels at 48 h after injury. However, the increased Bax/Bcl-2 ratio was reduced by treatment with l-cysteine. The effect of l-cysteine on the SAH-induced elevation in the Bax/Bcl-2 ratio was reversed by AOAA (Figures 5A,B).
Figure 5.

Effects of l-cysteine on Bax and Bcl-2 at the mRNA and protein levels. (A) The relative expression levels of Bax and Bcl-2 mRNA in the prefrontal cortex (PFC) were analyzed by semi-quantitative reverse transcription polymerase chain reaction. The densities of the protein bands were analyzed and normalized to β-actin (n = 3). (B) Representative Western blots showing the levels of Bax and Bcl-2 in the PFC and bar graphs showing the quantification of the protein levels of Bax and Bcl-2 (n = 3). The mRNA and protein levels were obtained from three independent experiments. The values represent the means ± SD. **p < 0.01, ***p < 0.001 subarachnoid hemorrhage (SAH) vs Sham, #p < 0.05, ###p < 0.001 SAH + l-Cys vs SAH, +p < 0.05, ++p < 0.01 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
l-Cysteine Increased the Expression of BDNF Following SAH-Induced Injury
To determine whether l-cysteine can affect production of neuroprotective factors, the BDNF concentration of the PFC was measured at 48 h after SAH. As shown in Figure 6A, the BDNF mRNA expression level was significantly lower at 48 h in the SAH group than that in the Sham group. l-Cysteine significantly increased the expression level of BDNF mRNA in the PFC 48 h post-SAH exposure (Figure 6A). Consistent with the changes in the mRNA, the SAH-induced decrease in the BDNF protein levels was also reversed by l-cysteine treatment, but the effect of l-cysteine was reversed by AOAA (Figure 6B).
Figure 6.

The effect of l-cysteine on brain-derived neurotrophic factor (BDNF) expression levels at the mRNA and protein levels. (A) The BDNF expression levels at the mRNA level in the prefrontal cortex was assessed by semi-quantitative reverse transcription polymerase chain reaction at 48 h after subarachnoid hemorrhage (SAH). Each value was normalized to β-actin. Bar graphs showing the quantification of the BDNF mRNA levels were generated by Image-Pro Plus 6.0 (n = 4). (B) The BDNF protein expression level was analyzed by Western blotting at 48 h after SAH, and β-actin was used to evaluate protein loading. The bar graphs showing the quantification of the protein levels of BDNF were generated by Image-Pro Plus 6.0 (n = 3). The values represent the mean ± SD. *p < 0.05, ***p < 0.001 SAH vs Sham, #p < 0.05, ###p < 0.001 SAH + l-Cys vs SAH, +p < 0.05, ++p < 0.01 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
Administration of l-Cysteine Improves CREB phosphorylation In Vivo
Phosphorylated CREB regulates the transcription of several genes that code for molecules involved in neuronal plasticity, including BDNF, tyrosine hydroxylase, and neural cell adhesion molecule; these molecules are associated with the stress response. Thus, we examined the CREB phosphorylation levels after SAH and l-cysteine treatment. As shown in Figure 7, phosphorylated CREB expression significantly decreased at 48 h after SAH compared to that in the Sham group. Treatment with l-cysteine significantly increased the expression level of phosphorylated CREB in the PFC at 48 h post-SAH exposure. Additionally, the effect of l-cysteine on the SAH-induced CREB phosphorylation levels was reversed by AOAA.
Figure 7.
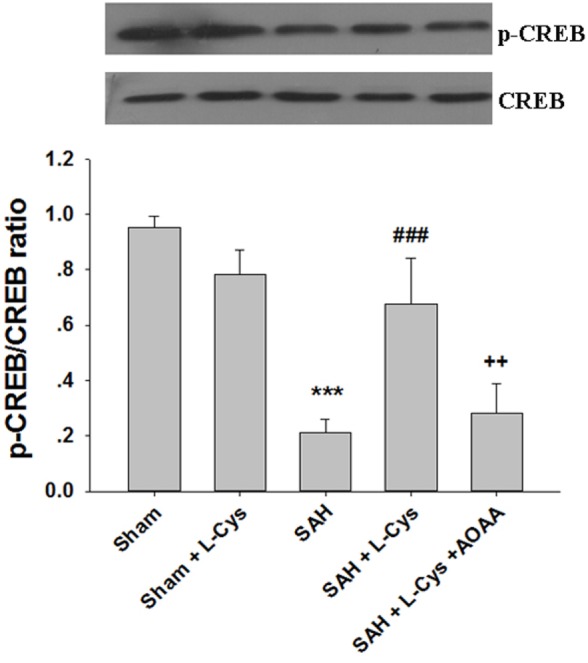
Effects of l-cysteine on CREB phosphorylation after subarachnoid hemorrhage (SAH). At 48 h after SAH, whole prefrontal cortex extracts were subjected to Western blot analysis using antibodies against phospho-cAMP response element binding protein (p-CREB) and CREB. Bar graphs showing the quantification of the expression levels of p-CREB/CREB were generated by Image-Pro Plus 6.0 (n = 3). The values represent the means ± SD. ***p < 0.001 SAH vs Sham, ###p < 0.001 SAH + l-Cys vs SAH, ++p < 0.001 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
Effects of l-Cysteine on Synaptic Structure and Expression of Synaptophysin and PSD95 after SAH
Neuronal damage, including synapse collapse, occurs after SAH; therefore, we investigated the morphological changes in the synapses of the PFC using TEM. Compared with the Sham and Sham + l-cysteine groups (Figure 8), the SAH group exhibited vague structural changes in the synapses, which included swollen borders and dark staining that indicated degeneration. In addition, the number of normal synapses decreased in the SAH group. l-cysteine treatment dramatically ameliorated the synaptic damage and upregulated the number of synapses in the SAH group, whereas the effects of l-cysteine were abrogated by AOAA.
Figure 8.

Effects of l-cysteine treatment on synaptic changes in the prefrontal cortex (PFC). Representative transmission electron microscopy images of the PFC from each group. The arrows indicate regular synaptic structures. The stars denote collapsed synapses. Scale bar = 500 nm.
Next, we measured the level of the presynaptic marker synaptophysin and the postsynaptic marker PSD95. Synaptophysin was significantly decreased at both the mRNA and protein levels in the SAH group. Treatment with l-cysteine significantly increased the expression of synaptophysin in the PFC at 48 h post-SAH exposure. However, compared with the Sham group, the mRNA and protein levels of PSD95 were significantly increased at 48 h in the SAH group (Figures 9A,B). l-cysteine significantly decreased the expression of PSD95 in the PFC at 48 h post-SAH exposure. AOAA reversed the effects of l-cysteine on the synaptophysin and PSD95 expression levels.
Figure 9.

Effects of l-cysteine treatment on synaptophysin and PSD95 expression levels in the prefrontal cortex (PFC). (A) The mRNA levels of synaptophysin and PSD95 were measured by semi-quantitative reverse transcription polymerase chain reaction. Each value was normalized to β-actin. Bar graphs showing the quantification of the mRNA levels of synaptophysin and PSD95 were generated by Image-Pro Plus 6.0 (n = 3). (B) At 48 h after subarachnoid hemorrhage (SAH), whole PFC extracts were subjected to Western blot analysis using antibodies against synaptophysin and PSD95. The bar graphs showing the quantification of the mRNA levels of synaptophysin and PSD95 were generated by Image-Pro Plus 6.0 (n = 3). The values represent the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 SAH vs Sham, #p < 0.05, ##p < 0.01 SAH + l-Cys vs SAH, +p < 0.05 SAH + l-Cys + AOAA vs SAH + l-Cys. AOAA, amino-oxyacetic acid.
Discussion
In the current research, we demonstrated that l-cysteine could enhance H2S levels in the brain via an interaction with CBS; additionally, l-cysteine played a neuroprotective role in SAH by ameliorating cerebral edema and neuronal apoptosis. Moreover, l-cysteine sensitized the CREB–BDNF pathway and upregulated the expression of proteins related to synaptic plasticity. The positive effects initiated by l-cysteine were significantly abrogated when the CBS antagonist AOAA was administered.
Cystathionine-β-synthase is a pyridoxal-5′-phosphate-dependent enzyme that catalyzes β-replacement in which the β-position of the substrate is substituted by a nucleophile YH (25) and transforms substrates to 2-mercaptoethanol and H2S (26, 27). A close association between mutations in several regions of the human CBS gene and mental disorders and vascular diseases has been identified (28). In the rat brain, CBS was more highly expressed than cystathionine γ lyase and was mainly responsible for H2S generation (7). l-cysteine, an amino acid containing the electronegative substituent -SH, is the preferred substrate for H2S, which accounts for 70% of H2S production (29). Li et al. demonstrated that l-cysteine administration upregulated the production of H2S, whereas AOAA markedly attenuated the effects of l-cysteine in a dose-dependent manner (30). In Kmamt’s study, administration of NaHS, a, H2S donor, reversed the decreased expression of CBS, an H2S-metabolizing enzyme (31). In our study, l-cysteine was first administered to the animals via intracerebroventricular injection at 30 min post-SAH and increased CBS activity and expression in the PFC, which is consistent with the H2S production levels. We suggest that l-cysteine provokes a potential feedback loop to increase CBS activity in response to SAH and increase H2S production to exert neuroprotective effects.
Cell apoptosis is a major characteristic of EBI, and the mitochondrial pathway may also be involved (32). Additionally, exogenous H2S was recently shown to protect against global and focal cerebral ischemia/reperfusion injury (33, 34). However, whether H2S can preserve neurons through l-cysteine metabolism after SAH remains unknown. In our study, we observed numerous TUNEL-positive cells in the CNS of the SAH group, which is consistent with Chen’s report (35). l-cysteine administration can reduce the number of apoptotic cells induced by SAH. Meanwhile, we further investigated the Bax/Bcl-2 ratio. The Bcl-2 family member Bax was markedly upregulated after SAH, which resulted in the release of cytochrome c to the cytosol (32). We found that the SAH-induced upregulation of the Bax/Bcl-2 ratio could be reversed by l-cysteine administration. Moreover, we analyzed the activation of caspase-3; cytochrome c release triggers the cleavage of the caspase-3 protein, which results in DNA fragmentation and apoptosis (36). Inhibition of cleaved caspase-3 could reduce neuronal loss in SAH models (37). Our data revealed that treatment with l-cysteine prevented the SAH-induced increase in cleaved caspase-3 in the PFC. Our findings suggest that l-cysteine could protect neurons from apoptosis after SAH. However, when AOAA was administered with l-cysteine, all the beneficial effects on apoptosis were abolished. Thus, we hypothesize that the neuroprotective effects of l-cysteine on SAH may be due to an increase in endogenous H2S.
Brain-derived neurotrophic factor is a growth factor and supports neuronal survival, plasticity and neurogenesis (38, 39). Moreover, BDNF is involved in the pathophysiology of SAH. For example, clinical evidence has shown that a BDNF polymorphism is associated with poor patient recovery from SAH (40, 41). Animal experiments have demonstrated that exogenous BDNF infusion or upregulation of its expression improves neurobehavioral outcomes after SAH (42, 43). Regarding the underlying mechanisms of the neuroprotective effect of BDNF on neuronal apoptosis, some studies have shown that these effects are dependent on the activation of the PI3K/Akt and/or ERK signaling cascade, which subsequently activates CREB phosphorylation and promotes neuronal survival (44, 45). Previous studies have reported that H2S promotes BDNF expression, and blocking the BDNF-TrkB pathway reverses the H2S-mediated neuroprotection against apoptosis and oxidative stress in neurons (46, 47). Moreover, H2S can activate the CREB signaling pathway and prevent ischemia-reperfusion injury in the brain (48). In our study, the expression levels of p-CREB and BDNF increased after l-cysteine administration, which suggests that H2S could activate the CREB signaling pathway and increase the expression of its downstream pro-survival gene, BDNF. Importantly, these findings raise the possibility that H2S exerts anti-apoptotic effects via upregulation of p-CREB and BDNF in the PFC. Considering that AOAA blocks CBS, an l-cysteine catalyst, and reduces H2S production, we hypothesize that l-cysteine simulates CREB–BDNF expression in the CNS via inducing H2S during SAH.
Recently, Shen et al. reported that neuronal damage, including synapse collapse, occurs after SAH (49). Synapses are critical structural units for transmitting information in the brain. Accumulating evidence has demonstrated that changes in synapse density are highly correlated with cognitive status (50, 51). Synaptophysin and PSD95 are reliable markers to indirectly evaluate the integrality and function of the synapses (52, 53). Synaptophysin is a marker of the presynaptic nerve terminal density, which is essential for vesicle fusion and the release of neurotransmitter (54). The decrease in synaptophysin in CNS diseases indicates a reduction in synaptic plasticity (55). PSD95 is a scaffold protein that anchors and organizes NMDA receptors and controls the number and size of dendritic spines (56). We demonstrated that injection of l-cysteine after SAH significantly attenuates synaptic damage by ameliorating structural degeneration and upregulating the number of healthy synapses. At the mRNA and protein levels, a decrease in synaptophysin occurs in SAH, and synaptophysin levels are improved by l-cysteine administration, which indicates the potential role of H2S in stimulating changes in synaptophysin levels. To our surprise, we observed that PSD95 was upregulated after SAH; in contrast, previous studies have shown that PSD95 expression is decreased in diverse brain diseases (57, 58). We additionally showed that l-cysteine could suppress PSD95 expression. We postulate that interactions between PSD95 and the NMDA receptor are increased after SAH, which leads to neuronal injury. However, AOAA did not block the effect of l-cysteine on PSD95 expression in SAH. The improvement induced by l-cysteine may not be achieved through the regulation of PSD95 by H2S as we expected.
There are several limitations to our study. First, we used AOAA to block CBS activity and l-cysteine function, but the CNS contains other enzymes that can produce H2S, such as 3-mercaptopyruvate sulfurtransferase, which need to be investigated in future studies (6). Second, although apoptosis is a major contributor to EBI, other factors, including cerebral vasospasm, inflammation and oxidative stress, may also be responsible for the development of EBI in SAH (2, 59). Determining whether l-cysteine has a beneficial effect on these factors and exploring the potential underlying mechanism of its effects will require further study. Third, how l-cysteine regulates the expression of synaptophysin and PSD95 and how these factors affect neurological function were not determined (31, 60). Finally, the route, timing and dosage of l-cysteine treatment need to be further elucidated.
In summary, l-cysteine treatment could alleviate the development of EBI induced by SAH through multiple mechanisms, including reducing cell apoptosis, upregulating BDNF–CREB expression, and improving synapse density, by activating the CBS/H2S system.
Ethics Statement
The International Guiding Principles for Animal Research, as stipulated by the CIOMS and adopted by the Laboratory Animal Center at Shandong University, were generally followed for all animal handling and care. Researchers handling animals were systematically trained following the Institutional Animal Care and Use Committee (IACUC) guidebook. Euthanasia for animal models was performed as instructed under “AVMA Guidelines for the Euthanasia of Animals: 2013 Edition.” All efforts were made to reduce the number of animals used and their suffering, in compliance with 3R principles. The protocol was approved by the Ethic Committee of Qilu Hospital.
Author Contributions
GL and ZW were involved in designing the study, interpreting the data and writing the manuscript; TL and LXW performed the majority of the experiments and contributed to the analysis of the data; QH, SL, XMB, and YKX were responsible for the animal model; TTZ and SSB were responsible for Western blotting; and XQG and SHW were responsible for preparing pathological sections. The authors have no conflicts of interest to declare.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by funding from the National Natural Science Foundation of China (No. 81571284, 81671213), the Fundamental Research Funds of Shandong University (2015JC008), and the Science and Technology Development Plan of Shandong Province (2013GSF11804).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fneur.2017.00176/full#supplementary-material.
Abbreviations
AOAA, amino-oxyacetic acid; BDNF, brain-derived neurotrophic factor; CBS, cystathionine-β-synthase; CNS, central nervous system; CREB, cAMP response element binding protein; p-CREB, phospho-cAMP response element binding protein; DAB, diaminobenzidine; DAPI, 4′,6-diamidino-2-phenylindole dihydrochloride; EBI, early brain injury; H2S, hydrogen sulfide; IHC, immunohistochemical staining; NNDPD, N,N-dimethyl-p-phenylenediamine sulfate; SAH, subarachnoid hemorrhage; TEM, transmission electron microscopy.
References
- 1.Bederson JB, Connolly ES, Jr, Batjer HH, Dacey RG, Dion JE, Diringer MN, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke (2009) 40:994–1025. 10.1161/STROKEAHA.108.191395 [DOI] [PubMed] [Google Scholar]
- 2.Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol (2012) 97:14–37. 10.1016/j.pneurobio.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids (2012) 42:231–46. 10.1007/s00726-011-0867-5 [DOI] [PubMed] [Google Scholar]
- 4.Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM, Jr, Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J (2004) 18:1246–8. 10.1096/fj.03-0971fje [DOI] [PubMed] [Google Scholar]
- 5.Kumar P, Maurya PK. l-Cysteine efflux in erythrocytes as a function of human age: correlation with reduced glutathione and total anti-oxidant potential. Rejuvenation Res (2013) 16:179–84. 10.1089/rej.2012.1394 [DOI] [PubMed] [Google Scholar]
- 6.Kimura H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem Int (2013) 63:492–7. 10.1016/j.neuint.2013.09.003 [DOI] [PubMed] [Google Scholar]
- 7.Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci (1996) 16:1066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, et al. Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br J Pharmacol (2013) 169:922–32. 10.1111/bph.12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev (2012) 92:791–896. 10.1152/physrev.00017.2011 [DOI] [PubMed] [Google Scholar]
- 10.Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J (2004) 18:1165–7. 10.1096/fj.04-1815fje [DOI] [PubMed] [Google Scholar]
- 11.Hu LF, Wong PT, Moore PK, Bian JS. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J Neurochem (2007) 100:1121–8. 10.1111/j.1471-4159.2006.04283.x [DOI] [PubMed] [Google Scholar]
- 12.Wang JF, Li Y, Song JN, Pang HG. Role of hydrogen sulfide in secondary neuronal injury. Neurochem Int (2014) 64:37–47. 10.1016/j.neuint.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 13.Zhang Q, Yuan L, Liu D, Wang J, Wang S, Zhang Q, et al. Hydrogen sulfide attenuates hypoxia-induced neurotoxicity through inhibiting microglial activation. Pharmacol Res (2014) 84:32–44. 10.1016/j.phrs.2014.04.009 [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Zhan J, Wang X, Gu J, Xie K, Zhang Q, et al. Sodium hydrosulfide prevents hypoxia-induced behavioral impairment in neonatal mice. Brain Res (2013) 1538:126–34. 10.1016/j.brainres.2013.09.043 [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Liu DX, Wang FW, Zhang Q, Du ZX, Zhan JM, et al. l-Cysteine promotes the proliferation and differentiation of neural stem cells via the CBS/H(2)S pathway. Neuroscience (2013) 237:106–17. 10.1016/j.neuroscience.2012.12.057 [DOI] [PubMed] [Google Scholar]
- 16.Roy A, Khan AH, Islam MT, Prieto MC, Majid DS. Interdependency of cystathione gamma-lyase and cystathione beta-synthase in hydrogen sulfide-induced blood pressure regulation in rats. Am J Hypertens (2012) 25:74–81. 10.1038/ajh.2011.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Y, Duan X, Li H, Dang B, Yin J, Wang Y, et al. Hydrogen sulfide ameliorates early brain injury following subarachnoid hemorrhage in rats. Mol Neurobiol (2016) 53:3646–57. 10.1007/s12035-015-9304-1 [DOI] [PubMed] [Google Scholar]
- 18.Li T, Liu H, Xue H, Zhang J, Han X, Yan S, et al. Neuroprotective effects of hydrogen sulfide against early brain injury and secondary cognitive deficits following subarachnoid hemorrhage. Brain Pathol (2016) 27:51–63. 10.1111/bpa.12361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke (1995) 26:627–34; discussion 635. 10.1161/01.STR.26.4.627 [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa Y, Suzuki H, Altay O, Zhang JH. Preservation of tropomyosin-related kinase B (TrkB) signaling by sodium orthovanadate attenuates early brain injury after subarachnoid hemorrhage in rats. Stroke (2011) 42:477–83. 10.1161/STROKEAHA.110.597344 [DOI] [PubMed] [Google Scholar]
- 21.Fujii M, Sherchan P, Krafft PR, Rolland WB, Soejima Y, Zhang JH. Cannabinoid type 2 receptor stimulation attenuates brain edema by reducing cerebral leukocyte infiltration following subarachnoid hemorrhage in rats. J Neurol Sci (2014) 342:101–6. 10.1016/j.jns.2014.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura H. Signaling of hydrogen sulfide and polysulfides. Antioxid Redox Signal (2015) 22:347–9. 10.1089/ars.2014.5869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H. Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J (2005) 19:1854–6. 10.1096/fj.05-3724fje [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto R, Otsuguro K, Yamaguchi S, Ito S. Neuronal regulation of expression of hydrogen sulfide-producing enzyme cystathionine beta-synthase in rat spinal cord astrocytes. Neurosci Res (2015) 97:52–9. 10.1016/j.neures.2015.03.003 [DOI] [PubMed] [Google Scholar]
- 25.Miles EW, Kraus JP. Cystathionine beta-synthase: structure, function, regulation, and location of homocystinuria-causing mutations. J Biol Chem (2004) 279:29871–4. 10.1074/jbc.R400005200 [DOI] [PubMed] [Google Scholar]
- 26.Braunstein AE, Goryachenkova E. The b-replacement-specific pyridoxal-P-dependent lyases. Adv Enzymol Relat Areas Mol Biol (1984) 56:1–89. [DOI] [PubMed] [Google Scholar]
- 27.Hill R, Spenser I. Pyridoxal Phosphate: Chemical, Biochemical and Medical Aspects. Dr. Dolphin DNY-L. New York: Wiley and Sons; (1986). p. 131–222. [Google Scholar]
- 28.Welsh M, Ramsey B, Accurso F, Cutting G, Scriver C, Beaudet A, et al. The Metabolic and Molecular Basis of Inherited Disease. 8th ed McGraw-Hill Professional; (2001). p. 5085–108. [Google Scholar]
- 29.McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids (2012) 42:199–205. 10.1007/s00726-011-0864-8 [DOI] [PubMed] [Google Scholar]
- 30.Xiao A, Li J, Liu T, Liu Z, Wei C, Xu X, et al. l-Cysteine enhances nutrient absorption via a cystathionine-beta-synthase-derived H2 S pathway in rodent jejunum. Clin Exp Pharmacol Physiol (2016) 43:562–8. 10.1111/1440-1681.12562 [DOI] [PubMed] [Google Scholar]
- 31.Kamat PK, Kyles P, Kalani A, Tyagi N. Hydrogen sulfide ameliorates homocysteine-induced Alzheimer’s disease-like pathology, blood-brain barrier disruption, and synaptic disorder. Mol Neurobiol (2016) 53:2451–67. 10.1007/s12035-015-9212-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cahill J, Calvert JW, Marcantonio S, Zhang JH. p53 may play an orchestrating role in apoptotic cell death after experimental subarachnoid hemorrhage. Neurosurgery (2007) 60:531–45; discussion 545. 10.1227/01.NEU.0000249287.99878.9B [DOI] [PubMed] [Google Scholar]
- 33.Gheibi S, Aboutaleb N, Khaksari M, Kalalian-Moghaddam H, Vakili A, Asadi Y, et al. Hydrogen sulfide protects the brain against ischemic reperfusion injury in a transient model of focal cerebral ischemia. J Mol Neurosci (2014) 54:264–70. 10.1007/s12031-014-0284-9 [DOI] [PubMed] [Google Scholar]
- 34.Jiang Z, Li C, Manuel ML, Yuan S, Kevil CG, McCarter KD, et al. Role of hydrogen sulfide in early blood-brain barrier disruption following transient focal cerebral ischemia. PLoS One (2015) 10:e0117982. 10.1371/journal.pone.0117982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Wang L, Wu C, Hu Q, Gu C, Yan F, et al. Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J Pineal Res (2014) 56:12–9. 10.1111/jpi.12086 [DOI] [PubMed] [Google Scholar]
- 36.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol (1999) 144:281–92. 10.1083/jcb.144.2.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke (2004) 35:2412–7. 10.1161/01.STR.0000141162.29864.e9 [DOI] [PubMed] [Google Scholar]
- 38.Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem (2003) 10:86–98. 10.1101/lm.54603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem (2008) 89:312–23. 10.1016/j.nlm.2007.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siironen J, Juvela S, Kanarek K, Vilkki J, Hernesniemi J, Lappalainen J. The Met allele of the BDNF Val66Met polymorphism predicts poor outcome among survivors of aneurysmal subarachnoid hemorrhage. Stroke (2007) 38:2858–60. 10.1161/STROKEAHA.107.485441 [DOI] [PubMed] [Google Scholar]
- 41.Vilkki J, Lappalainen J, Juvela S, Kanarek K, Hernesniemi JA, Siironen J. Relationship of the Met allele of the brain-derived neurotrophic factor Val66Met polymorphism to memory after aneurysmal subarachnoid hemorrhage. Neurosurgery (2008) 63:198–203; discussion 203. 10.1227/01.NEU.0000320382.21577.8E [DOI] [PubMed] [Google Scholar]
- 42.Tang J, Hu Q, Chen Y, Liu F, Zheng Y, Tang J, et al. Neuroprotective role of an N-acetyl serotonin derivative via activation of tropomyosin-related kinase receptor B after subarachnoid hemorrhage in a rat model. Neurobiol Dis (2015) 78:126–33. 10.1016/j.nbd.2015.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang ZY, Yang MF, Wang T, Li DW, Liu YL, Zhang JH, et al. Cysteamine alleviates early brain injury via reducing oxidative stress and apoptosis in a rat experimental subarachnoid hemorrhage model. Cell Mol Neurobiol (2015) 35:543–53. 10.1007/s10571-014-0150-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia Y, Wang CZ, Liu J, Anastasio NC, Johnson KM. Brain-derived neurotrophic factor prevents phencyclidine-induced apoptosis in developing brain by parallel activation of both the ERK and PI-3K/Akt pathways. Neuropharmacology (2010) 58:330–6. 10.1016/j.neuropharm.2009.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papadia S, Hardingham GE. The dichotomy of NMDA receptor signaling. Neuroscientist (2007) 13:572–9. 10.1177/1073858407305833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang JM, Zhou CF, Gao SL, Tian Y, Wang CY, Wang L, et al. BDNF-TrkB pathway mediates neuroprotection of hydrogen sulfide against formaldehyde-induced toxicity to PC12 cells. PLoS One (2014) 10:e0119478. 10.1371/journal.pone.0119478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei HJ, Xu JH, Li MH, Tang JP, Zou W, Zhang P, et al. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol Sin (2014) 35:707–15. 10.1038/aps.2013.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai HB, Ji X, Zhu SH, Hu YM, Zhang LD, Miao XL, et al. Hydrogen sulphide and mild hypothermia activate the CREB signaling pathway and prevent ischemia-reperfusion injury. BMC Anesthesiol (2015) 15:119. 10.1186/s12871-015-0097-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen H, Chen Z, Wang Y, Gao A, Li H, Cui Y, et al. Role of neurexin-1beta and neuroligin-1 in cognitive dysfunction after subarachnoid hemorrhage in rats. Stroke (2015) 46:2607–15. 10.1161/STROKEAHA.115.009729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WG, Lou W, et al. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci (2010) 30:7507–15. 10.1523/JNEUROSCI.6410-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen KS, Masliah E, Mallory M, Gage FH. Synaptic loss in cognitively impaired aged rats is ameliorated by chronic human nerve growth factor infusion. Neuroscience (1995) 68:19–27. 10.1016/0306-4522(95)00099-5 [DOI] [PubMed] [Google Scholar]
- 52.Glantz LA, Gilmore JH, Hamer RM, Lieberman JA, Jarskog LF. Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience (2007) 149:582–91. 10.1016/j.neuroscience.2007.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Luo J, Zhang M, Yao W, Ma X, Yu SY. Effects of curcumin on chronic, unpredictable, mild, stress-induced depressive-like behaviour and structural plasticity in the lateral amygdala of rats. Int J Neuropsychopharmacol (2014) 17:793–806. 10.1017/S1461145713001661 [DOI] [PubMed] [Google Scholar]
- 54.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science (1993) 259:780–5. 10.1126/science.8430330 [DOI] [PubMed] [Google Scholar]
- 55.Cunha GM, Canas PM, Oliveira CR, Cunha RA. Increased density and synapto-protective effect of adenosine A2A receptors upon sub-chronic restraint stress. Neuroscience (2006) 141:1775–81. 10.1016/j.neuroscience.2006.05.024 [DOI] [PubMed] [Google Scholar]
- 56.El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science (2000) 290:1364–8. 10.1126/science.290.5495.1364 [DOI] [PubMed] [Google Scholar]
- 57.Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ. Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis (2011) 24:547–57. 10.3233/JAD-2011-101782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, et al. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell (2012) 151:1581–94. 10.1016/j.cell.2012.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res (2006) 28:399–414. 10.1179/016164106X115008 [DOI] [PubMed] [Google Scholar]
- 60.Kamat PK, Kalani A, Givvimani S, Sathnur P, Tyagi S, Tyagi N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience (2013) 252:302–19. 10.1016/j.neuroscience.2013.07.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.