

ABSTRACT
PCR is effective in detecting bacterial DNA in samples, but it is unable to differentiate viable bacteria from inactivated cells or free DNA fragments. New PCR-based analytical strategies have been developed to address this limitation. Molecular viability testing (MVT) correlates bacterial viability with the ability to rapidly synthesize species-specific rRNA precursors (pre-rRNA) in response to brief nutritional stimulation. Previous studies demonstrated that MVT can assess bacterial inactivation by chlorine, serum, and low-temperature pasteurization. Here, we demonstrate that MVT can detect inactivation of Escherichia coli, Aeromonas hydrophila, and Enterococcus faecalis cells by UV irradiation. Some UV-inactivated E. coli cells transiently retained the ability to synthesize pre-rRNA postirradiation (generating false-positive MVT results), but this activity ceased within 1 h following UV exposure. Viable but transiently undetectable (by culture) E. coli cells were consistently detected by MVT. An alternative viability testing method, viability PCR (vPCR), correlates viability with cell envelope integrity. This method did not distinguish viable bacteria from UV-inactivated bacteria under some conditions, indicating that the inactivated cells retained intact cell envelopes. MVT holds promise as a means to rapidly assess microbial inactivation by UV treatment.
IMPORTANCE UV irradiation is increasingly being used to disinfect water, food, and other materials for human use. Confirming the effectiveness of UV disinfection remains a challenging task. In particular, microbiological methods that rely on rapid detection of microbial DNA can yield misleading results, due to the detection of remnant DNA associated with dead microbial cells. This report describes a novel method that rapidly distinguishes living microbial cells from dead microbial cells after UV disinfection.
KEYWORDS: molecular viability testing (MVT), disinfection, viability PCR (vPCR), propidium monoazide (PMA), drinking water, viable but nonculturable (VBNC), ratiometric pre-rRNA analysis, UV irradiation, bacterial inactivation
INTRODUCTION
Assessing the viability of microbial cells (defined as the capacity to form progeny) in samples is critically important but challenging for microbiologists (1, 2). Microbiological cultures require viability but underestimate microbial diversity because only a fraction of species can be cultured (3). Furthermore, culturing can be time-consuming (1 to 30 days, depending on species). PCR is a fast, sensitive, and specific alternative to culturing methods. However, traditional PCR cannot distinguish viable cells from nonviable cells or from free nucleic acids in the samples.
Viability PCR (vPCR) is a PCR-based strategy that selectively detects viable microbial cells. In vPCR, a membrane-impermeant DNA-binding compound, propidium monoazide (PMA), selectively associates with free DNA and DNA in cells with compromised cell membranes. Upon photoactivation, PMA covalently binds DNA and prevents amplification by PCR (4). Viable cells with intact cell membranes exclude PMA and retain strong quantitative PCR (qPCR) signals. This method is very effective in improving the specificity of PCR-based detection of viable cells (5). Moreover, it is highly versatile and adaptable, because it can be applied to virtually any PCR target sequence. However, its performance varies with sample and disinfection conditions (1, 6–9). Since vPCR correlates viability with cell membrane integrity, inactivated cells that retain intact cell membranes can yield false-positive results (6, 9–12).
Assays for bacterial rRNA precursors (pre-rRNA) are useful alternatives to vPCR in some applications (13–15). Pre-rRNAs are intermediates in rRNA synthesis with leader and tail fragments that are enzymatically removed to yield mature rRNA. In growing or nutritionally stimulated bacteria, pre-rRNAs can account for ≥25% of total cellular rRNA (16), making them considerably easier to detect than mRNAs. Pre-rRNA pools are drained when growth slows and are rapidly replenished when viable cells (but not dead cells) are given fresh nutrients (17–19). Growth-related changes in pre-rRNA copy numbers far exceed those for mature rRNA (16, 17). Pre-rRNA sequences are hypervariable and species specific, such that pre-rRNA-targeted PCRs can detect pre-rRNA synthesis of individual bacterial species within complex samples (13, 14). Like other structural RNA sequences, however, they are generally well conserved within bacterial species (13–16, 20, 21).
Pre-rRNA synthesis in response to nutritional stimulation is exploited in a method termed molecular viability testing (MVT) (13–15). In MVT, a sample is split into two aliquots, one of which is nutritionally stimulated by the addition of bacteriological culture medium. If viable cells of a targeted species are present in the sample, then pre-rRNA (measured by reverse transcription [RT]-qPCR) increases in the stimulated aliquot, relative to the control (nonstimulated) aliquot. Because nonviable cells cannot catalyze this change, MVT selectively detects viable bacteria. Pre-rRNA stimulation is very rapid, requiring exposure to nutrients for 1 to 2 generation times or less (1 to 3 h for most species). All or nearly all bacteria synthesize pre-rRNA upon nutritional stimulation, allowing the successful application of MVT to multiple diverse species (13–15). MVT is also more sensitive than standard (static) qPCR assays for DNA (14). Sensitivity is enhanced both by the dynamic nature of the method and by the high copy number of pre-rRNA in stimulated bacteria.
Previous reports demonstrated that MVT was effective in differentiating viable cells of diverse bacterial species from cells that had been inactivated by chlorine (15), serum exposure (13), or low-temperature pasteurization (14). The current study asked whether MVT could detect bacterial inactivation by UV irradiation, an increasingly significant method of disinfecting drinking water and other materials (22–24). UV radiation primarily damages nucleic acids directly by pyrimidine dimerization, thereby inactivating bacteria while leaving cell envelopes intact (23). As a result, vPCR can be confounded by UV inactivation (1, 6, 10). Therefore, we tested the hypothesis that MVT, which measures biosynthetic responses to an environmental stimulus rather than cell membrane integrity, could rapidly assess bacterial inactivation under such conditions. The method was applied to Gram-negative and Gram-positive water quality indicator species (Escherichia coli and Enterococcus faecalis, respectively) and to a Gram-negative waterborne pathogen (Aeromonas hydrophila). This report also presents evidence that MVT can detect viable E. coli cells that are transiently undetectable by culture or in viable-but-nonculturable (VBNC) states (25, 26).
RESULTS
MVT detection of bacterial inactivation by UV irradiation.
Following strategies used previously to assess vPCR as an analytical tool for assessing UV inactivation (6), initial experiments used high-density cell suspensions to extend the range of fold inactivation and thereby maximize the resolving power. E. coli and A. hydrophila cells were exposed to UV radiation for periods ranging from 3 s to 5 min. At every time point examined, MVT, along with vPCR as a comparative method, was conducted on samples; standard microbiological plating (colony counting) served as a control method (Fig. 1). DNA was amplified for vPCR using the same primer set as used to amplify pre-RNA in MVT. The functionality of vPCR in our hands was confirmed by applying it to heat-killed cells (data not shown).
FIG 1.
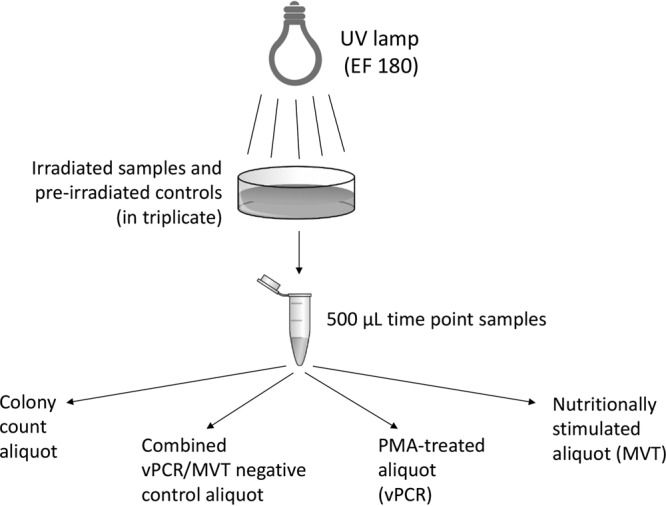
Flow diagram of the experimental design. Experiments comparing vPCR and MVT readouts for UV-irradiated bacteria are depicted.
UV intensity (irradiance) at 254 nm was measured under the experimental conditions (at the top center of the cell suspension) with a calibrated UVP UVX radiometer and an UVX-25 probe. The intensity ranged from 1,296 μW/cm2 (3 s on) to 1,870 μW/cm2 (2 to 5 min on). UV fluence under these conditions ranged from 4.1 mJ/cm2 (3 s) to 539.3 mJ/cm2 (5 min). Little variation (<10%) in irradiance or fluence over four separate measurements was observed over 4 weeks. After stabilization, the intensity was also measured at 18 points spanning the space occupied by a cell suspension (9 points on the top and 9 points on the bottom). Intensities at the bottom were 9.4% to 12.4% lower than those at the top, while lateral variation within each plane was <2%.
Under our experimental conditions, cells of both species were completely inactivated within 1 min of UV exposure, as measured by CFU plating. Plating, MVT, and vPCR results for both species are summarized in Fig. 2. In one A. hydrophila experiment (Fig. 2A), MVT results agreed with culture results at every exposure time point. As in previous studies (13, 14), samples were considered positive by MVT when all three replicates exhibited differences in quantification cycle (Cq) values between nonstimulated and stimulated aliquots (ΔCq values) of >1 Cq unit (dashed lines in Fig. 2). MVT results remained positive until after 10 s of UV exposure, despite a >4-log-unit reduction in viability. By the 30-s exposure time point, all viability detectable by plate counting was lost and MVT results were negative (ΔCq values of ≤1). Another experiment yielded similar results, with the exception of the 10-s time point, when a false-negative MVT result was obtained despite ∼0.1% viability (relative to nonirradiated cells) by culture (Fig. 2B). For E. coli, MVT ΔCq values remained positive (ΔCq values of >1) until the cells had been irradiated for 5 min, despite plating results that indicated complete inactivation between the 30-s and 2-min time points (Fig. 2C). A repeat time course experiment with E. coli (Fig. 2D) yielded greater concordance between MVT and plating results, with ΔCq values decreasing below 1 in some replicates starting at 30 s of irradiation. Viability PCR was unable to distinguish viable from inactivated cells for either organism (Fig. 2A to D).
FIG 2.

MVT and vPCR of UV-treated A. hydrophila and E. coli. Triplicate cell suspensions in ATW (A. hydrophila) (A and B) or PBS (E. coli) (C and D) at 1 × 108 CFU/ml were separately exposed to a time course of UV irradiation. Equal portions of each time point sample were subjected to MVT (white bars, left axes), vPCR (gray bars, left axes), and plating (triangles, right axes). Culture results (triangles) are displayed as percent viability relative to unexposed cells (time zero). Black triangles indicate no detected colonies (below the limit of detection). MVT (RT-qPCR) and vPCR (qPCR) results are both expressed as ΔCq values (left axes). For MVT, positive ΔCq (unstimulated minus stimulated) values of >1 (dashed lines) indicate viable cells. For vPCR, elevated ΔCq (with PMA minus without PMA) values indicate inactivated cells with compromised membranes.
Short-lived pre-rRNA synthesis in inactivated cells.
To better understand the ability of the E. coli cells shown in Fig. 2C to synthesize pre-rRNA after UV inactivation, we asked whether UV-inactivated cells lose this ability over time postirradiation. E. coli cells were irradiated for 45 s (Fig. 3A) or 1 min (Fig. 3B), and time point samples were taken at intervals of up to 120 h post-UV exposure. These exposures resulted in complete inactivation of the bacteria. Time point samples were analyzed by MVT as well as vPCR. The ability to synthesize pre-rRNA upon nutritional stimulation was short-lived following irradiation, persisting for less than 1 h postexposure. Pre-UV ΔCq values differed between experiments, but the time courses of viability loss by MVT were similar. Samples tested immediately after irradiation (time zero) exhibited diminished but positive MVT signals (ΔCq values of >1), while time point samples taken ≥1 h postirradiation were negative. Viability PCR was unable to detect UV inactivation even 5 days postirradiation.
FIG 3.
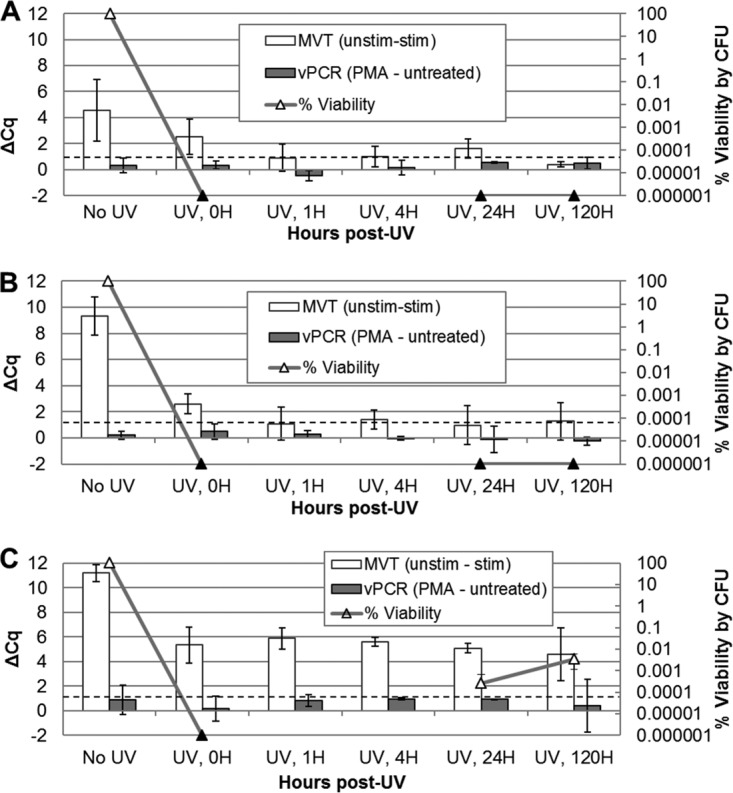
Postirradiation time course for E. coli. Triplicate E. coli suspensions in PBS, at an estimated density of 1 × 108 CFU/ml, were separately irradiated with UV light for 45 s (A and C) or 1 min (B). Cell suspensions were incubated at room temperature in PBS after UV exposure. Aliquots were taken for MVT (white bars), vPCR (gray bars), and plating (triangles) prior to irradiation (No UV) and at time points (0, 1, 4, 24, and 120 h) following irradiation. MVT (RT-qPCR) and vPCR (qPCR) data are both expressed as ΔCq values (left axes). For MVT, positive ΔCq (unstimulated minus stimulated) values of >1 (dashed lines) indicate viable cells. For vPCR, elevated ΔCq (with PMA minus without PMA) values indicate inactivated cells with compromised membranes. Black triangles indicate no detected colonies (no cultures were performed at the 1- and 4-hour time points). The results in panels A and B demonstrate a short-lived capacity to synthesize pre-rRNA after UV inactivation. The results in panel C suggest that transiently undetectable (by culture) cells were detected by MVT.
Transiently undetectable cells identified by MVT.
A third run of the experiments shown in Fig. 3A and B resulted in the generation of cells that were transiently undetectable by culture (Fig. 3C). Culturability of the cell suspensions fell below the detectable level immediately following irradiation, as in the first two experiments, but recovered by 24 h postirradiation (no CFU plating was done at the 1- and 4-hour time points). Colonies growing after 24 h were confirmed to be E. coli by qPCR (data not shown), using the qPCR primers and probe presented in Table 1. Uniquely in this experiment, MVT results remained positive throughout the time course. These findings suggested that these cell populations retained the ability to synthesize pre-rRNA while being unable to form colonies in detectable numbers at the intermediate time points.
TABLE 1.
Primers and probes
| Target organism | Forward PCR primer | Reverse PCR primer | Hydrolysis probea |
|---|---|---|---|
| A. hydrophila | ATTTGAATCAAGCAATCTGTG | GTTCAATCTGAGCCATGATC | 5′-6-FAM-TGGGCACTCACAGCATCGAGCATC-TAMRA-3′ |
| E. coli | TGCTCTTTAACAATTTATCAGACAATC | GACATTACTCACCCGTCC | 5′-6-FAM-TGGCTCAGA/ZEN/TTGAACGCTGGCGG-3IABkFQ-3′ |
| E. faecalis | AGCAAACAAATTGAGCTTAACA | GGAGGAAAGAAGCGTTCG | 5′-6-FAM-TTTGATCCTGGCTCAGGACGAACG-BHQ1a-3′ |
6-FAM, 6-carboxyfluorescein; TAMRA, carboxytetramethylrhodamine; 3IABkFQ, 3′ Iowa Black fluorescent quencher; BHQ, black hole quencher; ZEN, internal fluorescent quencher (Integrated DNA Technologies).
In order to better understand the phenomenon observed in Fig. 3C, additional UV irradiations of E. coli were performed in attempts to generate similar populations of transiently nonculturable viable cells. Cell suspensions were irradiated nine times in triplicate under the conditions used for Fig. 3 and were plated for detection of culturable cells, if any. Time point samples were taken and MVT analyses were conducted retrospectively in experiments in which transient nonculturability was observed. Transiently nonculturable (within our limits of detection) populations were observed in two of the nine experiments (Fig. 4). In those experiments, viable cells were undetectable by culture immediately following irradiation (viability, <10−7) but individual replicates regained culturability between 1 and 24 h after irradiation. It is not known whether this delayed ability to form colonies represented recovery of VBNC cells, proliferation of viable cells present in undetectable numbers, or some combination of the two. Colonies were confirmed to be E. coli by qPCR (data not shown), using the primers and probe presented in Table 1. These cell populations were positive by MVT at every time point. Analysis of individual replicates from the experiment shown in Fig. 4A showed that replicates with slowly recovering populations (nonculturable up to 4 h postirradiation) were also positive by MVT (data not shown), although natural sampling variations could account for the differences in CFU counts observed between replicates.
FIG 4.

Postirradiation time course for transiently undetectable (by culture) cells. Triplicate (A) and quadruplicate (B) E. coli suspensions in PBS at 1 × 108 CFU/ml were separately irradiated with UV light for 45 to 50 s. Cell suspensions were incubated at room temperature in PBS after UV exposure. Aliquots were taken for MVT (white bars) and plating (triangles) before (No UV) and at time points (0, 1, 4, 24, and 120 h) following irradiation. MVT (RT-qPCR) data are expressed as ΔCq values (left axes). For MVT, ΔCq (unstimulated minus stimulated) values of >1 (dashed lines) indicate viable cells. Black triangles indicate no detected colonies (below the limit of detection).
MVT and vPCR conducted with stirred, moderate-density suspensions of Gram-negative and Gram-positive bacteria.
The experiments described above used high-density cultures to extend the range of percent inactivation values and to maximize the resolving power. Experimental UV inactivations more commonly use lower cell densities in stirred cell suspensions. Therefore, we conducted additional experiments using 100-fold lower cell densities and actively stirred suspensions during irradiation. We also determined the efficacy of the MVT method when applied to E. coli and a Gram-positive water quality indicator organism, i.e., Enterococcus faecalis. Two triplicate experiments were conducted with each organism, using a 1-h postirradiation holding period, as in Fig. 3.
Under these conditions, E. coli cells were inactivated within 5 to 15 s and E. faecalis cells within 30 s, as measured by CFU plating. In the first E. coli experiment (Fig. 5A), MVT results matched CFU results at all time points except transiently at 15 s, when false-positive (relative to plating) MVT results were observed. In contrast to the higher-density suspensions used above and in a previous evaluation (6), this experiment showed evidence of a vPCR response, in that ΔCq values were elevated in UV-inactivated cells, relative to unexposed (time zero) cells (Fig. 5A). A repeat experiment with somewhat different time points yielded similar results for MVT, i.e., MVT positivity matched culture positivity at all time points except transiently at 5 to 15 s (Fig. 5B). In that experiment, however, the vPCR signal remained low throughout the time course.
FIG 5.

MVT and vPCR of UV-treated (moderate-density) stirred suspensions of E. coli and E. faecalis. Triplicate cell suspensions in PBS (E. coli) (A and B) or PBS with 0.1% Tween 20 (E. faecalis) (C and D), at 1 × 106 CFU/ml, were separately exposed to a time course of UV irradiation during active stirring. Equal portions of each time point sample were subjected to MVT (white bars, left axes), vPCR (gray bars, left axes), and plating (triangles, right axes). Culture results (triangles) are presented as percent viability relative to unexposed cells (time zero). Black triangles indicate no detected colonies (below the limit of detection). MVT (RT-qPCR) and vPCR (qPCR) results are both expressed as ΔCq values (left axes). For MVT, ΔCq (unstimulated minus stimulated) values of >1 indicate viable cells. For vPCR, elevated ΔCq (with PMA minus without PMA) values indicate inactivated cells with compromised membranes.
In the first E. faecalis experiment, MVT results were concordant with CFU results except transiently at 10 s, when false-negative MVT results were observed for one replicate (Fig. 5C). Similar results were seen with MVT in the second experiment (Fig. 5D). In both experiments with E. faecalis, there was inconsistent evidence of a vPCR response, suggesting that vPCR could potentially be used under these conditions with further optimization.
DISCUSSION
This study asked whether MVT can assess the viability of bacteria exposed to UV radiation. Cells inactivated by irradiation can remain intact and impermeable, confounding permeability-based viability tests such as vPCR and LIVE/DEAD staining (1, 6, 10). Given that pre-rRNA synthesis requires multiple factors in addition to cell envelope integrity, we hypothesized that MVT could distinguish viable bacteria from UV-inactivated bacteria. With the exception of transient false-negative or false-positive results at intermediate points in the time course experiments, the results supported this hypothesis. As shown previously (13, 15), the magnitude of pre-rRNA synthesis in viable cells is sufficient to enable their detection even when they are greatly outnumbered by inactivated cells. E. coli retained a short-lived ability to synthesize pre-rRNA after UV inactivation. However, this biosynthetic capacity was lost within 1 h postirradiation, a time frame short enough to allow the realistic application of MVT in most UV disinfection contexts.
In all UV irradiation experiments with high-density cell suspensions, vPCR failed to distinguish viable from inactivated bacteria, even 5 days post-UV exposure. This may reflect the persistence of intact cell envelopes in UV-inactivated bacteria. Prolonged cell envelope integrity after irradiation is consistent with other observations (27–29), although high doses of UV (particularly UV-C) radiation have been found to compromise cell membranes (30). In contrast to the high-cell-density experiments reported here and elsewhere (6), there was evidence of a vPCR response in experiments with moderate cell densities. Thus, vPCR may become applicable with further optimization. Successful application of vPCR generally requires optimization for specific bacterial targets and experimental conditions and the identification of distinct threshold values of ΔCq for each condition (1, 4). In contrast, although experience with MVT remains relatively limited, a uniform ΔCq threshold of 1.0 was used throughout this study and in all previous studies (1, 13–15). This default value is recommended as a starting point when applying the method to other organisms and conditions.
In three experiments, the viability of E. coli cells fell below the plate detection limit immediately following UV exposure and then started to recover within 24 h postexposure. The cell suspensions retained their ability to synthesize pre-rRNA (and thus to produce positive MVT results) during and after recovery from their nonculturable state. In a background of ∼107 inactivated cells, fewer than 10 viable cells (accounting for sampling variations) could not be expected to produce detectable amounts of pre-rRNA (15), suggesting that some cells that did not form colonies at early time points might have retained their abilities to synthesize macromolecules. All experiments with cell populations that lost and regained culturability (n = 10 individual disinfections) were positive by MVT, while all experiments with irreversibly inactivated cell populations (n = 6 disinfections) were negative by MVT (Fisher's exact test, P = 1.2 × 10−4). This suggests that the species-specific MVT method might serve as a viability test for transiently unculturable or VBNC cells. If this is confirmed in additional work, then MVT could help improve our understanding of the potentially important but poorly understood physiological state termed VBNC (26).
In summary, MVT can assess UV disinfection of bacteria, with the caveat that a short postirradiation period (≤1 h for E. coli) may be needed for some inactivated cells to become reliably MVT negative. Additional work is needed to assess whether MVT can detect inactivation by other means that have narrow immediate effects on microbial cells, such as solar disinfection and the inhibition of DNA, RNA, or protein synthesis by antibiotics (1, 14).
MATERIALS AND METHODS
Bacteriological cultures.
E. coli (ATCC 25922) and A. hydrophila (ATCC 7966) were grown to the early stationary phase in 25-ml overnight broth cultures, with shaking at 150 rpm, in 250-ml baffled glass flasks. E. faecalis (ATCC 29212) was grown overnight in 2-ml broth cultures, in 14-ml round-bottom tubes. E. coli was grown in Luria broth (LB) at 37°C, E. faecalis in brain heart infusion (BHI) broth at 37°C, and A. hydrophila in Trypticase soy broth (TSB) at 28°C. CFU plating was done on LB agar (E. coli), sheep's blood agar (E. faecalis), or Trypticase soy agar (TSA) (A. hydrophila), and cells were incubated overnight at 37°C (LB and blood agar) or 28°C (TSA). These media were chosen because they supported robust growth of each species. Limits of detection for CFU plating were determined by plating untreated cell suspensions.
UV inactivation.
Two UV inactivation protocols were used. In one of them, E. coli and A. hydrophila cells grown as described above were enumerated by spectrophotometry and resuspended at final densities of approximately 108 CFU/ml in phosphate-buffered saline (PBS) and autoclaved tap water (ATW), respectively. High cell densities were used to maximize the resolving power of disinfection measurements, enabling detection of up to 107-fold inactivation. UV disinfections were carried out as described previously (6). Fifteen-milliliter aliquots of cell suspensions, in triplicate, were transferred to disposable 60-mm petri dishes and irradiated with shortwave UV light (254 nm) using a Spectroline germicidal UV lamp (model EF 180) placed 20 cm above the uncovered suspensions. Samples (500 μl) were obtained at time points before, during, and after irradiation and were kept on ice in the dark until analysis.
The second UV inactivation protocol was similar but used lower densities (106 CFU/ml) of actively stirred E. coli and E. faecalis cells in PBS and ATW with 0.1% Tween 20, respectively. The Tween 20 supplement reduced cell loss during serial dilution.
Viability testing by MVT and vPCR.
In most experiments, MVT, vPCR, and colony counting were applied to all samples. MVT and vPCR both involve splitting a sample into two aliquots, one of which is kept as a nontreated control while the other is subjected to a specialized treatment (nutritional stimulation in MVT and exposure to PMA in vPCR). To enable direct comparisons between the two methods, the experimental design depicted in Fig. 1 was used. Each 500-μl time point sample was separated into 100-μl aliquots as follows: an aliquot for MVT nutritional stimulation, an aliquot for PMA treatment, a single MVT/PMA control aliquot (not nutritionally stimulated and not PMA treated), and a viability plating (colony counting) aliquot. MVT/PMA control aliquots were pelleted by centrifugation, supernatants were aspirated, and pellets were frozen at −80°C until DNA/RNA extraction. As described previously (6), PMA treatment aliquots were incubated for 5 min in the dark with 50 μM PMA (Biotium, Inc., Hayward, CA) and were photo-cross-linked for 2 min on ice with a GE FCW 650W halogen bulb (model 41672). PMA-treated cells were then pelleted, aspirated, and stored at −80°C as described above. MVT nutritional stimulation aliquots were treated by adding the 100-μl cell aliquots directly to 900 μl of prewarmed LB (E. coli, at 37°C), TSB (A. hydrophila, at 28°C), or BHI broth (E. faecalis, at 37°C) in 14-ml loose-capped round-bottom tubes and incubating the mixtures, with shaking at 150 rpm, for 30 min (E. coli and A. hydrophila) or 60 min (E. faecalis). Cells were then pelleted, aspirated, and stored at −80°C as described above. For colony counting, serial dilutions of the 100-μl aliquots were plated onto LB (E. coli), TSA (A. hydrophila), or blood agar (E. faecalis) and incubated overnight to quantify the percent viability, relative to the measured density of viable bacteria before treatment.
In initial experiments, samples were taken at time points before and during irradiation (Fig. 1). In subsequent experiments with E. coli, downstream effects of UV irradiation were assessed after treatment. For these experiments, triplicate 15-ml cell suspensions were disinfected for 45 or 60 s as described above. Following disinfection, cells were incubated in closed petri dishes at room temperature. Samples (500 μl) were obtained immediately (time zero) and 1, 4, 24, and 120 h after irradiation. Time point samples were tested by MVT, vPCR, and colony counting as described above. In later experiments exploring the detection of transiently undetectable (by culture) viable cells with MVT, irradiations were 45 to 50 s, time point samples were taken 1, 4, 24, 48, and 144 h postirradiation, and no vPCR was conducted.
MVT design and interpretation.
MVT measures a change in bacterial physiology, specifically increased pre-rRNA copy numbers in response to nutritional stimulation. The measurement is made by comparing quantification cycle (Cq) values generated by RT-qPCR analysis of samples before and after nutritional stimulation. When RT-qPCR is used to measure RNA, smaller Cq values indicate greater RNA abundance in the samples. As in previous studies (13–15), RT-qPCR primers straddled the junction between the 5′ terminus of the mature rRNA (16S) and the pre-rRNA leader region (external transcribed spacer [ETS]), such that intact pre-rRNA molecules were required as templates. Each MVT measurement was performed in triplicate, and samples were considered positive by MVT when all three stimulated replicates exhibited pre-rRNA values that were >1 Cq unit lower than those for all three control replicates (ΔCq values of >1.0). For each biological replicate (separate irradiation), ΔCq values were calculated from cDNA for MVT (unstimulated minus stimulated) and from DNA for vPCR (PMA-treated minus untreated). All results presented are means and standard deviations calculated for triplicate experiments.
Nucleic acid extraction and RT-qPCR.
DNA (for vPCR) and RNA (for MVT) were simultaneously extracted with an Epicentre MasterPure complete DNA and RNA purification kit, using the furnished protocols. Total nucleic acid (TNA) was eluted in 30 μl of Tris-EDTA (TE) buffer (10 mM Tris and 1 mM EDTA, pH 8.0), from which 10 μl was removed for DNA measurement. RNA was purified from the remaining 20 μl by DNase I treatment and reprecipitation, as directed in the included protocol, and then was resuspended in the same volume. In experiments either exploring the detection of UV-induced transiently undetectable (by culture) cell populations or using lower cell densities, DNA and RNA (for MVT) were extracted using a Qiagen AllPrep minikit, with on-column DNase purification of RNA, according to the manufacturer's furnished protocols.
Purified RNA was first converted to cDNA by reverse transcription (RT) with a Promega ImProm-II system, using 2 μl of the template and 1 μM 16S-specific oligonucleotide (5′-ATTCCGATTAACGCTTGCAC-3′). Purified genomic DNA and converted cDNA were measured in separate reactions by qPCR (using the same primer/probe set) utilizing SsoFast, SsoAdvanced, or iTaq probe mixtures (Bio-Rad) with ROX passive reference dye. In each 20-μl reaction mixture, there was 2 μl template, 0.4 μM (each) forward and reverse primers, and 0.2 μM hydrolysis probe (see Table 1 for oligonucleotide sequences). Reactions (including 5- to 7-log-unit standard curves) were run on an Applied Biosystems StepOnePlus system under the following reaction conditions: 95°C for 5 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Pre-rRNA from experiments exploring lower-density cell suspensions and transiently unculturable cells was amplified using the Thermo Verso 1-step RT-qPCR kit under the following reaction conditions: 50°C for 30 min, 95°C for 15 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Thresholds were set automatically by the StepOnePlus software (or manually adjusted when necessary) in the exponential range, and results were exported to a spreadsheet for analysis.
ACKNOWLEDGMENTS
Portions of this work were supported by grant 7423357 from the Washington Life Sciences Discovery Fund.
We are grateful to Julie Do for providing oligonucleotides.
REFERENCES
- 1.Cangelosi GA, Meschke JS. 2014. Dead or alive: molecular assessment of microbial viability. Appl Environ Microbiol 80:5884–5891. doi: 10.1128/AEM.01763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keer JT, Birch L. 2003. Molecular methods for the assessment of bacterial viability. J Microbiol Methods 53:175–183. doi: 10.1016/S0167-7012(03)00025-3. [DOI] [PubMed] [Google Scholar]
- 3.Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu Rev Microbiol 57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 4.Nocker A, Camper AK. 2009. Novel approaches toward preferential detection of viable cells using nucleic acid amplification techniques. FEMS Microbiol Lett 291:137–142. doi: 10.1111/j.1574-6968.2008.01429.x. [DOI] [PubMed] [Google Scholar]
- 5.Gensberger ET, Polt M, Konrad-Köszler M, Kinner P, Sessitsch A, Kostić T. 2014. Evaluation of quantitative PCR combined with PMA treatment for molecular assessment of microbial water quality. Water Res 67:367–376. doi: 10.1016/j.watres.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 6.Nocker A, Sossa KE, Camper AK. 2007. Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR. J Microbiol Methods 70:252–260. doi: 10.1016/j.mimet.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Gedalanga PB, Olson BH. 2009. Development of a quantitative PCR method to differentiate between viable and nonviable bacteria in environmental water samples. Appl Microbiol Biotechnol 82:587–596. doi: 10.1007/s00253-008-1846-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kralik P, Babak V, Dziedzinska R. 2014. Repeated cycles of chemical and physical disinfection and their influence on Mycobacterium avium subsp. paratuberculosis viability measured by propidium monoazide F57 quantitative real time PCR. Vet J 201:359–364. doi: 10.1016/j.tvjl.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 9.Schmidlin M, Alt M, Brodmann P, Bagutti C. 2010. Insufficient distinction between DNA from viable and nonviable Staphylococcus aureus cells in wipe-samples by use of propidium monoazide-PCR. Appl Biosaf 15:180–185. doi: 10.1177/153567601001500404. [DOI] [Google Scholar]
- 10.Banihashemi A, Dyke MI, Huck PM. 2012. Long-amplicon propidium monoazide-PCR enumeration assay to detect viable Campylobacter and Salmonella. J Appl Microbiol 113:863–873. doi: 10.1111/j.1365-2672.2012.05382.x. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Badoni M, Gill CO. 2011. Use of propidium monoazide and quantitative PCR for differentiation of viable Escherichia coli from E. coli killed by mild or pasteurizing heat treatments. Food Microbiol 28:1478–1482. doi: 10.1016/j.fm.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Løvdal T, Hovda MB, Björkblom B, Møller SG. 2011. Propidium monoazide combined with real-time quantitative PCR underestimates heat-killed Listeria innocua. J Microbiol Methods 85:164–169. doi: 10.1016/j.mimet.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 13.Weigel KM, Jones KL, Do JS, Melton Witt J, Chung J-H, Valcke C, Cangelosi GA. 2013. Molecular viability testing of bacterial pathogens from a complex human sample matrix. PLoS One 8:e54886. doi: 10.1371/journal.pone.0054886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Do JS, Weigel KM, Meschke JS, Cangelosi GA. 2014. Biosynthetic enhancement of the detection of bacteria by the polymerase chain reaction. PLoS One 9:e86433. doi: 10.1371/journal.pone.0086433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cangelosi GA, Weigel KM, Lefthand-Begay C, Meschke JS. 2010. Molecular detection of viable bacterial pathogens in water by ratiometric pre-rRNA analysis. Appl Environ Microbiol 76:960–962. doi: 10.1128/AEM.01810-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oerther DB, Pernthaler J, Schramm A, Amann R, Raskin L. 2000. Monitoring precursor 16S rRNAs of Acinetobacter spp. in activated sludge wastewater treatment systems. Appl Environ Microbiol 66:2154–2165. doi: 10.1128/AEM.66.5.2154-2165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cangelosi GA, Brabant WH. 1997. Depletion of pre-16S rRNA in starved Escherichia coli cells. J Bacteriol 179:4457–4463. doi: 10.1128/jb.179.14.4457-4463.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mackow ER, Chang FN. 1985. Processing of precursor ribosomal RNA and the presence of a modified ribosome assembly scheme in Escherichia coli relaxed strain. FEBS Lett 182:407–412. doi: 10.1016/0014-5793(85)80343-4. [DOI] [PubMed] [Google Scholar]
- 19.Srivastava AK, Schlessinger D. 1990. Mechanism and regulation of bacterial ribosomal RNA processing. Annu Rev Microbiol 44:105–129. doi: 10.1146/annurev.mi.44.100190.000541. [DOI] [PubMed] [Google Scholar]
- 20.Stroot PG, Oerther DB. 2003. Elevated precursor 16S rRNA levels suggest the presence of growth inhibitors in wastewater. Water Sci Technol 47:241–250. [PubMed] [Google Scholar]
- 21.Cangelosi GA, Hamlin AM, Marin R, Scholin CA. 1997. Detection of stable pre-rRNA in toxigenic Pseudo-nitzschia species. Appl Environ Microbiol 63:4859–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ngwenya N, Ncube EJ, Parsons J. 2013. Recent advances in drinking water disinfection: successes and challenges. Rev Environ Contam Toxicol 222:111–170. doi: 10.1007/978-1-4614-4717-7_4. [DOI] [PubMed] [Google Scholar]
- 23.Cutler TD, Zimmerman JJ. 2011. Ultraviolet irradiation and the mechanisms underlying its inactivation of infectious agents. Anim Heal Res Rev 12:15–23. doi: 10.1017/S1466252311000016. [DOI] [PubMed] [Google Scholar]
- 24.Nerandzic MM, Fisher CW, Donskey CJ. 2014. Sorting through the wealth of options: comparative evaluation of two ultraviolet disinfection systems. PLoS One 9:e107444. doi: 10.1371/journal.pone.0107444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byrd JJ, Xu HS, Colwell RR. 1991. Viable but nonculturable bacteria in drinking water. Appl Environ Microbiol 57:875–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Mendis N, Trigui H, Oliver JD, Faucher SP. 2014. The importance of the viable but non-culturable state in human bacterial pathogens. Front Microbiol 5:258. doi: 10.3389/fmicb.2014.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bosshard F, Berney M, Scheifele M, Weilenmann HU, Egli T. 2009. Solar disinfection (SODIS) and subsequent dark storage of Salmonella typhimurium and Shigella flexneri monitored by flow cytometry. Microbiology 155:1310–1317. doi: 10.1099/mic.0.024794-0. [DOI] [PubMed] [Google Scholar]
- 28.Bosshard F, Bucheli M, Meur Y, Egli T. 2010. The respiratory chain is the cell's Achilles' heel during UVA inactivation in Escherichia coli. Microbiology 156:2006–2015. doi: 10.1099/mic.0.038471-0. [DOI] [PubMed] [Google Scholar]
- 29.Cho M, Kim J, Kim JY, Yoon J, Kim JH. 2010. Mechanisms of Escherichia coli inactivation by several disinfectants. Water Res 44:3410–3418. doi: 10.1016/j.watres.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 30.Pigeot-Rémy S, Simonet F, Atlan D, Lazzaroni JC, Guillard C. 2012. Bactericidal efficiency and mode of action: a comparative study of photochemistry and photocatalysis. Water Res 46:3208–3218. doi: 10.1016/j.watres.2012.03.019. [DOI] [PubMed] [Google Scholar]