

ABSTRACT
After oral exposure, the early replication of certain prion strains upon stromal cell-derived follicular dendritic cells (FDC) in the Peyer's patches in the small intestine is essential for the efficient spread of disease to the brain. However, little is known of how prions are initially conveyed from the gut lumen to establish infection on FDC. Our previous data suggest that mononuclear phagocytes such as CD11c+ conventional dendritic cells play an important role in the initial propagation of prions from the gut lumen into Peyer's patches. However, whether these cells conveyed orally acquired prions toward FDC within Peyer's patches was not known. The chemokine CXCL13 is expressed by FDC and follicular stromal cells and modulates the homing of CXCR5-expressing cells toward the FDC-containing B cell follicles. Here, novel compound transgenic mice were created in which a CXCR5 deficiency was specifically restricted to CD11c+ cells. These mice were used to determine whether CXCR5-expressing conventional dendritic cells propagate prions toward FDC after oral exposure. Our data show that in the specific absence of CXCR5-expressing conventional dendritic cells the early accumulation of prions upon FDC in Peyer's patches and the spleen was impaired, and disease susceptibility significantly reduced. These data suggest that CXCR5-expressing conventional dendritic cells play an important role in the efficient propagation of orally administered prions toward FDC within Peyer's patches in order to establish host infection.
IMPORTANCE Many natural prion diseases are acquired by oral consumption of contaminated food or pasture. Once the prions reach the brain they cause extensive neurodegeneration, which ultimately leads to death. In order for the prions to efficiently spread from the gut to the brain, they first replicate upon follicular dendritic cells within intestinal Peyer's patches. How the prions are first delivered to follicular dendritic cells to establish infection was unknown. Understanding this process is important since treatments which prevent prions from infecting follicular dendritic cells can block their spread to the brain. We created mice in which mobile conventional dendritic cells were unable to migrate toward follicular dendritic cells. In these mice the early accumulation of prions on follicular dendritic cells was impaired and oral prion disease susceptibility was reduced. This suggests that prions exploit conventional dendritic cells to facilitate their initial delivery toward follicular dendritic cells to establish host infection.
KEYWORDS: prions, chemokines, intestine, dendritic cells, Peyer's patches, gastrointestinal infection, transmissible spongiform encephalopathy
INTRODUCTION
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a unique group of subacute neurodegenerative diseases that affect humans and some domesticated and free-ranging animal species. Prion diseases are characterized by the accumulation of aggregations of PrPSc, abnormally folded isoforms of the cellular prion protein (PrPC), in affected tissues. Prion infectivity copurifies with PrPSc and constitutes the major component of the infectious agent (1). Many prion diseases, including natural sheep scrapie, bovine spongiform encephalopathy, chronic wasting disease in cervid species, and kuru and variant Creutzfeldt-Jakob disease in humans, are acquired by peripheral exposure, such as by oral consumption of prion-contaminated food or pasture.
After ingestion many prion isolates initially accumulate and replicate first upon follicular dendritic cells (FDC) within the gut-associated lymphoid tissues (GALT) in the small intestine, such as the Peyer's patches, before they spread to the nervous system (termed neuroinvasion) (2–9). After their replication upon FDC studies in experimental rodents show that the prions subsequently infect neurons of both the sympathetic and parasympathetic nervous systems and spread along these to the central nervous system (CNS), where they ultimately cause neurodegeneration (10–12).
Mononuclear phagocytes arise from precursors in the bone marrow and comprise a heterogeneous population of monocytes, conventional dendritic cells (DC), and tissue macrophages. The different mononuclear phagocyte populations display a diverse range of roles during prion disease (13). For example, tissue macrophages appear to aid the sequestration and clearance of prions (14, 15), and microglia help protect the brain against prion-induced neuropathology (16). Conventional DC are a distinct lineage from the stromal derived FDC (17, 18) and are strategically located throughout the body to sample the local environment for pathogens and their antigens. After antigen uptake, these cells typically undergo a degree of maturation and migrate toward the draining lymphoid tissue to initiate a specific immune response. Some conventional DC populations can retain antigens in their native states and transfer them intact to naive B cells in order to initiate a specific antibody response (19). The migratory characteristics of conventional DC are exploited by some pathogens to facilitate their delivery to lymphoid tissues (20–23). Independent studies have shown that the early replication of prions upon FDC in the draining lymphoid tissue is impeded in the transient absence of conventional DC at the time of exposure (24–27). This suggests that prions may also exploit these cells to establish host infection after peripheral exposure, perhaps using them as “Trojan horses.” Whether conventional DC propagate prions toward FDC within the B cell follicles is not known. Treatments that prevent the uptake and propagation of prions to FDC can significantly delay disease pathogenesis and reduce susceptibility (24–29). A thorough understanding of the cellular mechanisms used by prions to establish infection upon FDC in the GALT may help to identify novel targets for prophylactic intervention.
Chemokines attract lymphocytes and leukocytes to lymphoid tissues and control their positioning within them. The chemokine CXCL13 is expressed by FDC and follicular stromal cells in the B-cell follicles of lymphoid tissues and mediates the homing of CXCR5-expressing cells toward them (30, 31). The migration of certain populations of conventional DC toward the FDC-containing B-cell follicles is also mediated by CXCL13-CXCR5 signaling (32–34). We therefore reasoned that if conventional DC were important for the efficient propagation of prions toward FDC in order to establish host infection, this activity would be impeded and disease susceptibility reduced in mice which specifically lacked CXCR5-expressing conventional DC. Since mice that lack Peyer's patches are refractory to oral prion infection (3, 5, 6, 35), CXCR5−/− mice and CXCL13−/− mice were unsuitable for use here because they also lack most Peyer's patches and certain lymph nodes (31, 36). The few Peyer's patches that do develop in these mice are much smaller, and their microarchitecture is grossly disturbed since the B cells are unable to form organized follicles (31, 36). Furthermore, as a consequence of the disturbed microarchitecture in the spleens of CXCR5−/− mice, prion neuroinvasion after intraperitoneal exposure occurs at a higher rate because their FDC are abnormally superimposed over sympathetic nerves (37). Therefore, in the present study a unique compound transgenic mouse model was created in which Cxcr5 was specifically ablated in CD11c+ conventional DC. These CXCR5ΔDC mice were then used to test the hypothesis that conventional DC play an important role in the efficient propagation of prions toward FDC within the B cell follicles of Peyer's patches after oral exposure.
RESULTS
Conditional deletion of CXCR5 in CD11c+ cells.
To enable conditional deletion of Cxcr5 in specific cell populations without affecting the CXCL13-CXCR5-dependent events required for normal lymphoid tissue development, mice with a conditional Cxcr5 allele were created by introducing loxP sites flanking exon 2. Expression of Cre recombinase under the control of the Itgax locus (which encodes CD11c) in CD11c-Cre mice (38) has been used in many studies to conditionally delete the expression of target genes in conventional DC (38–40). Homozygous CXCR5F/F mice were therefore crossbred to CD11c-Cre mice to generate mice specifically lacking CXCR5 expression in CD11c+ conventional DC, termed CXCR5ΔDC mice here.
CD11c+ and CD11c− cells were enriched from the spleens of CXCR5ΔDC mice. The CD11c− cells were further sorted based on their expression on CD11b, B220, and CD90.2 to broadly represent tissue macrophages, B cells and T cells, respectively. Reverse transcription-PCR (RT-PCR) analysis confirmed the expression of Cre only in mRNA derived from splenic CD11c+ cells (Fig. 1a). Further PCR analysis confirmed that in CXCR5ΔDC mice Cre recombinase-mediated recombination of the Cxcr5 allele had only occurred in the genomic DNA of CD11c+ cells and was absent in each of the CD11c− cell populations studied (Fig. 1b). These data show that in CXCR5ΔDC mice, Cre recombinase-mediated recombination of Cxcr5 is restricted to CD11c+ conventional DC.
FIG 1.
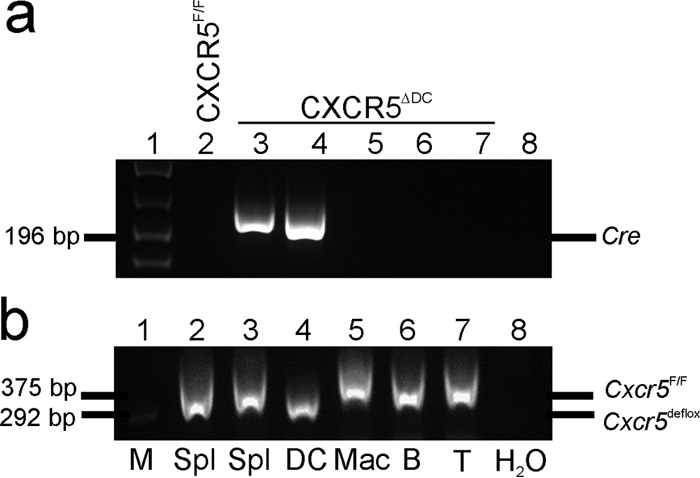
Conditional deletion of Cxcr5 in CD11c+ cells. CD11c+ and CD11c− cells were enriched from the spleens of CXCR5ΔDC mice. The CD11c− cells were further sorted based on their expression on CD11b, B220, and CD90.2 to broadly represent tissue macrophages, B cells, and T cells, respectively. (a) RT-PCR analysis confirmed the expression of Cre only in mRNA derived from splenic CD11c+ cells. (b) Analysis of genomic DNA confirmed that Cre recombinase-mediated recombination of the Cxcr5 allele had only occurred in CD11c+ cells, as demonstrated by presence of the lower Cxcr5de-flox band. M, DNA size markers; Spl, splenocytes; DC, CD11c+ conventional DC; Mac, CD11c− macrophages; B, B cells; T, T cells.
Conventional DC-specific CXCR5-deficiency does not affect secondary lymphoid tissue formation.
Next, groups of CXCR5ΔDC mice and CXCR5F/F (control) mice were injected intraperitoneally with Chicago Sky Blue 6B ink and analyzed 7 day later. Over the exposure period, the dye becomes concentrated within secondary lymphoid organs, enabling their macroscopic detection postmortem. The majority of the murine secondary lymphoid tissues develop consistently, whereas the lumbar aortic lymph nodes and lateral iliac lymph nodes are inconsistently present. As anticipated, the incidence and frequency of the secondary lymphoid tissues in CXCR5F/F mice was equivalent to those of nontransgenic wild-type mice (41). The secondary lymphoid tissues in CXCR5ΔDC mice were also present at similar incidences and frequencies to CXCR5F/F control mice (Table 1), unlike those in independently generated lines of CXCR5−/− mice and CXCL13−/− mice (31, 36) (Table 2). These data show that a conventional DC-restricted CXCR5 deficiency does not impact lymphoid tissue organogenesis.
TABLE 1.
Comparison of the formation and frequency of secondary lymphoid tissues in CXCR5ΔDC mice and CXCR5F/F (control) micea
| Secondary lymphoid tissueb | CXCR5F/F mice |
CXCR5ΔDC mice |
||
|---|---|---|---|---|
| Incidence | No. present (range) | Incidence | No. present (range) | |
| Spleen | 6/6 | 1 | 8/8 | 1 |
| Mandibular LN | 6/6 | 2 | 8/8 | 2 |
| Accessory mandibular LN | 6/6 | 2 | 8/8 | 2 |
| Superficial parotid LN | 6/6 | 2 | 8/8 | 2 |
| Cranial deep cervical LN | 6/6 | 2 | 8/8 | 2 |
| Proper axillary LN (brachial) | 6/6 | 5 (4–6) | 8/8 | 5 (4–6) |
| Accessory axillary LN | 6/6 | 5 (2–7) | 8/8 | 5 (3–6) |
| Subiliac LN (inguinal) | 6/6 | 2 | 8/8 | 2 |
| Sciatic LN | 6/6 | 2 | 8/8 | 2 |
| Popliteal LN | 6/6 | 2 | 8/8 | 2 |
| Cranial mediastinal LN | 6/6 | 4 | 7/8 | 4 |
| Tracheobronchial LN | 6/6 | 1 | 8/8 | 1 |
| Caudal mediastinal LN | 6/6 | 1 | 8/8 | 1 |
| Gastric LN | 6/6 | 1 | 7/8 | 1 (0–1) |
| Pancreaticoduodenal LN | 6/6 | 1 | 7/8 | 1 (0–1) |
| Jejunal LN (mesenteric) | 6/6 | 5 (4–6) | 8/8 | 5 (5–6) |
| Colic LN | 5/6 | 1 (0–2) | 7/8 | 1 (0–3) |
| Caudal mesenteric LN | 6/6 | 1 | 8/8 | 1 |
| Renal LN | 6/6 | 2 (1–3) | 8/8 | 2 |
| Lumbar aortic LN* | 4/6 | 2 (0–2) | 1/8 | 1 (0–1) |
| Lateral iliac LN* | 5/6 | 1 (0–1) | 2/8 | 1 (0–1) |
| Medial iliac LN | 6/6 | 2 (1–2) | 8/8 | 2 |
| External iliac LN | 2/6 | 1 (0–1) | 6/8 | 1 (0–1) |
| Peyer’s patches | 6/6 | 6 (5–9) | 8/8 | 6 (5–7) |
| Cecal patch (follicles) | 6/6 | 5 (3–8) | 7/8 | 2 (0–5) |
“Incidence” indicates the number of mice in which the tissue of interest was detectable/the number of mice tested; “no. present (range)” indicates the number of tissues (minimum to maximum) detectable in each mouse strain.
LN, lymph nodes. *, Inconsistently present in wild-type mice.
TABLE 2.
Comparison of secondary lymphoid tissue formation in CXCR5ΔDC mice, CXCR5F/F (control) mice, and independent lines of CXCL13−/− mice and CXCR5−/− micea
| Secondary lymphoid tissue | Incidence |
|||||
|---|---|---|---|---|---|---|
| Present study |
Ansel et al. (31)b |
|||||
| CXCR5F/F C57BL/6 cells | CXCR5ΔDC C57BL/6 cells | CXCL13+/± B6/129 cells | CXCL13−/− B6/129 cells | CXCR5−/− B6/129 cells | CXCR5−/− 129 cells | |
| Superficial parotid LN | 6/6 | 8/8 | 47/47 | 42/46 | 5/5 | 9/10 |
| Cranial deep cervical LN | 6/6 | 8/8 | 14/16 | 1/14 | 0/5 | 0/10 |
| Proper axillary (brachial) LN | 6/6 | 8/8 | 42/42 | 1/42 | 2/5 | 0/10 |
| Accessory axillary LN | 6/6 | 8/8 | 42/42 | 1/42 | 0/5 | 0/10 |
| Subiliac (inguinal) LN | 6/6 | 8/8 | 41/41 | 0/41 | 0/5 | 0/10 |
| Popliteal LN | 6/6 | 8/8 | 15/19 | 0/18 | 0/5 | 10/10 |
| Jejunal (mesenteric) LN | 6/6 | 8/8 | 50/50 | 50/50 | 5/5 | 10/10 |
| Renal LN | 6/6 | 8/8 | 12/15 | 0/12 | 0/5 | 3/9 |
| Medial iliac LN | 6/6 | 8/8 | 38/38 | 0/41 | 0/5 | 0/10 |
| Peyer’s patchesc | 6/6 (6, 5–9) | 8/8 (6, 5–7) | 31/31 (2 × 3–6 29 × > 6) | 16/37 (21 × 0, 11 × 1–2, 5 × 3–6) | 4/5 (1 × 0, 2 × 1–2, 1 × 3–6, 1 × > 6) | 9/9 (5 × 1–2, 3 × 3–6, 1 × > 6) |
The mouse strain background is specified in each column subheading. “Incidence” is expressed as the number of mice in which the tissue of interest was detectable/the number of mice tested. LN, lymph nodes.
Data are derived from a study by Ansel et al. (31).
The “median number of Peyer′s patches/mouse, range” is indicated within parentheses for the present study. For the study by Ansel et al., the values indicate “n × 3–6”, where n is the number of mice with Peyer’s patches within the ranges indicated.
Conventional DC from CXCR5ΔDC mice have impaired chemotaxis toward CXCL13.
The chemokine CXCL13 is expressed by FDC and follicular stromal cells in the B-cell follicles of lymphoid tissues and mediates the homing of CXCR5-expressing cells toward them (30, 31). Ex vivo chemotaxis assays confirmed that the migration of CD11c+ conventional DC from CXCR5ΔDC mice toward CXCL13 was significantly impeded compared to conventional DC from CXCR5F/F control mice (Fig. 2; P < 0.024). In contrast, the chemotaxis of B cells (B220+ cells) from CXCR5ΔDC mice toward CXCL13 was equivalent to that observed from cells from CXCR5F/F mice. The ability of conventional DC to migrate toward the chemokine CCL21 (which signals via CCR7) was also similar in cells from each mouse group.
FIG 2.

CD11c+ conventional DC from CXCR5ΔDC mice have impaired chemotaxis toward CXCL13. Ex vivo chemotaxis of mesenteric lymph node (MLN) cells from CXCR5ΔDC mice and CXCR5F/F control mice toward CXCL13 or CCL21 (200 ng/ml). The number of CD11c+ cells, CD3+ lymphocytes (T cells) and B220+ lymphocytes (B cells) which had migrated into the lower chamber after 24 h was determined by flow cytometry. Med., medium alone was used as a control. The ability of CD11c+ conventional DC from CXCR5ΔDC mice to migrate toward CXCL13 was significantly impeded. *, P < 0.024 (n = 5 wells/treatment).
Lymphoid tissue microarchitecture in CXCR5ΔDC mice.
The sampling of prions across the intestinal epithelium by M cells, and their subsequent early replication upon PrPC-expressing FDC is obligatory for efficient neuroinvasion after oral exposure (4–6, 29, 42). We therefore determined whether the development of these cells was affected in the lymphoid tissues of CXCR5ΔDC mice. Whole-mount immunostaining using the mature M-cell marker glycoprotein 2 (GP2 [43, 44]) revealed similar densities of GP2+ M cells in the follicle-associated epithelia (FAE) overlying the Peyer's patches and CXCR5ΔDC mice and CXCR5F/F control mice (Fig. 3A and B). The size of the FAE was also equivalent (Fig. 3C). Unlike the disturbed distribution of FDC in the spleens of CXCL13−/− mice (37), those in the lymphoid tissues of CXCR5ΔDC mice formed distinct networks. Furthermore, immunohistochemistry (IHC) and morphometric analysis suggested that FDC size (the area occupied by CD35+ immunostaining) was similar in the mesenteric lymph nodes (MLN) and spleens from CXCR5ΔDC and CXCR5F/F mice (Fig. 3H and K, respectively), although those in the Peyer's patches of CXCR5ΔDC mice were slightly smaller (Fig. 3E). However, the abundance of PrPC+ immunostaining upon FDC in Peyer's patches and MLN from each mouse group was similar (Fig. 3F and I, respectively), although a marginal reduction was in observed in the spleens of CXCR5ΔDC mice (Fig. 3L).
FIG 3.

Comparison of M cell and follicular dendritic cell (FDC) status in the secondary lymphoid tissues of CXCR5ΔDC mice and CXCR5F/F control mice. (A) Peyer's patches were whole-mount immunostained to detect M cells (GP2+ cells, green). F-actin (blue) was used as a counterstain. The broken line indicates the boundary of the follicle associated epithelium (FAE). V, villi. (B) The number of GP2+ M cells/FAE was similar in Peyer's patches from each mouse group (P = 0.710). (C) Morphometric analysis suggested the size of the FAE area was also similar in each mouse group (P = 0.265 [Student t test]; data were derived from 2 to 20 FAE/mouse [n = 6 to 8 mice/group]). IHC comparisons of CD35 (magenta) and PrPC (green) expression by FDC in the Peyer's patches (D), mesenteric lymph nodes (MLN) (G), and spleens (J) of CXCR5F/F and CXCR5ΔDC mice are shown. The sizes of the FDC in the Peyer's patches (E), MLN (H), and (K) spleens of mice from each group were estimated by morphometric analysis of the area of CD35+ immunostaining. The abundance of PrPC expressed by the FDC in Peyer's patches (F), MLN (I), or spleens (L) from each mouse group was estimated by morphometric analysis of the number of PrPC+ pixels within each FDC network. Data were derived from 2 to 10 FDC/mouse (n = 6 to 8 mice/group).
Prion accumulation in lymphoid tissues is reduced in orally exposed CXCR5ΔDC mice.
In order to determine the effects of conventional DC-specific CXCR5-deficiency on oral prion pathogenesis, groups of CXCR5ΔDC mice and CXCR5F/F mice were orally exposed to ME7 scrapie prions and Peyer's patches, MLN, and spleens collected at intervals afterwards (n = 4/group). Prion disease-specific, abnormal accumulations of PrP (referred to as PrPd) characteristically present only in prion-affected tissues were detected by IHC (4–6, 29, 45–48). Paraffin-embedded tissue (PET) blot analysis of adjacent membrane-bound sections was used to confirm that these PrPd aggregates contained prion disease-specific, relatively proteinase K (PK)-resistant PrPSc (49).
As anticipated, by 70 days postinfection (dpi), abundant PrPSc accumulations were detected in association with FDC (CD21/35+ cells) in the majority of the Peyer's patches from CXCR5F/F control mice (Fig. 4A). By 105 dpi, heavy FDC-associated PrPSc accumulations were detected in all the Peyer's patches of CXCR5F/F mice (Fig. 4B and C). However, the incidence of the PrPSc accumulation in Peyer's patches of CXCR5ΔDC mice was reduced at each of these times after prion exposure (Fig. 4A to C).
FIG 4.

The early accumulation of prions in the Peyer's patches is delayed in CXCR5ΔDC mice. Mice were orally exposed to ME7 scrapie prions; Peyer's patches were collected at 70 and 105 dpi and analyzed by IHC and PET immunoblot analysis. At 70 (A) and 105 (B) dpi, the frequencies of FDC networks containing PrPSc in the Peyer's patches from each mouse group were compared (n = 4 mice/group). Horizontal bar, median. (C) IHC analysis showed that at 105 dpi, high levels of disease-specific PrP (PrPd; brown, middle row, arrows) were detected in association with FDC (CD21/35+ cells; brown, upper row) in Peyer's patches from CXCR5F/F control mice. Sections were counterstained with hematoxylin to detect cell nuclei (blue). Analysis of adjacent sections by PET immunoblot analysis confirmed the presence of prion-specific PK-resistant PrPSc (blue/black). In contrast, although PrPSc was detectable in association with FDC in some of the tissues from orally exposed CXCR5ΔDC mice (right-hand column), many FDC in the Peyer's patches of CXCR5ΔDC mice lacked PrPSc accumulation (middle column). Scale bar, 100 μm.
Within weeks of oral exposure, high levels of ME7 scrapie prions first accumulate upon FDC in the Peyer's patches and are subsequently disseminated via the blood and lymph to most other lymphoid tissues (4–6, 24, 29, 50). In the spleens of orally exposed CXCR5F/F control mice, some FDC-associated PrPSc accumulations were first evident at 70 dpi (Fig. 5A to C). However, the abundance of these FDC-associated PrPSc accumulations was reduced in the spleens of CXCR5ΔDC mice at 70 and 105 dpi (Fig. 5A to C). To compare the prion infectivity levels within these tissues, spleen homogenates were prepared and injected intracerebrally (i.c.) into groups of tga20 indicator mice (n = 4 recipient tga20 mice/spleen homogenate). High levels of prion infectivity were detected in the majority of the spleens collected from the CXCR5F/F control mice at 70 dpi and had increased in magnitude by 105 dpi (Fig. 5D). In contrast, significantly lower levels of prion infectivity were detected in the spleens of orally exposed CXCR5ΔDC mice (Fig. 5D; 70 dpi, P < 0.0004; 105 dpi, P < 0.009; log-rank [Mantel-Cox] test). Only trace levels of prion infectivity were detected in three of four spleens from the CXCR5ΔDC mice analyzed at 70 dpi and in two of the four spleens analyzed at 105 dpi.
FIG 5.

Comparison of prion accumulation in the spleens and MLN of orally exposed CXCR5F/F and CXCR5ΔDC mice. Mice were orally exposed to ME7 scrapie prions, and spleens and MLN were collected at 70 and 105 dpi. At 70 (A) and 105 (B) dpi, the frequencies of FDC networks containing PrPSc in the spleens from CXCR5ΔDC mice were reduced compared to CXCR5F/F control mice (n = 4 mice/group). Horizontal bar, median. (C) IHC analysis showed that at 105 dpi high levels of disease-specific PrP (PrPd; brown, middle row, arrow) were detected in association with FDC (CD21/35+ cells; brown, upper row) in spleens from CXCR5F/F control mice. Sections were counterstained with hematoxylin to detect cell nuclei (blue). Analysis of adjacent sections by PET immunoblot analysis confirmed the presence of prion-specific PK-resistant PrPSc (blue/black). In contrast, PrPSc was not detectable in association with the majority of the FDC in spleens from orally exposed CXCR5ΔDC mice. (D) Prion infectivity levels were assayed in spleens from CXCR5F/F and CXCR5ΔDC mice (n = 4 spleens/group) collected at 70 and 105 dpi. Prion infectivity titers (log10 ID50/g tissue) were determined by the injection of tissue homogenates into groups of tga20 indicator mice (n = 4 recipient mice/spleen). Each symbol represents data derived from an individual spleen. Data below the horizontal line indicate disease incidence in the recipient mice <100% and considered to contain trace levels of prion infectivity. IHC and PET blot analysis suggested that the frequencies of FDC networks containing PrPSc in the MLN of CXCR5ΔDC and CXCR5F/F control mice were similar at 70 (E) and 105 (F) dpi (n = 4 mice/group). (G) Analysis of adjacent sections showed that at 105 dpi high levels of PrPSc (blue/black, upper row, arrows) were detected in association with FDC (CD21/35+ cells; brown, low row) in MLN from each mouse group. Sections were counterstained with hematoxylin to detect cell nuclei (blue).
Together, these data show that in the specific absence of CXCR5-expressing conventional DC at the time of oral exposure, the early accumulation of prions upon FDC in the Peyer's patches and spleen is significantly delayed. However, abundant levels of PrPSc were evident in association with FDC in the MLN of CXCR5ΔDC and CXCR5F/F mice at each time point after oral exposure to ME7 scrapie prions (Fig. 5E to G). This implied that the accumulation of prions upon FDC in the MLN was unaffected by an absence of CXCR5-expressing conventional DC.
Oral prion disease susceptibility is reduced in the specific absence of CXCR5-expressing conventional DC.
Since oral exposure to a limiting dose of ME7 scrapie prions typically yields a disease incidence of <100% in wild-type (control) mice, it was used here to enable the effects of conventional DC-restricted CXCR5-deficiency on survival time and prion disease susceptibility to be determined. As anticipated, the majority of the orally exposed CXCR5F/F (control) mice succumbed to clinical prion disease (Fig. 6A; mean, 376 ± 12 days; median, 382 days; n = 6/8). In contrast, disease susceptibility was significantly reduced in the orally exposed CXCR5ΔDC mice, since only two of seven mice succumbed to clinical prion disease with individual survival times of 343 and 371 days: the remaining five mice did not develop clinical signs of prion disease up to at least 501 dpi (Fig. 6A; P < 0.023 [Fisher exact test]). All of the brains from the mice in each group that developed clinical signs of prion disease had the characteristic spongiform pathology (vacuolation), disease-specific PrP accumulation, astrogliosis, and microgliosis and typically associated with terminal infection with ME7 scrapie prions (Fig. 6B and C). The distribution and severity of the spongiform pathology was also similar in the brains of all the clinically affected mice (Fig. 6D), indicating that conventional DC-specific CXCR5 deficiency did not alter the course of CNS prion disease once neuroinvasion had occurred. No histopathological signs of prion disease were detected in the brains of any of the clinically negative mice.
FIG 6.

Oral prion disease susceptibility is reduced in CXCR5ΔDC mice. (A) Disease susceptibility after oral exposure of CXCR5ΔDC (red) and CXCR5F/F control (blue) mice to a limiting dose of prions (P < 0.023 [Fisher exact test]. (B) High levels of spongiform pathology (hematoxylin and eosin staining [H&E]), heavy accumulations of disease-specific PrP, (PrPd; brown), reactive astrocytes expressing GFAP (brown), and active microglia expressing IBA1 (brown) were detected in the brains of all orally exposed mice with clinical prion disease. None of these histopathological signs of prion disease were detected in the brains of any of the clinically negative mice up to at least 501 days after oral exposure. Clin., clinical prion disease status; pos., clinically positive; neg., clinically negative; individual survival times are shown. Sections were counterstained with hematoxylin to detect cell nuclei (blue). (C) Immunoblot analysis of brain tissue homogenates confirmed the presence of high levels of prion-specific, relatively PK-resistant PrPSc within the brains of the clinically affected mice from each group. However, no PrPSc was detected in the brains of any of the clinically negative CXCR5ΔDC mice. Samples were treated in the presence (lower panel) or absence (upper panel) of PK before electrophoresis. After PK treatment, a typical three-band pattern was observed between molecular mass value of 20 to 30 kDa, representing unglycosylated, monoglycosylated, and diglycosylated isomers of PrP (in order of increasing molecular mass). (D) The severity and distribution of the spongiform pathology (vacuolation) within each brain was scored on a scale of 1 to 5 in nine gray-matter areas: G1, dorsal medulla; G2, cerebellar cortex; G3, superior colliculus; G4, hypothalamus; G5, thalamus; G6, hippocampus; G7, septum; G8, retrosplenial and adjacent motor cortex; and G9, cingulate and adjacent motor cortex. Each point represents the mean vacuolation score ± SE.
After oral exposure, high levels of ME7 scrapie prions are maintained upon FDC in lymphoid tissues until the terminal stages of disease (5, 6, 24, 29). Here, FDC-associated PrPSc accumulations were detected in the Peyer's patches, MLN, and spleens of all clinically affected CXCR5ΔDC and CXCR5F/F mice (Fig. 7). In comparison, no evidence of PrPSc accumulation was observed in any of the tissues from the clinically negative mice (Fig. 7). These data indicate that all of the clinically negative mice were free of prions in their lymphoid tissues and brains and were highly unlikely to succumb clinical prion disease after substantially extended survival times. Together, these data show that CXCR5-expressing conventional DC are essential for efficient prion neuroinvasion after oral exposure.
FIG 7.

Prion accumulation in the lymphoid tissues of CXCR5ΔDC and CXCR5F/F mice at the terminal stage of disease. Mice were orally exposed to ME7 scrapie prions, and Peyer's patches, MLN, and spleens were collected from all clinically affected mice and from those free of clinical signs of prion disease at the end of the experiment at 501 dpi. Clin., clinical prion disease status; pos., clinically positive; neg. clinically negative. Individual survival times are shown. High levels of PrPSc (PET immunoblot; black, arrows) were detected in association with follicular dendritic cells (CD21/35+ cells; brown, arrows) in the Peyer's patches, MLN, and spleens from all clinically affected mice. In contrast, no PrPSc was detected in tissues from any of the clinically negative survivors at 501 dpi. Sections were counterstained with hematoxylin to detect cell nuclei (blue). CXCR5F/F Clin. pos, n = 6 mice; CXCR5F/F Clin. neg, n = 2 mice; CXCR5ΔDC Clin. pos, n = 2 mice; CXCR5ΔDC Clin. neg, n = 5 mice.
Conventional DC-specific CXCR5 deficiency does not influence prion disease susceptibility when infection is established directly within the CNS.
When groups of CXCR5ΔDC and CXCR5F/F mice were injected i.c. with ME7 scrapie prions directly into the CNS, all mice succumbed to clinical disease with similar survival times (CXCR5ΔDC mice, 157 ± 2 days, n = 4/4; CXCR5fl mice, 162 ± 3 days, n = 4/4; P = 0.302). Histopathological analysis of the brains from each group of clinically affected mice revealed the characteristic neuropathology and PrPSc accumulation associated with terminal infection with ME7 scrapie prions (Fig. 8A and B). The distribution and severity of the spongiform pathology was also similar in the brains of the clinically affected CXCR5ΔDC and CXCR5F/F mice (Fig. 8C). Therefore, a CXCR5 deficiency specifically in conventional DC did not affect prion disease pathogenesis or susceptibility when the infection was established directly within the CNS. These data are consistent with the independent observation that prion propagation within the CNS is not affected in CXCR5−/− mice (37).
FIG 8.

Neuropathological comparison of brains from clinically affected CXCR5ΔDC and CXCR5F/F mice after injection of prion directly into the CNS. CXCR5ΔDC and CXCR5F/F mice (n = 4/group) were injected i.c. with ME7 scrapie prions, and brains were collected at the terminal stage of disease. (A) Histopathological analysis revealed the typical spongiform pathology (H&E, top row), heavy accumulations of prion disease-specific PrPd (brown, second row), reactive astrocytes expressing GFAP (brown, third row), and active microglia expressing IBA1 (brown, bottom row) in the brains of all clinically affected mice from each group. Sections were counterstained with hematoxylin to detect cell nuclei (blue). (B) Immunoblot analysis of brain tissue homogenates confirmed the presence of high levels of prion-specific, relatively PK-resistant PrPSc within the brains of mice from each group. Samples were treated in the presence (+) or absence (−) of PK before electrophoresis. After PK treatment, a typical three-band pattern was observed between molecular mass values of 20 to 30 kDa, representing unglycosylated, monoglycosylated, and diglycosylated isomers of PrP (in order of increasing molecular mass). (C) The severity and distribution of the spongiform pathology (vacuolation) within the brains of all clinically affected CXCR5ΔDC and CXCR5F/F mice was similar. The severity of the vacuolation in each brain was scored on a scale of 1 to 5 in the following gray matter regions: G1, dorsal medulla; G2, cerebellar cortex; G3, superior colliculus; G4, hypothalamus; G5, thalamus; G6, hippocampus; G7, septum; G8, retrosplenial and adjacent motor cortex; and G9, cingulate and adjacent motor cortex. Each point represents the mean vacuolation score ± the SE.
Splenectomy before oral prion exposure does not influence disease susceptibility.
The data presented above show that the early accumulation of prions in the Peyer's patches (Fig. 4) and spleens (Fig. 5) of orally exposed CXCR5ΔDC mice was impaired. Although early prion replication upon FDC in the Peyer's patches in the small intestine is essential for efficient neuroinvasion after oral exposure (5, 6, 45), we considered it plausible that the effects observed here on disease susceptibility were due to impaired neuroinvasion from the spleen. If the spleen did play an important role in prion neuroinvasion after oral exposure, we reasoned that splenectomy before prion exposure would similarly impede neuroinvasion and reduce disease susceptibility. To test this hypothesis, wild-type mice were surgically splenectomized and 8 days later orally exposed to a 0.1% dose of ME7 scrapie prions as described above. A parallel group of mice were sham operated before oral prion exposure as a control. Our data show that splenectomy did not influence prion disease pathogenesis or susceptibility as mice from each treatment group succumbed to clinical disease with similar survival times and disease incidences (Fig. 9A; splenectomized mice, mean = 371 ± 13 days, median = 364 days, n = 6/11; sham-operated mice, mean = 367 ± 11 days, median = 364 days, n = 7/11 [P = 1.0, Fisher exact test]). The distribution and severity of the spongiform pathology were also similar in the brains of all the clinically affected splenectomized and sham-operated mice (Fig. 9B). These data clearly show that the spleen does not play an important role in neuroinvasion after oral prion exposure. This suggests that the major effects observed on oral prion disease pathogenesis and susceptibility in CXCR5ΔDC mice were not due to impaired neuroinvasion from their spleens.
FIG 9.
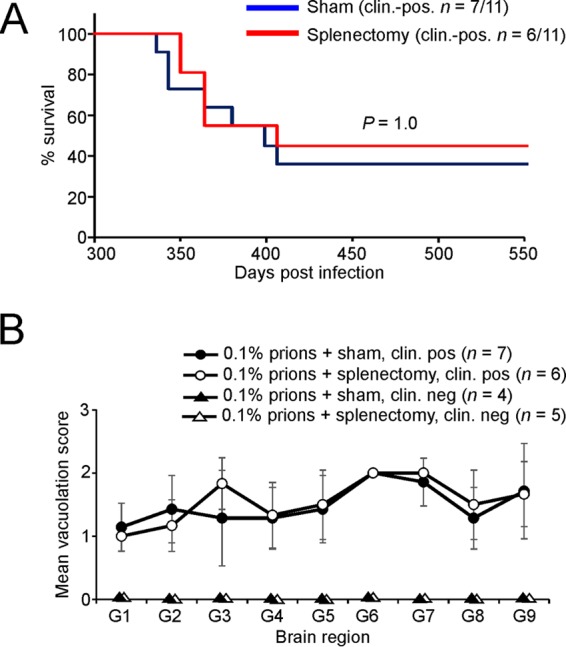
Splenectomy before oral prion exposure does not influence disease susceptibility. C57BL/Dk wild-type mice were surgically splenectomized, or sham operated (as a control), and 8 days later orally exposed to a 0.1% dose of ME7 scrapie prions (n = 11/group). (A) Comparison of disease susceptibility of splenectomized (red) and sham-operated mice (blue) to oral prion infection (P = 1.0 [Fisher exact test]). (B) The severity and distribution of the spongiform pathology (vacuolation) within the brains of all clinically affected splenectomized (open circles) and sham-operated mice (closed circles) was similar. The severity of the vacuolation in each brain was scored on a scale of 1 to 5 in the following gray-matter regions: G1, dorsal medulla; G2, cerebellar cortex; G3, superior colliculus; G4, hypothalamus; G5, thalamus; G6, hippocampus; G7, septum; G8, retrosplenial and adjacent motor cortex; and G9, cingulate and adjacent motor cortex. Each point represents the mean vacuolation score ± the SE.
DISCUSSION
The early replication of prions upon FDC in Peyer's patches is essential for efficient neuroinvasion from the small intestine after oral exposure (3–6, 35). The cellular mechanism by which orally acquired prions are initially conveyed to FDC in order to establish host infection was uncertain. Production of the chemokine CXCL13 by FDC and follicular stromal cells plays an important role in attracting CXCR5-expressing cells toward the FDC-containing B-cell follicles of secondary lymphoid organs (31, 36, 51). Here, a unique compound transgenic mouse model was created in which CXCR5 deficiency was restricted to CD11c+ conventional DC. In the specific absence of CXCR5-expressing conventional DC in CXCR5ΔDC mice, the early accumulation of prions upon FDC in the Peyer's patches and spleen was reduced, significantly reducing disease susceptibility. Our data suggest that CXCR5-expressing conventional DC are required for the efficient delivery of orally acquired prions to FDC in the Peyer's patches of the small intestine in order to achieve host infection.
The data presented here help advance our understanding of how orally acquired prions utilize an elegant cellular relay to establish host infection from the lumen of the small intestine. Prions are first transported across the gut epithelium by M cells in the FAE overlying the Peyer's patches (29, 45, 52, 53). This initial transport of prions across the gut epithelium by M cells is essential to establish infection in Peyer's patches (29, 45). Antigens and particles that have been transported across the gut epithelium by M cells are released into their underlying basolateral pockets where they are sampled by mononuclear phagocytes (54). Our data suggest that prions are subsequently acquired by conventional DC (24, 55) and propagated by them in a CXCL13-CXCR5-dependent manner toward FDC within the B-cell follicles of Peyer's patches. The prions are then acquired by FDC and amplified upon their surfaces above the threshold required to achieve neuroinvasion (4–6, 42, 55). Studies from experimental mice indicate that prions can establish infection in enteric nerves within 21 days after oral exposure (4, 55). How the prions spread between the FDC and enteric nerves is not known, but a role for conventional DC in this process has been proposed (56–58).
Whether these apparent prion propagation activities are restricted to certain conventional DC populations is uncertain, but the specific depletion of CD8+ CD11c+ cells does not influence oral prion disease pathogenesis, implying that CD8+ conventional DC do not play a role (26). Indeed, within Peyer's patches CD8+ conventional DC are rarely encountered within the subepithelial dome region immediately beneath the M-cell-containing FAE. Instead, these cells are mostly localized within the T-cell-rich interfollicular regions (59). Splenic plasmacytoid DC also accumulate high levels of infectious prions during infection (60) but are unlikely to play a significant role in prion propagation since they do not migrate in the lymphatics during the steady state or after activation (61).
Whether the initial uptake of prions by conventional DC involves a specific receptor is uncertain. Most mononuclear phagocyte populations express cellular PrPC (62–64), but the propagation of prions from peripheral exposure sites to FDC is not affected by the absence of PrPC expression in hematopoietic cells (42, 48, 65–67). Conventional DC, like the FDC in the B-cell follicles (28, 68–71), can acquire prions after their opsonization by complement components such as C1q and C3 (72, 73). Depending on their location, subset, and activation status, certain populations of conventional DC express a variety of complement receptors (CR), including CR1 (CD35), CR2 (CD21), CR4 (CD11c/CD18), calreticulin, CD93, and SIGN-R1 (CD209b), but it is uncertain whether they also facilitate the complement-mediated uptake of prions by these cells (72, 73). However, SIGN-R1 is unlikely to play a role since its transient downregulation before prion exposure does not influence disease pathogenesis (74). The possibility also cannot be excluded that conventional DC simply acquire prions nonspecifically as they constitutively sample their microenvironments via fluid-phase micropinocytosis.
Although the data in the present study suggest that CXCR5-expressing conventional DC are required for the efficient propagation of prions to FDC in Peyer's patches, it is not known how they are transferred between these cell populations. Follicular B cells within the subcapsular sinus of lymph nodes acquire lymph-borne immune complexes via their CR and deliver them to FDC. The higher immune-complex-binding affinity of FDC appears to enable them to strip the B cells of their immune-complex cargo as they migrate into the follicles (75–77). Conventional DC can retain PrPSc on both the cell surface and in intracellular compartments (73). Since the expression level of CR1 and CR2 on FDC is greater than the surrounding lymphocytes and leukocytes, it is plausible that complement-opsonized prions are stripped from the surfaces of conventional DC by FDC in a similar manner. Alternatively, it is plausible that conventional DC might indirectly deliver infectious prions toward FDC. Since CR-expressing marginal zone B cells in the spleen can shuttle complement-opsonized antigens to the FDC (78), B cells may similarly strip opsonized prions from conventional DC in the vicinity of the B cell follicle and deliver them to the FDC.
Tunneling nanotubes (TNT) are thin, membrane-bound cylinders of cytoplasm that can connect neighboring cells to enable cell-to-cell communication and the intercellular transfer of plasma membrane or cytoplasmic components. These TNT structures are exploited by HIV-1 as a means of intercellular transfer between T cells (79) and to shuttle virus-encoded immunosuppressive factors from infected macrophages to B cells to suppress the humoral response (80). In vitro coculture studies suggest prions may also transfer between conventional DC to neurons via endolysosomal vesicles within TNT (57, 58, 81, 82). Infectious prions may also be released from infected cells in the form of small endosome-derived vesicles termed exosomes (83) and, in doing so, enhance their ability to infect neighboring cells (60). Whether the transfer of prions between conventional DC and FDC occurs in vivo via one or a combination of the above processes remains to be determined.
We have previously shown that although the transient depletion of CD11c+ cells impedes the accumulation of prions in Peyer's patches and reduces disease susceptibility, a small number of mice did develop clinical disease (24). In the present study, we observed a similar effect on prion disease pathogenesis in the absence of CXCR5-expressing conventional DC: prion accumulation was impeded in the Peyer's patches and spleens of orally exposed CXCR5ΔDC mice, and disease susceptibility was reduced, but a small number of mice eventually succumbed to clinical prion disease. These studies suggest that conventional DC provide an efficient route by which prions are initially conveyed to FDC. However, in their absence a limited amount of prions are able to avoid clearance by cells such as tissues macrophages (14, 15) and reach the FDC via alternative and less efficient routes (27, 73, 84).
Studies in experimental mice show that after oral exposure, prions accumulate first in the Peyer's patches in the small intestine and then spread via the blood and lymph to most other secondary lymphoid tissues, including the spleen (4–6, 24, 50). Analysis of the pathogenesis of natural sheep scrapie suggests a similar temporal distribution (9). In the present study, the early accumulation of prions within the MLN of CXCR5ΔDC mice was not impaired, suggesting that the prions had established infection within the MLN independently of CXCR5-expressing conventional DC. Although prions also replicate upon FDC in the MLN soon after oral exposure, our studies in mice have shown that the MLN do not influence prion neuroinvasion from the intestine (5, 6, 45).
We also showed that early prion replication in the spleens of CXCR5ΔDC mice was significantly reduced. This raised the possibility that the significantly reduced prion disease susceptibility observed in these mice was, at least in part, due to impaired neuroinvasion from their spleens. However, splenectomy of conventional mice prior to oral exposure with ME7 scrapie prions did not influence survival times or disease susceptibility. These data are consistent with an earlier study using mouse-passaged 139A scrapie prions which also suggested that the spleen does not play a role in prion disease pathogenesis after intragastric exposure (85), and our subsequent demonstrations that the GALT in the small intestine are the important sites of early prion accumulation and neuroinvasion after oral exposure (5, 6).
Many studies have defined conventional DC based on their expression of distinct cell surface markers such as the integrin CD11c. Murine conventional DC express CD11c highly, and this has been used as a reliable marker for these cells in a wide range of studies. However, the expression of this integrin is not restricted to conventional DC since certain macrophage populations and activated monocytes can also express CD11c to some degree (86, 87). The recent discovery that expression of the zinc finger transcription factor ZBTB46 is restricted to conventional DC (88, 89) provides an excellent opportunity to create conditional knockout mouse models to further define the separate roles of conventional DC and tissue macrophages in prion disease pathogenesis (90).
In conclusion, our data demonstrate that prions exploit conventional DC in order to facilitate their efficient propagation to FDC in Peyer's patches in order to establish host infection. The status of conventional DC is dramatically influenced by microbial stimuli and inflammatory conditions in the intestine and can enhance the uptake of certain pathogens from the gut lumen (91–93). Treatments that prevent the initial uptake and accumulation of prions in Peyer's patches can significantly reduce disease susceptibility (4, 24, 29). Therefore, a thorough understanding of the cellular and molecular mechanisms which prions exploit to establish infection upon FDC in the GALT may help to identify important factors that influence disease susceptibility or novel targets for prophylactic intervention.
MATERIALS AND METHODS
Mice.
The following mouse strains were used in this study where indicated: CD11c-Cre (38); and tga20, which overexpress PrPC (94). Cxcr5F/F mice were produced by Ozgene (Bentley DC, Australia) and created by introducing loxP sites flanking exon 2 of the Cxcr5 gene via homologous recombination. All mice were bred and maintained on a C57BL/6J mice background and maintained under specific-pathogen-free conditions. In some studies C57BL/Dk mice were also used, where indicated. All studies and regulatory licenses were approved by University of Edinburgh's ethics committee and carried out under the authority of a UK Home Office Project License.
Genotype confirmation by PCR analysis.
CD11c+ cells were positively enriched from spleens by magnetic antibody cell sorting using CD11c microbeads according to the manufacturer's instructions (Miltenyi Biotec, Bisley, UK). The cells in the CD11c− flowthrough fraction were further sorted using CD11b microbeads to enrich monocytes/macrophages, CD45R (B220) microbeads to enrich B cells, and CD90.2 microbeads to enrich T cells. DNA was extracted from each cell populations using a DNeasy blood and tissue kit (Qiagen, Crawley, UK) according to the manufacturer's instructions. RNA was extracted using RNA-Bee (AMS Biotechnology, Abingdon, UK), and cDNA was synthesized using a Superscript first-strand synthesis kit (Invitrogen). Where indicated, genomic or cDNA samples were analyzed for the presence of Cre, Cxcr5F, and recombined Cxcr5F (Cxcr5de-flox) using the following primers: Cre (5′-CGAGTGATGAGGTTCGCAAGAACC-3′ and 5′-GCTAAGTGCCTTCTCTACACCTGC-3′) and Cxcr5F and recombined Cxcr5flox (Cxcr5de-flox; 5′-AGGAGGCCATTTCCTCAGTT-3′, 5′-GGCTTAGGGATTGCAGTCAG-3′, and 5′-TTCCTTAGAGCCTGGAAAAGG-3′).
Macroscopic analysis of secondary lymphoid tissues.
Mice were injected intraperitoneally with 300 μl of 1% Chicago Sky Blue 6B in sterile phosphate-buffered saline. Mice were culled 7 days later, and the presence or absence of secondary lymphoid organs was determined macroscopically (41).
Chemotaxis assays.
Ex vivo chemotaxis assays were performed as described previously (33, 95). Briefly, MLN cells in RPMI medium with 5% fetal calf serum (Invitrogen) were placed in the upper chamber of a 5-μm Transwell insert in a 24-well plate (Corning, Corning, NY). The lower chamber contained 200 ng of either CXCL13 or CCL21 (Peprotech, London, UK)/ml. Five technical replicates were performed. After 24 h, the upper chamber was discarded, and the cells in the lower chamber were collected and immunostained with anti-CD3 (clone 17A2) to detect T cells, anti-B220 (clone RA3-2GB) to detect B cells, and anti-CD11c (clone N418; BioLegend, London, UK) and resuspended in 500 μl of fluorescence-activated cell sorting buffer. The number of migrated cells was counted for 60 s using a LSRFortessa flow cytometer (BD Biosciences).
Prion exposure and disease monitoring.
For oral exposure, mice were fed individual food pellets dosed with 50 μl of a 0.1% (wt/vol) dilution of scrapie brain homogenate (containing approximately 2.5 × 103 i.c. 50% infectious dose [ID50] units) prepared from mice terminally affected with ME7 scrapie prions according to our standard protocol (4–6, 24, 29, 96). During the dosing period mice were individually housed in bedding- and food-free cages with water provided ad libitum. A single prion-dosed food pellet was then placed in the cage. The mice were returned to their original cages (with bedding and food ad libitum) as soon as the food pellet was observed to have been completely ingested. The use of bedding- and additional food-free cages ensured easy monitoring of consumption of the prion-contaminated food pellet. For i.c. exposure, mice were injected with 20 μl of a 1% dilution of scrapie brain homogenate. After prion exposure, mice were coded and assessed weekly for signs of clinical disease and culled at a standard clinical endpoint. The clinical endpoint of disease was determined by rating the severity of clinical signs of prion disease exhibited by the mice. The mice were clinically scored as “unaffected,” “possibly affected,” or “definitely affected” according to standard criteria that typically present in mice with terminal ME7 scrapie prion disease. Clinical signs following infection with the ME7 scrapie agent may include weight-loss, starry coat, hunched and jumpy behavior (at early onset) progressing to limited movement, upright tail, wet genitals, decreased awareness, discharge from eyes and/or blinking eyes, and ataxia of the hind legs. The clinical endpoint of disease was defined in one of the following ways: (i) the day on which a mouse received a second consecutive “definite” rating, (ii) the day on which a mouse received a third “definite” rating within four consecutive weeks, or (iii) the day on which a mouse was culled in extremis. Survival times were recorded for mice that did not develop clinical signs of disease during the observation period. Prion diagnosis was confirmed by histopathological assessment of vacuolation in the brain. For the construction of lesion profiles, vacuolar changes were scored in nine distinct gray-matter regions of the brain, as described previously (97).
For bioassay of prion infectivity, individual spleens were prepared as 1% (wt/vol) homogenates. For each tissue homogenate, groups of tga20 indicator mice (n = 4/homogenate) were injected i.c. with 20 μl of homogenate. The prion infectivity titer in each sample was determined from the mean incubation period in the indicator mice by reference to a dose/incubation period response curve for ME7 scrapie-infected spleen tissue serially titrated in tga20 mice using the following relationship: y = 9.4533 to 0.0595x (where y is the log ID50 U/20 μl of homogenate, and x is the incubation period [R2 = 0.9562]). Since the expression level of cellular PrPC controls the prion disease incubation period, tga20 mice overexpressing PrPC are extremely useful as indicator mice in scrapie agent infectivity bioassays because they succumb to disease with much shorter incubation times than conventional mouse strains (94).
Splenectomy.
Groups of C57BL/Dk mice were surgically splenectomized and orally exposed to prions 8 days later. Briefly, an ∼1-cm incision was made in the left upper quadrant of the abdominal wall, and the spleen was identified. Blood supply to the organ was tied off with sutures before removal. The incision site was then also closed with sutures. Isoflurane anesthesia was used throughout the surgical procedure. After surgery, the mice were closely monitored throughout the recovery period and given buprenorphine hydrochloride as an analgesic. Sham-operated mice (laparotomy incision and suture repair with no spleen removal) were used as a control.
IHC and immunofluorescent analyses.
Whole-mount immunostaining was performed as previously described (29). Briefly, Peyer's patches were fixed with BD Cytofix/Cytoperm (BD Biosciences, Oxford, UK), and subsequently immunostained with rat anti-mouse GP2 monoclonal antibody (MAb; MBL International, Woburn, MA). After the addition of primary antibody, the tissues were stained with Alexa Fluor 488-conjugated anti-rat IgG antibody (Invitrogen, Paisley, UK) and Alexa Fluor 647-conjugated phalloidin to detect f-actin (Invitrogen).
Intestines, MLN, and spleens were also removed and snap-frozen at the temperature of liquid nitrogen. Serial frozen sections (6 μm in thickness) were cut on a cryostat and immunostained with the following antibodies: FDC were visualized by staining with MAb 8C12 to detect CR1 (CD35; BD Biosciences) and cellular PrPC was detected using PrP-specific polyclonal antibody (pAb) 1B3 (98). Where appropriate, sections were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) to detect cell nuclei (Life Technologies).
For the detection of disease-specific PrP (PrPd) in intestines, MLN, spleens, and brains, tissues were fixed in periodate-lysine-paraformaldehyde fixative and embedded in paraffin wax. Sections (thickness, 6 μm) were deparaffinized and pretreated to enhance the detection of PrPd by hydrated autoclaving (15 min, 121°C, hydration) and subsequent immersion in formic acid (98%) for 5 min. The sections were then immunostained with 1B3 PrP-specific pAb. For the detection of astrocytes, brain sections were immunostained with anti-glial fibrillary acidic protein (GFAP; Dako, Ely, UK). For the detection of microglia, deparaffinized brain sections were first pretreated with citrate buffer and subsequently immunostained with anti-ionized calcium-binding adaptor molecule 1 (Iba-1; Wako Chemicals GmbH, Neuss, Germany). For the detection of FDC in intestines, MLN, and spleens, deparaffinized sections were first pretreated with Target Retrieval Solution (Dako) and then immunostained with anti-CD21/35 (clone 7G6; BD Biosciences). PET immunoblot analysis was used to confirm the PrPd detected by IHC was proteinase K-resistant PrPSc (49). Membranes were subsequently immunostained with 1B3 PrP-specific pAb.
For light microscopy, following the addition of primary antibodies, biotin-conjugated species-specific secondary antibodies (Stratech, Soham, UK) were applied, and immunolabeling was revealed using horseradish peroxidase-conjugated avidin-biotin complex (ABC kit; Vector Laboratories, Peterborough, United Kingdom) and visualized with DAB (diaminobenzidine; Sigma). Sections were counterstained with hematoxylin to distinguish cell nuclei. For fluorescence microscopy, following the addition of primary antibody, streptavidin-conjugated or species-specific secondary antibodies coupled to Alexa Fluor 488 (green), Alexa Fluor 594 (red), or Alexa Fluor 647 (blue) dyes (Life Technologies) were used. The sections were subsequently mounted in fluorescent mounting medium (Dako). Images of whole-mount immunostained intestinal pieces and cryosections were obtained using a Zeiss LSM710 confocal microscope (Zeiss, Welwyn Garden City, UK).
Image analysis.
For morphometric analysis, images were analyzed using ImageJ software (http://rsb.info.nih.gov/ij/) as described on coded sections (42, 45, 99). Background intensity thresholds were first applied using an ImageJ macro which measures pixel intensity across all immunostained and nonstained areas of the images. The obtained pixel intensity threshold value was then applied in all subsequent analyses. Next, the number of pixels of each color (black, red, green, yellow, etc.) were automatically counted. For these analyses, data are presented as the proportion of positively stained pixels for a given IHC marker per total number of pixels (all colors) in the specific area of interest. In each instance, typically six images were analyzed per mouse, from tissues from multiple mice per group (n = 6 to 8 mice/group). Details for all of the sample sizes for each parameter analyzed are provided in the figure legends.
Immunoblot detection of PrPSc.
Brain homogenates (10% weight/volume) were prepared in NP-40 lysis buffer (1% NP-40, 0.5% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-HCl [pH 7.5]) and incubated at 37°C for 1 h with proteinase K (PK) at 20 μg/ml. Digestions were halted by addition of 1 mM phenylmethylsulfonyl fluoride. Samples were then subjected to electrophoresis through 12% Tris-glycine polyacrylamide gels (Nupage; Life Technologies) and transferred to polyvinylidene difluoride membranes by semidry blotting. PrP was detected using anti-mouse PrP-specific MAb 7A12 (100), followed by horseradish peroxidase-conjugated goat anti-mouse antibody (Jackson ImmunoResearch) and visualized chemiluminescence (BM chemiluminescent substrate kit; Roche, Burgess Hill, UK).
Statistical analyses.
Unless indicated otherwise, data are presented as means ± the standard errors (SE), and significant differences between samples in different groups were evaluated by Student t test. In instances where there was evidence of nonnormality, data were analyzed by nonparametric analysis of variance (Kruskal-Wallis test) with Dunn's multiple comparison post hoc test. P values of <0.05 were accepted as significant.
ACKNOWLEDGMENTS
We thank Bob Fleming, Dave Davies, Fraser Laing, Sally Carpenter, Kris Hogan, and the Pathology Services Group (University of Edinburgh, Edinburgh, UK) for excellent technical support. Splenectomy experiments were originally undertaken by Christine Farquhar and Patricia McBride (Neuropathogenesis Unit, Edinburgh, United Kingdom).
This study was supported by project funding (grants BB/F019726/1 and 201/BS41057) and institute strategic programme grant funding (grant BB/J004332/1) from the Biotechnology and Biological Sciences Research Council (BBSRC).
REFERENCES
- 1.Legname G, Baskakov IV, Nguyen H-OB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. 2004. Synthetic mammalian prions. Science 305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 2.Horiuchi M, Yamazaki N, Ikeda T, Ishiguro N, Shinagawa M. 1995. A cellular isoform of prion protein (PrPc) exists in many non-neuronal tissues of sheep. J Gen Virol 76:2583–2587. doi: 10.1099/0022-1317-76-10-2583. [DOI] [PubMed] [Google Scholar]
- 3.Prinz M, Huber G, Macpherson AJS, Heppner FL, Glatzel M, Eugster H-P, Wagner N, Aguzzi A. 2003. Oral prion infection requires normal numbers of Peyer's patches but not of enteric lymphocytes. Am J Pathol 162:1103–1111. doi: 10.1016/S0002-9440(10)63907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mabbott NA, Young J, McConnell I, Bruce ME. 2003. Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. J Virol 77:6845–6854. doi: 10.1128/JVI.77.12.6845-6854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glaysher BR, Mabbott NA. 2007. Role of the GALT in scrapie agent neuroinvasion from the intestine. J Immunol 178:3757–3766. doi: 10.4049/jimmunol.178.6.3757. [DOI] [PubMed] [Google Scholar]
- 6.Donaldson DS, Else KJ, Mabbott NA. 2015. The gut-associated lymphoid tissues in the small intestine, not the large intestine, play a major role in oral prion disease pathogenesis. J Virol 15:9532–9547. doi: 10.1128/JVI.01544-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreoletti O, Berthon P, Marc D, Sarradin P, Grosclaude J, van Keulen L, Schelcher F, Elsen J-M, Lantier F. 2000. Early accumulation of PrPSc in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J Gen Virol 81:3115–3126. doi: 10.1099/0022-1317-81-12-3115. [DOI] [PubMed] [Google Scholar]
- 8.Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O'Rourke KI, Hoover EA. 1999. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J Gen Virol 80:2757–2764. doi: 10.1099/0022-1317-80-10-2757. [DOI] [PubMed] [Google Scholar]
- 9.van Keulen LJ, Schreuder BE, Vromans ME, Langeveld JP, Smits MA. 2000. Pathogenesis of natural scrapie in sheep. Arch Virol Suppl 16:57–71. [DOI] [PubMed] [Google Scholar]
- 10.Beekes M, McBride PA. 2000. Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci Lett 278:181–184. doi: 10.1016/S0304-3940(99)00934-9. [DOI] [PubMed] [Google Scholar]
- 11.McBride PA, Schulz-Shaeffer WJ, Donaldson M, Bruce M, Diringer H, Kretzschmar HA, Beekes M. 2001. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J Virol 75:9320–9327. doi: 10.1128/JVI.75.19.9320-9327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glatzel M, Heppner FL, Albers KM, Aguzzi A. 2001. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 31:25–34. doi: 10.1016/S0896-6273(01)00331-2. [DOI] [PubMed] [Google Scholar]
- 13.Mabbott NA, Bradford BM. 2015. The good, the bad, and the ugly of dendritic cells during prion disease. J Immunol Res 2015:168574. doi: 10.1155/2015/168574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beringue V, Demoy M, Lasmezas CI, Gouritin B, Weingarten C, Deslys J-P, Andreux JP, Couvreur P, Dormont D. 2000. Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J Pathol 190:495–502. doi:. [DOI] [PubMed] [Google Scholar]
- 15.Maignien T, Shakweh M, Calvo P, Marcé D, Salès N, Fattal E, Deslys JP, Couvreur P, Lasmézas CI. 2005. Role of gut macrophages in mice orally contaminated with scrapie or BSE. Int J Pharmaceutics 298:293–304. doi: 10.1016/j.ijpharm.2005.02.042. [DOI] [PubMed] [Google Scholar]
- 16.Zhu C, Hermann US, Falsig J, Abakumova I, Nuvolone M, Schwarz P, Frauenknecht K, Rushing EJ, Aguzzi A. 2016. A neuroprotective role for microglia during prion diseases. J Exp Med 213:1047–1059. doi: 10.1084/jem.20151000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, Schwarz P, Armulik A, Browning JL, Tallquist M, Buch T, Oliveira-Martins JB, Zhu C, Hermann M, Wagner U, Brink R, Heikenwalder M, Aguzzi A. 2012. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell 150:194–206. doi: 10.1016/j.cell.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mabbott NA, Bailie JK, Kobayashi A, Donaldson DS, Ohmori H, Yoon S-O, Freedman AS, Freeman TC, Summers KM. 2011. Expression of mesenchyme-specific gene signatures by follicular dendritic cells: insights from the meta-analysis of microarray data from multiple mouse cell populations. Immunology 133:482–498. doi: 10.1111/j.1365-2567.2011.03461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wykes M, Pombo A, Jenkins C, MacPherson GG. 1998. Dendritic cells interact directly with Naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J Immunol 161:1313–1319. [PubMed] [Google Scholar]
- 20.Wu S-JL, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, Louder MK, Filgueira L, Marovich MA, Wong HK, Blauvelt A, Murphy GS, Robb ML, Innes BL, Birx DL, Hayes CG, Schlesinger Frankel S. 2000. Human skin Langerhans cells are targets of dengue virus infection. Nat Med 6:816–820. doi: 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- 21.Ho L-J, Wang J-J, Shaio M-F, Kao C-L, Chang D-M, Han S-W, Lai J-H. 2001. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J Immunol 166:1499–1506. doi: 10.4049/jimmunol.166.3.1499. [DOI] [PubMed] [Google Scholar]
- 22.Steinman RM, Granelli-Piperno A, Pope M, Trumpfheller C, Ignatius R, Arrode G, Racz P, Tenner-Racz K. 2003. The interaction of immunodeficiency viruses with dendritic cells. Curr Top Microbiol Immunol 276:1–30. [DOI] [PubMed] [Google Scholar]
- 23.Ho AW, Prabhu N, Betts RJ, Ge MQ, Dai X, Hutchinson PE, Lew FC, Wong KL, Hanson BJ, Macary PA, Kemeny DM. 2011. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J Immunol 187:6011–6021. doi: 10.4049/jimmunol.1100987. [DOI] [PubMed] [Google Scholar]
- 24.Raymond CR, Aucouturier P, Mabbott NA. 2007. In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J Immunol 179:7758–7766. doi: 10.4049/jimmunol.179.11.7758. [DOI] [PubMed] [Google Scholar]
- 25.Cordier-Dirikoc S, Chabry J. 2008. Temporary depletion of CD11c+ dendritic cells delays lymphoinvasion after intraperitoneal scrapie infection. J Virol 82:8933–8936. doi: 10.1128/JVI.02440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sethi S, Kerksiek KM, Brocker T, Kretzschmar H. 2007. Role of the CD8+ dendritic cell subset in transmission of prions. J Virol 81:4877–4880. doi: 10.1128/JVI.02345-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wathne GJ, Kissenpfennig A, Malissen B, Zurzolo C, Mabbott NA. 2012. Determining the role of mononuclear phagocytes in prion neuroinvasion from the skin. J Leukoc Biol 91:817–828. doi: 10.1189/jlb.1211633. [DOI] [PubMed] [Google Scholar]
- 28.Mabbott NA, Bruce ME, Botto M, Walport MJ, Pepys MB. 2001. Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat Med 7:485–487. doi: 10.1038/86562. [DOI] [PubMed] [Google Scholar]
- 29.Donaldson DS, Kobayashi A, Ohno H, Yagita H, Williams IR, Mabbott NA. 2012. M cell depletion blocks oral prion disease pathogenesis. Mucosal Immunol 5:216–225. doi: 10.1038/mi.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. 1998. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature 391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 31.Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster J. 2000. A chemokine-driven feedback loop organizes lymphoid follicles. Nature 406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 32.Yu P, Wang Y, Chin RK, Martinez-Pomares L, Gordon S, Kosco-Vilbois MH, Cyster J, Fu Y-X. 2002. B cells control the migration of a subset of dendritic cells into B cell follicles via CXC chemokine ligand 13 in a lymphotoxin-dependent fashion. J Immunol 168:5117–5123. doi: 10.4049/jimmunol.168.10.5117. [DOI] [PubMed] [Google Scholar]
- 33.Saeki H, Wu M, Olasz E, Hwang ST. 2000. A migratory population of skin-derived dendritic cells expresses CXCR5, responds to B lymphocyte chemoattractant in vitro, and colocalizes to B cell zones in lymph nodes in vivo. Eur J Immunol 30:2808–2814. doi:. [DOI] [PubMed] [Google Scholar]
- 34.Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, Lund FE. 2012. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat Immunol 13:681–690. doi: 10.1038/ni.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horiuchi M, Furuoka H, Kitamura N, Shinagawa M. 2006. Alymphoplasia mice are resistant to prion infection via oral route. Jpn J Vet Res 53:149–157. [PubMed] [Google Scholar]
- 36.Förster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. 1996. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 87:1037–1047. doi: 10.1016/S0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 37.Prinz M, Heikenwalder M, Junt T, Schwarz P, Glatzel M, Heppner FL, Fu Y-X, Lipp M, Aguzzi A. 2003. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 425:957–962. doi: 10.1038/nature02072. [DOI] [PubMed] [Google Scholar]
- 38.Caton ML, Smith-Raska MR, Reizis B. 2007. Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J Exp Med 204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, van Praet J, Branco-Madeira F, Janssens S, Reizis B, Elewaut D, Beyaert R, Hammad H, Lambrecht BN. 2011. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 35:82–96. doi: 10.1016/j.immuni.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Ihara S, Hirata Y, Serizawa T, Suzuki N, Sakitani K, Kinoshita H, Hayakawa Y, Nakagawa H, Ijichi H, Tateishi K, Koike K. 2016. TGF-β signaling in dendritic cells governs colonic homeostasis by controlling epithelial differentiation and the lumenal microbiota. J Immunol 196:4603–4613. doi: 10.4049/jimmunol.1502548. [DOI] [PubMed] [Google Scholar]
- 41.Van den Broek W, Derore A, Simoens P. 2006. Anatomy and nomenclature of murine lymph nodes: descriptive study and nomenclatory standardization in BALB/cAnNCrl mice. J Immunol Met 312:12–19. doi: 10.1016/j.jim.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 42.McCulloch L, Brown KL, Bradford BM, Hopkins J, Bailey M, Rajewsky K, Manson JC, Mabbott NA. 2011. Follicular dendritic cell-specific prion protein (PrPC) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog 7:e1002402. doi: 10.1371/journal.ppat.1002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanaya T, Hase K, Takahashi D, Fukuda S, Hoshino K, Sasaki I, Hemmi H, Knoop KA, Kumar N, Sato M, Katsuno T, Yokosuka O, Toyooka K, Nakai K, Sakamoto A, Kitahara Y, Jinnohara T, McSorley SJ, Kaisho T, Williams IR, Ohno H. 2012. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat Immunol 13:729–736. doi: 10.1038/ni.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, Yabashi A, Waguri S, Nakato G, Kimura S, Murakami T, Iimura M, Hamura K, Fukuoka S-I, Lowe AW, Itoh K, Kiyono H, Ohno H. 2009. Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune responses. Nature 462:226–231. doi: 10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- 45.Donaldson DS, Sehgal A, Rios D, Williams IR, Mabbott NA. 2016. Increased abundance of M cells in the gut epithelium dramatically enhances oral prion disease susceptibility. PLoS Pathog 12:e1006075. doi: 10.1371/journal.ppat.1006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McBride P, Eikelenboom P, Kraal G, Fraser H, Bruce ME. 1992. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J Pathol 168:413–418. doi: 10.1002/path.1711680412. [DOI] [PubMed] [Google Scholar]
- 47.Mabbott NA, Mackay F, Minns F, Bruce ME. 2000. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat Med 6:719–720. doi: 10.1038/77401. [DOI] [PubMed] [Google Scholar]
- 48.Brown KL, Stewart K, Ritchie D, Mabbott NA, Williams A, Fraser H, Morrison WI, Bruce ME. 1999. Scrapie replication in lymphoid tissues depends on PrP-expressing follicular dendritic cells. Nat Med 5:1308–1312. doi: 10.1038/15264. [DOI] [PubMed] [Google Scholar]
- 49.Schulz-Schaeffer WJ, Tschoke S, Kranefuss N, Drose W, Hause-Reitner D, Giese A, Groschup MH, Kretzschmar HA. 2000. The paraffin-embedded tissue blot detects PrPsc early in the incubation time in prion diseases. Am J Pathol 156:51–56. doi: 10.1016/S0002-9440(10)64705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mok SW, Proia RL, Brinkmann V, Mabbott NA. 2012. B cell-specific S1PR1 deficiency blocks prion dissemination between secondary lymphoid organs. J Immunol 188:5032–5040. doi: 10.4049/jimmunol.1200349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt TH, Bannard O, Gray EE, Cyster JG. 2013. CXCR4 promotes B cell egress from Peyer's patches. J Exp Med 210:1099–1107. doi: 10.1084/jem.20122574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heppner FL, Christ AD, Klein MA, Prinz M, Fried M, Kraehenbuhl J-P, Aguzzi A. 2001. Transepithelial prion transport by M cells. Nat Med 7:976–977. doi: 10.1038/nm0901-976. [DOI] [PubMed] [Google Scholar]
- 53.Miyazawa K, Kanaya T, Takakura I, Tanaka S, Hondo T, Watanabe H, Rose MT, Kitazawa H, Yamaguchi T, Katamine S, Nishida N, Aso H. 2010. Transcytosis of murine-adapted bovine spongiform encephalopathy agents in an in vitro bovine M cell model. J Virol 84:12285–12291. doi: 10.1128/JVI.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakhon OS, Ross B, Gusti V, Pham AJ, Vu K, lo DD. 2015. M cell-derived vesicles suggest a unique pathway for transepithelial antigen delivery. Tissues Barriers 3:e1004975. doi: 10.1080/21688370.2015.1004975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kujala P, Raymond C, Romeijn M, Godsave SF, van Kasteren SI HW, Prusiner SB, Mabbott NA, Peters PJ. 2011. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog 7:e1002449. doi: 10.1371/journal.ppat.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aucouturier P, Geissmann F, Damotte D, Saborio GP, Meeker HC, Kascsak R, Kascsak R, Carp RI, Wisniewski T. 2001. Infected splenic dendritic cells are sufficient for prion transmission to the CNS in mouse scrapie. J Clin Invest 108:703–708. doi: 10.1172/JCI200113155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chanouard N, de Chaumont F, Martino A, Enninga J, Olivio-Marin J-C, Männel D, Zurzolo C. 2009. Prions hijack tunneling nanotubes for intercellular spread. Nat Cell Biol 11:328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 58.Langevin C, Gousset K, Costanzo M, Richard-Le Goff O, Zurzolo C. 2010. Characterization of the role of dendritic cells in prion transfer to primary neurons. Biochem J 431:189–198. doi: 10.1042/BJ20100698. [DOI] [PubMed] [Google Scholar]
- 59.Iwasaki A, Kelsalla BA. 2000. Localization of distinct Peyer's patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3, MIP-3β, and secondary lymphoid organ chemokine. J Exp Med 191:1381–1394. doi: 10.1084/jem.191.8.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castro-Seoane R, Hummerich H, Sweeting T, Tattum MH, Lineham JM, Fernandez de Marco M, Brandner S, Collinge J, Klöhn PC. 2012. Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog 8:e1002538. doi: 10.1371/journal.ppat.1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yrlid U, Cerovic V, Milling S, Jenkins CD, Zhang J, Crocker PR, Klavinskis LS, MacPherson GG. 2006. Plasmacytoid dendritic cells do not migrate in intestinal or hepatic lymph. J Immunol 177:6115–6121. doi: 10.4049/jimmunol.177.9.6115. [DOI] [PubMed] [Google Scholar]
- 62.Burthem J, Urban B, Pain A, Roberts DJ. 2001. The normal cellular prion protein is strongly expressed by myeloid dendritic cells. Blood 98:3733–3738. doi: 10.1182/blood.V98.13.3733. [DOI] [PubMed] [Google Scholar]
- 63.Cordier-Dirikoc S, Zsürger N, Cazareth J, Ménard B, Chabry J. 2008. Expression profiles of prion and doppel proteins and of their receptors in mouse splenocytes. Eur J Immunol 38:1–11. doi: 10.1002/eji.200790062. [DOI] [PubMed] [Google Scholar]
- 64.Miyazawa K, Kanaya T, Tanaka S, Takakura I, Watanabe K, Ohwada S, Kitazawa H, Rose MT, Sakaguchi S, Katamine S, Yamaguchi T, Aso H. 2007. Immunohistochemical characterization of cell types expressing the cellular prion protein in the small intestine of cattle and mice. Histochem Cell Biol 127:291–301. doi: 10.1007/s00418-006-0250-x. [DOI] [PubMed] [Google Scholar]
- 65.Klein MA, Frigg R, Raeber AJ, Flechsig E, Hegyi I, Zinkernagel RM, Weissmann C, Aguzzi A. 1998. PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat Med 4:1429–1433. doi: 10.1038/4022. [DOI] [PubMed] [Google Scholar]
- 66.Mohan J, Brown KL, Farquhar CF, Bruce ME, Mabbott NA. 2004. Scrapie transmission following exposure through the skin is dependent on follicular dendritic cells in lymphoid tissues. J Dermatol Sci 35:101–111. doi: 10.1016/j.jdermsci.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 67.Loeuillet C, Lemaire-Vielle C, Naquet P, Cesbron-Delauw M-F, Gagnon J, Cesbron J-Y. 2010. Prion replication in the hematopoietic compartment is not required for neuroinvasion in scrapie mouse model. PLoS One 5:e13166. doi: 10.1371/journal.pone.0013166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, Carroll MC, Verbeek JS, Botto M, Walport MJ, Molina H, Kalinke U, Acha-Orbea H, Aguzzi A. 2001. Complement facilitates early prion pathogenesis. Nat Med 7:488–492. doi: 10.1038/86567. [DOI] [PubMed] [Google Scholar]
- 69.Zabel MD, Heikenwalder M, Prinz M, Arright I, Schwarz P, Kranich J, Von Teichman A, Haas KM, Zeller N, Tedder TF, Weis JH, Aguzzi A. 2007. Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J Immunol 179:6144–6152. doi: 10.4049/jimmunol.179.9.6144. [DOI] [PubMed] [Google Scholar]
- 70.Michel B, Ferguson A, Johnson T, Bender H, Meyerett-Reid C, Pulford B, von Teichman A, Seelig D, Weiss JH, Telling GC, Aguzzi A, Zabel MD. 2012. Genetic depletion of complement receptors CD21/35 prevents terminal prion disease in a mouse model of chronic wasting disease. J Immunol 189:4520–4527. doi: 10.4049/jimmunol.1201579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Michel B, Ferguson A, Johnson T, Bender H, Meyerett-Reid C, Wycoff AC, Pulford B, Telling GC, Zabel MD. 2013. Complement protein C3 exacerbates prion disease in a mouse model of chronic wasting disease. Int Immunol 25:697–702. doi: 10.1093/intimm/dxt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flores-Lagnarica A, Sebti Y, Mitchell DA, Sim RB, MacPherson GG. 2009. Scrapie pathogenesis: the role of complement C1q in scrapie agent uptake by conventional dendritic cells. J Immunol 182:1305–1313. doi: 10.4049/jimmunol.182.3.1305. [DOI] [PubMed] [Google Scholar]
- 73.Michel B, Meyerett-Reid C, Johnson T, Ferguson A, Wycoff C, Pulford B, Bender H, Avery A, Telling G, Dow S, Zabel MD. 2012. Incunabular immunological events in prion trafficking. Sci Rep 2:440. doi: 10.1038/srep00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bradford BM, Brown KL, Mabbott NA. 2016. Prion pathogenesis is unaltered following down-regulation of SIGN-R1. Virology 497:337–345. doi: 10.1016/j.virol.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phan TG, Grigorova I, Okada T, Cyster JG. 2007. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol 8:992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- 76.Carrasco YR, Batista FD. 2007. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 27:1–12. doi: 10.1016/j.immuni.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 77.Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. 2009. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol 10:786–793. doi: 10.1038/ni.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cinamon G, Zachariah MA, Lam OM, Foss FW Jr, Cyster JG. 2008. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol 9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K, Oddos S, Eissmann P, Brodsky FM, Hopkins C, Onfelt B, Sattentau Q, Davis DM. 2008. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol 10:211–219. doi: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- 80.Xu W, Santini PA, Sullivan JS, He B, Shan M, Ball SC, Dyer WB, Ketas TJ, Chadburn A, Cohen-Gould L, Knowles DM, Chiu A, Sanders RW, Chen K, Cerutti A. 2009. HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic conduits and intestinal B cells via long-range intercellular conduits. Nat Immunol 10:1008–1017. doi: 10.1038/ni.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu S, Victoria GS, Marzo L, Ghosh R, Zurzolo C. 2015. Prion aggregates transfer through tunneling nanotubes in endocytic vesicles. Prion 9:125–135. doi: 10.1080/19336896.2015.1025189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tanaka Y, Sadaike T, Inoshima Y, Ishiguro N. 2012. Characterisation of PrPSc transmission from immune cells to neuronal cells. Cell Immunol 279:145–150. doi: 10.1016/j.cellimm.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 83.Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G. 2004. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 101:9683–9688. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gossner A, Hunter N, Hopkins J. 2005. Role of lymph-borne cells in the early stages of scrapie agent dissemination from the skin. Vet Immunol Immunopathol 109:267–278. doi: 10.1016/j.vetimm.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 85.Kimberlin RH, Walker CA. 1989. Pathogenesis of scrapie in mice after intragastric infection. Virus Res 12:213–220. doi: 10.1016/0168-1702(89)90040-3. [DOI] [PubMed] [Google Scholar]
- 86.Bradford BM, Sester D, Hume DA, Mabbott NA. 2011. Defining the anatomical localisation of subsets of the murine mononuclear phagocyte system using integrin alpha X (Itgax, CD11c) and colony stimulating factor 1 receptor (Csf1r, CD115) expression fails to discriminate dendritic cells from macrophages. Immunobiology 216:1228–1237. doi: 10.1016/j.imbio.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 87.Hume DA, Mabbott N, Raza S, Freeman TC. 2013. Can DCs be distinguished from macrophages by molecular signatures. Nat Immunol 14:187–189. doi: 10.1038/ni.2516. [DOI] [PubMed] [Google Scholar]
- 88.Meredith MM, Liu K, Kamphorst AO, Idoyaga J, Yamane A, Guermonprez P, Rihn S, Yoa KH, Silvia IT, Oliviera TY, Skokos D, Casellas R, Nussenzweig MC. 2012. Zinc finger transcription factor zDC is a negative regulator required to prevent activation of classical dendritic cells in the steady state. J Exp Med 209:1583–1593. doi: 10.1084/jem.20121003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Satpathy AT, Wumesh KC, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM. 2012. Zbt46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 209:1135–1152. doi: 10.1084/jem.20120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Loschko J, Rieke GJ, Schreiber HA, Meredith MM, Yao K-H, Guermonprez P, Nussenzweig A. 2016. Inducible targeting of cDC and their subsets in vivo. J Immunol Met 434:32–38. doi: 10.1016/j.jim.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl J-P, Ricciardi-Castagnoli P. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 92.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, et al. 2005. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 93.Vallon-Eberhard A, Landsman L, Yogev N, Verrier B, Jung S. 2006. Transepithelial pathogen uptake into the small intestinal lamina propria. J Immunol 176:2465–2469. doi: 10.4049/jimmunol.176.4.2465. [DOI] [PubMed] [Google Scholar]
- 94.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 95.Saeki H, Moore AM, Brown MJ, Hwang ST. 1999. Cutting Edge: Secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol 162:2472–2475. [PubMed] [Google Scholar]
- 96.Brown KL, Wathne GJ, Sales J, Bruce ME, Mabbott NA. 2009. The effects of host age on follicular dendritic cell status dramatically impair scrapie agent neuroinvasion in aged mice. J Immunol 183:5199–5207. doi: 10.4049/jimmunol.0802695. [DOI] [PubMed] [Google Scholar]
- 97.Fraser H, Dickinson AG. 1968. The sequential development of the brain lesions of scrapie in three strains of mice. J Comp Pathol 78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- 98.Farquhar CF, Somerville RA, Ritchie LA. 1989. Postmortem immunodiagnosis of scrapie and bovine spongiform encephalopathy. J Virol Met 24:215–222. doi: 10.1016/0166-0934(89)90023-2. [DOI] [PubMed] [Google Scholar]
- 99.Inman CF, Rees LEN, Barker E, Haverson K, Stokes CR, Bailey M. 2005. Validation of computer-assisted, pixel-based analysis of multiple-colour immunofluorescence histology. J Immunol Met 302:156–167. doi: 10.1016/j.jim.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 100.Yin S, Pham N, Yu S, Li C, Wong P, Chang B, Kang S-C, Biasini E, Tien P, Harris DA, Sy M-S. 2007. Human prion proteins with pathogenic mutations share common conformational changes resulting in enhanced binding to glycosaminoglycans. Proc Natl Acad Sci U S A 104:7546–7551. doi: 10.1073/pnas.0610827104. [DOI] [PMC free article] [PubMed] [Google Scholar]