

ABSTRACT
Poxvirus virion biogenesis is a complex, multistep process, starting with the formation of crescent-shaped viral membranes, followed by their enclosure of the viral core to form spherical immature virions. Crescent formation requires a group of proteins that are highly conserved among poxviruses, including A6 and A11 of vaccinia virus (VACV). To gain a better understanding of the molecular function of A6, we established a HeLa cell line that inducibly expressed VACV-A6, which allowed us to construct VACV mutants with an A6 deletion or mutation. As expected, the A6 deletion mutant of VACV failed to replicate in noncomplementing cell lines with defects in crescent formation and A11 localization. Surprisingly, a VACV mutant that had A6 replaced with a close ortholog from the Yaba-like disease virus YLDV-97 also failed to replicate. This mutant, however, developed crescents and had normal A11 localization despite failing to form immature virions. Limited proteolysis of the recombinant A6 protein identified an N domain and a C domain of approximately 121 and 251 residues, respectively. Various chimeras of VACV-A6 and YLDV-97 were constructed, but only one that precisely combined the N domain of VACV-A6 and the C domain of YLDV-97 supported VACV replication albeit at a reduced efficiency. Our results show that VACV-A6 has a two-domain architecture and functions in both crescent formation and its enclosure to form immature virions. While a cognate N domain is not required for crescent formation, it is required for virion formation, suggesting that interactions of the N domain with cognate viral proteins may be critical for virion assembly.
IMPORTANCE Poxviruses are unique among enveloped viruses in that they acquire their primary envelope not through budding from cellular membranes but by forming and extending crescent membranes. The crescents are highly unusual, open-ended membranes, and their origin and biogenesis have perplexed virologists for decades. A group of five viral proteins were recently identified as being essential for crescent formation, including the A6 protein of vaccinia virus. It is thus important to understand the structure and function of A6 in order to solve the long-standing mystery of poxvirus membrane biogenesis. Here, we established an experimental system that allowed the genetic manipulation of the essential A6L gene. By studying A6 mutant viruses, we found that A6 plays an essential role not only in the formation of crescents but also in their subsequent enclosure to form immature virions. We defined the domain architecture of A6 and suggested that one of its two domains cooperates with cognate viral proteins.
KEYWORDS: poxvirus, vaccinia virus, virion morphogenesis
INTRODUCTION
Poxviruses are a large family of complex DNA viruses that replicate entirely in the cytoplasm of infected cells (1). Eight genera of poxviruses infect a variety of vertebrate hosts, causing human or animal diseases. Among them, the orthopoxvirus genus has the greatest impact on human health, as it includes variola virus, which caused smallpox, and vaccinia virus (VACV), which serves as a smallpox vaccine. Yaba-like disease virus (YLDV) belongs to the yatapoxvirus genus, which causes mild febrile illness in monkeys and humans (2). The poxvirus genome has around 200 open reading frames (ORFs), approximately 90 of which are conserved in all vertebrate poxviruses (3). The conserved ORFs encode proteins that play essential roles in virus entry, viral gene transcription, genome replication, and virion morphogenesis (3).
Poxvirus virion morphogenesis is a complex process involving a series of intermediate steps discernible by electron microscopy (reviewed in reference 4). In cytoplasmic “viral factory” areas, electron-dense viroplasms, consisting of viral core proteins, appear first. At the periphery of the viroplasms, crescent-shaped viral membranes develop next. The crescents are highly unusual, open-ended membranes (5, 6). The extension of the crescent membranes leads to their enclosure of the viral core, generating spherical immature virions (IVs). IVs undergo additional transformations, including proteolytic processing of the core proteins, to become infectious mature virions (MVs).
The origin and biogenesis of the crescent membranes are among the least-understood aspects of poxvirus biology. Recent studies suggest that crescents may derive from the endoplasmic reticulum (ER) (5, 7). However, studies with brefeldin A (8) and dominant negative Sar1 (9) indicate that transport from the Golgi membrane to the ER and from the ER to post-ER compartments is not necessary for this process. Genetic studies with VACV revealed a number of viral proteins that are essential for crescent formation, including A11 (10), H7 (11), L2 (12), A6 (13), and A30.5 (14). These VACV proteins, collectively termed viral membrane assembly proteins (VMAPs) (reviewed in reference 15), are conserved in all vertebrate poxviruses. VACV mutants deficient in VMAP have a similar phenotype of arresting virion assembly prior to the development of normal crescent membranes.
Very little is known about the structure and function of the VMAPs. H7 is the only VMAP whose structure has been determined (24). Structure-function analysis of H7 showed that it binds phosphoinositides and that binding is essential for poxvirus membrane biogenesis (24). A6, the focus of the present study, localizes in the cytoplasm of infected cells and is packaged in the virion core to a minor extent (17). A VACV mutant that repressed A6 expression was defective at crescent membrane formation, with large masses of viral core proteins accumulating in the viral factory and many viral membrane proteins accumulating in the ER and the ER-Golgi intermediate compartment (13). A11, which specifically associates with viral membranes in viral factories during normal viral replication (18, 19), mislocalized to the cytoplasm when A6 expression was repressed (18).
To gain some understanding of the molecular basis for A6 function, we established an experimental system that allowed us to construct VACVs with an A6 deletion or mutation. The analysis of these mutants showed that the function of A6 is also required after the crescent membranes are formed. We defined the A6 domain architecture and suggested that its N domain is involved in cooperation with other VACV proteins for virion membrane assembly.
RESULTS
Establishment of a HeLa cell line that inducibly expresses A6.
To allow genetic manipulation of the essential A6L gene in VACV, we first set out to establish a complementing cell line for VACV defective in A6. We initially tried to make a cell line that constitutively expressed A6, but our attempts failed. Reasoning that constitutive A6 expression may be toxic to the cells, we then tried to establish a cell line that inducibly expressed A6. We first made a HeLa cell line that stably expressed the tetracycline (Tet) repressor (TetR) from the transposon Tn10. We then succeeded in stably incorporating a plasmid containing an A6 ORF under the control of a Tet-regulated promoter into the HeLa cell line. The final HeLa cell line (referred to here as HeLa-iA6) indeed expressed a significant amount of the A6 protein only in the presence of Tet, and the level was similar to that of VACV-expressed A6 in regular HeLa cells (Fig. 1A). In HeLa-iA6 cells, A6 localized throughout the cytoplasm (Fig. 1B), similar to the A6 localization in VACV-infected cells (17). HeLa-iA6 cells could support plaque formation for an A6-inducible VACV, iA6 (13), under the conditions that repressed the expression of VACV-encoded A6 (data not shown), suggesting that HeLa-iA6 cells could complement the replication of VACV defective in A6. The induction of A6 expression in HeLa-iA6 cells for a few days did not have any perceivable negative effect on cell growth or morphology (data not shown).
FIG 1.

Characterization of a HeLa cell line that inducibly expresses the VACV-A6 protein. (A) Western blot analysis of lysates from a HeLa cell line that inducibly expresses VACV-A6 (HeLa-iA6) and from regular HeLa cells infected with VACV WR. HeLa-iA6 cells were grown in either the presence (+Tet) or the absence (−Tet) of 100 ng/ml Tet for 12 h. Regular HeLa cells were infected with VACV (+WR) at an MOI of 5 PFU/cell for 12 h. Anti-A6 MAb 10F1 (top) and anti-Hsp70 (bottom) were used as the primary antibodies. (B) Immunofluorescence analysis of HeLa-iA6 cells that were grown in the presence of Tet for 12 h. The cells were permeabilized and stained with the DNA dye (DAPI) (left) and anti-A6 MAb 10F1 (A6) (right).
Construction of VACVs with an A6 deletion or mutation.
We then used HeLa-iA6 cells to propagate viruses that were derived from homologous recombination between wild-type (WT) VACV WR and a plasmid containing an A6-flanking sequence and Escherichia coli β-glucuronidase (GUS) (Fig. 2A). Viruses that expressed GUS were plaque purified on HeLa-iA6 cells in the presence of Tet (Fig. 2B). Recombinant viruses that had GUS in place of the A6 ORF were isolated and confirmed by PCR analysis of the viral genome and Western blotting for A6 (data not shown). The recombinant viruses (referred to here as vΔA6) replicated in HeLa-iA6 cells only in the presence of Tet, increasing the titer more than 50-fold after 48 h of infection (Fig. 2C).
FIG 2.
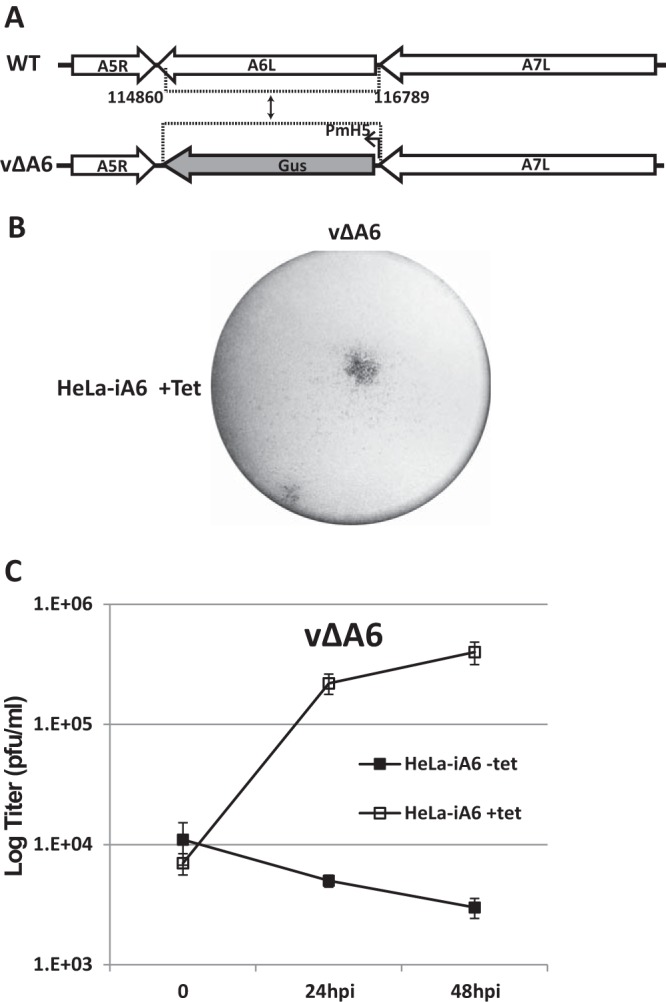
Generation and characterization of an A6 deletion mutant of VACV (vΔA6). (A) Schematic representation of the A6L locus of WT VACV WR and vΔA6. The A6L ORF in WT VACV WR was replaced with the GUS ORF regulated by the modified VACV H5 promoter (PmH5) through homologous recombination, and the resulting VACV vΔA6 mutant was isolated from HeLa-iA6 cells. (B) Plaque morphology of vΔA6 on HeLa-iA6 cells that were grown in the presence of Tet. The cells were stained with X-Gluc (5-bromo-4-chloro-3-indolyl-β-d-glucuronide) after infection with the viruses for 48 h. The original blue staining is shown in black. (C) Growth curve of vΔA6 in HeLa-iA6 cells in the presence or absence of Tet. HeLa-iA6 cells were infected with vΔA6 at an MOI of 0.5 PFU/cell. For infection with Tet, Tet was added to the cell culture medium 12 h before infection and maintained during infection. After 0, 24, and 48 hpi, the cells were harvested, and virus titers under both conditions were determined by a plaque assay on HeLa-iA6 cells that were grown with Tet-containing medium.
We used vΔA6 as the parental virus to construct VACVs with various mutations in A6 (Fig. 3A). The mutants were generated through homologous recombination with a plasmid containing the mutant allele and green fluorescent protein (GFP), and the recombinants were plaque purified on HeLa-iA6 cells. All the mutants and their phenotypes in noncomplementing cells are summarized together in Fig. 3B and C and described individually below.
FIG 3.
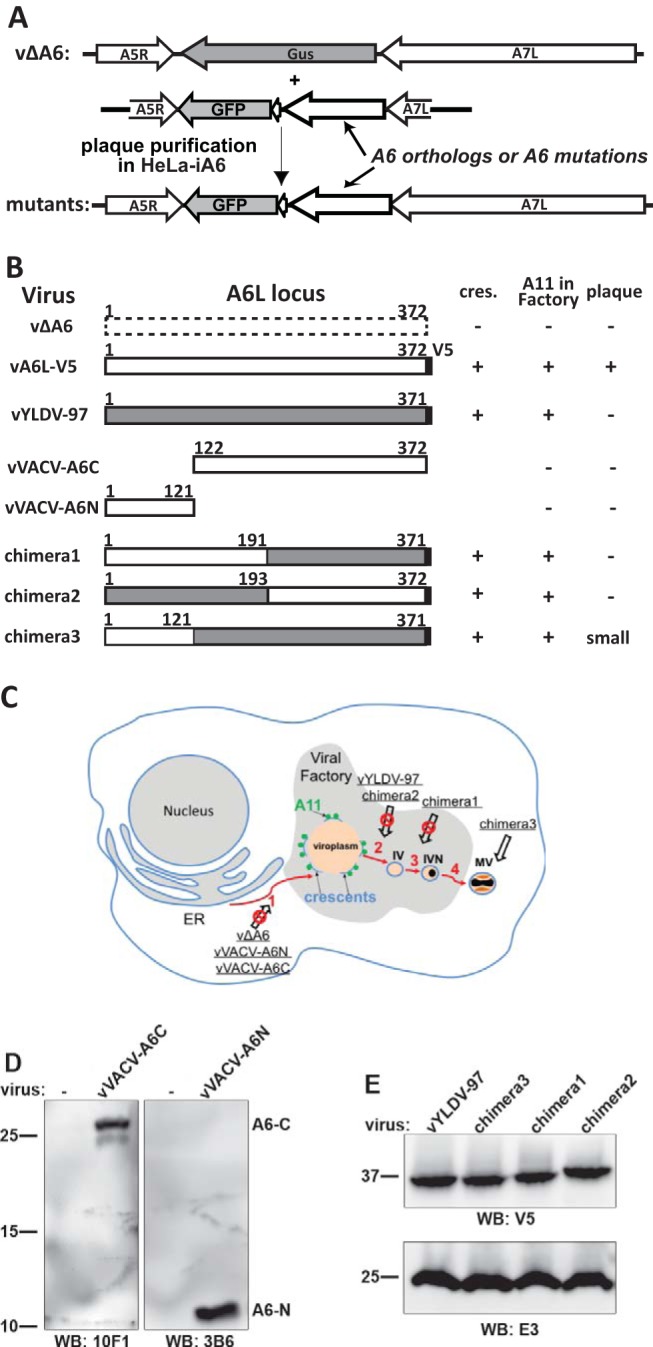
Summary of A6 mutant generation and phenotypic characterization. (A) Experimental scheme for making A6 mutant VACVs. Homologous recombination of vΔA6 with a plasmid containing orthologs or mutations of A6 and the GFP gene resulted in recombinant viruses that expressed an A6 variant under the control of the A6L promoter. The recombinant viruses were plaque purified on HeLa-iA6 cells. (B) Summary of A6 mutants and their phenotypes in noncomplementing cells. vA6L-V5, a VACV expressing A6 with a C-terminal V5 tag, was reported previously (17). VACV-A6, YLDV-97, and the V5 tag are represented by white, gray, and black boxes, respectively. The amino acid residue numbers are indicated. − and + indicate the phenotypes of the mutants in noncomplementing cells, including TEM detection of crescents (cres.) in BS-C-1 cells (Fig. 4), IFA of A11 localization in BHK cells (Fig. 5 and 7), and plaque formation on BS-C-1 cells (Fig. 8). (C) Summary of the phenotypes of A6 mutants in the context of a VACV morphogenesis model. The red arrows indicate the sequential steps of virion morphogenesis. (1) The crescent membranes (light blue lines) develop at the periphery of viroplasms (orange). A11 proteins (green spheres) associate with viral membranes around the viroplasms. (2 and 3) Crescents enclose viroplasms to become immature virions (IV) and immature virions with DNA-containing nucleoids (IVN). (4) immature virions with DNA-containing nucleoids mature into infectious mature virions (MV). The A6 mutants (underlined) and the step at which their virion assembly was arrested (crossed-out arrow) are indicated in the model. (D and E) Western blot (WB) analysis of A6 variants expressed by the mutant VACVs. HeLa cells were infected with the indicated mutants for 12 h, and the cell lysates were analyzed by Western blotting with the indicated antibodies. The sizes of the molecular mass markers (in kilodaltons) are shown.
A close A6 ortholog from YLDV failed to substitute for A6 function in VACV.
Since A6 has no obvious motif to guide mutation analysis, we decided to use A6 orthologs from other poxviruses as “mutagenesis” already performed by nature and to test their functionality in VACV replication. A6 is highly conserved among vertebrate poxviruses (17), so all A6 orthologs are expected to have similar structures and to perform the same function for their respective virus. However, divergent evolution, while preserving important interactions among the cognate viral proteins, may have rendered noncognate viral proteins not compatible for interactions. ORF97 of YLDV represents one of the closest A6 orthologs outside the orthopoxvirus genus, with nearly 60% amino acid identity. We constructed a VACV mutant that had A6L replaced with YLDV-97. The mutant, vYLDV-97, expressed YLDV-97 with a C-terminal V5 tag under the control of the natural A6L promoter. A C-terminal V5 tag was previously shown not to affect the function of A6 in VACV replication (17). vYLDV-97 replicated in HeLa-iA6 cells in the presence of Tet, but it failed to replicate in noncomplementing cell lines such as HeLa and BS-C-1 cells (see below), suggesting that some important interactions among VACV proteins are lost by replacing A6 with YLDV-97.
Transmission electron microscopy (TEM) and immunofluorescence analysis (IFA) were used to examine the defect of vYLDV-97 in comparison to vΔA6. TEM of vΔA6-infected BS-C-1 cells (Fig. 4B) showed very large, electron-dense viroplasms, which were aggregates of viral core proteins (10). There were no crescents around the viroplasms. Thus, similarly to iA6 VACV under nonpermissive conditions (13), vΔA6 has a defect in crescent formation. The phenotype of vΔA6, however, is more stringent than that of iA6, as iA6 formed short crescents around some viroplasms (13). In contrast to vΔA6, vYLDV-97-infected cells showed crescents around all viroplasms (Fig. 4C), and the viroplasms were visibly smaller than those in vΔA6-infected cells. Occasionally, some spherical immature virions were also observed (data not shown).
FIG 4.

TEM of BS-C-1 cells infected with VACV. The cells were infected with WT VACV WR (A), vΔA6 (B), vYLDV-97 (C), and chimeras 1 to 3 (D and E) at an MOI of 1 PFU per cell. Twenty-four hours after infection, the cells were fixed and prepared for TEM. C, crescent; V, viroplasm; IV, immature virion; IVN, IV with nucleoid.
vYLDV-97 expressed GFP, which was previously shown to accumulate in viroplasms and could be used as a marker for viroplasms in IFA (11, 13). IFA of infected baby hamster kidney (BHK) cells confirmed that the viroplasms in vYLDV-97-infected cells (GFP-positive granules in Fig. 5A to C) were more numerous and smaller than those in vΔA6-infected cells (empty holes in DNA factories in Fig. 5D). More importantly, in vYLDV-97-infected cells, A11 proteins localized exclusively in viral factories around the viroplasms, which were also surrounded by viral membrane proteins such as A13 (Fig. 5A and C). In contrast, A11 proteins localized throughout the cytoplasm of vΔA6-infected cells (Fig. 5D). Previously, it was shown that A11 specifically associated with viral membranes (18, 19), and this association required A6 function (18). V5-tagged YLDV-97 localized throughout the cells, but some of the proteins accumulated around the viroplasms (Fig. 5B). Altogether, the data showed that vYLDV-97 progressed further in virion assembly than did vΔA6, suggesting that YLDV-97 is functional at least for the formation of the initial crescent membranes but is defective at a later step. Since the localization of A11 clearly distinguishes the phenotypes of vYLDV-97 and vΔA6, it will be used as a more convenient marker for the progression of virion assembly in subsequent experiments.
FIG 5.

IFA of BHK cells infected with VACV. BHK cells were infected with vYLDV-97 (A to C) or vΔA6 (D and E) at an MOI of 0.5 PFU/cell. After 8 h, the infected cells were fixed, permeabilized, and stained with MAbs for A13 (11F7) (29), A11 (10G11) (18), or the V5 epitope, followed by DAPI and goat anti-mouse IgG coupled to Cy3. vYLDV-97 but not vΔA6 also expressed GFP. The fluorescence signal from DAPI is shown in white in the first column. Note the difference in A11 localization between panels A and D. F, viral factory.
A6 has a two-domain architecture.
In an effort to pinpoint the region of YLDV-97 that renders it not fully compatible for VACV virion assembly, we created different chimeras of VACV-A6 and YLDV-97 and tested their functionality in VACV replication. Our initial strategy of simply dividing the protein sequences in halves and making chimeras of different halves did not result in any viruses that could form plaques on noncomplementing cells. Therefore, we probed the A6 domain architecture by subjecting the purified recombinant A6 protein to limited proteolysis, which is a well-established technique for defining protein domains (20). After overnight incubation of the ∼43-kDa full-length A6 protein with either trypsin or chymotrypsin, an ∼28-kDa protein fragment remained stable (Fig. 6A). N-terminal peptide sequencing and mass spectrometry analysis revealed that the stable fragment from trypsin or chymotrypsin digestion contained amino acids (aa) 124 to 372 or aa 122 to 372, respectively. There was also a smaller fragment of <16 kDa after chymotrypsin digestion. Although it was not analyzed further, this fragment likely represents aa 1 to 121, which has a molecular mass of ∼14 kDa. Indeed, both fragments spanning aa 1 to 121 and aa 122 to 372 of A6 were readily expressed and purified in a recombinant form from prokaryotic cells, and the purified proteins were stable (Fig. 6B). We refer to aa 1 to 121 and aa 122 to 372 of A6 as the N and C domains, respectively.
FIG 6.
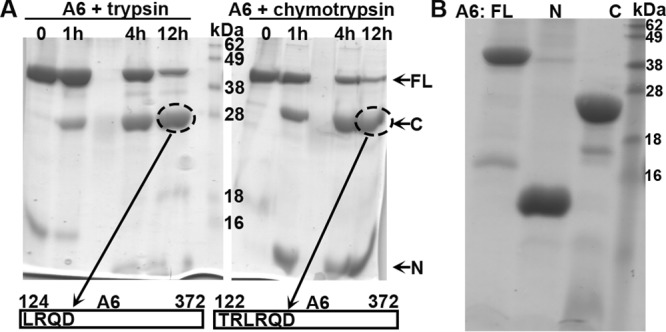
(A) Analysis of A6 domain architecture by limited proteolysis. The purified recombinant A6 protein at 2 mg/ml was mixed with trypsin (left) or chymotrypsin (right) at a 1,000:1 mass ratio and incubated at room temperature. Samples at various time points (indicated) were analyzed by SDS-PAGE. The Coomassie blue-stained gel image is shown in black. Note the stable fragment of about 28 kDa (circled) after overnight proteolysis in both cases. The fragments were excised from the gel and subjected to tandem mass spectrometry and N-terminal peptide sequencing analysis. The N-terminal sequences of the fragments were determined to be LRQD and TRLR, which are the starting sequences for A6 aa 124 to 372 and aa 122 to 372, respectively. (B). Purification of recombinant full-length (FL) A6, the N domain of A6 (N) (aa 1 to 121), and the C domain of A6 (C) (aa 122 to 372). The recombinant A6 proteins were expressed as a SUMO fusion with an N-terminal 6×His tag in E. coli and purified as described in Materials and Methods. The purified A6 proteins with the tag removed were analyzed by SDS-PAGE and stained with Coomassie blue (shown in black).
A cognate A6 N domain is required for virion assembly.
To test whether individual domains of A6 have partial function in virion assembly, we constructed VACV mutants with only the A6 N domain or C domain (Fig. 3). The mutants (vVACV-A6N and vVACV-A6C, respectively) expressed either the N domain or the C domain in infected cells, as detected by Western blotting with two different anti-A6 monoclonal antibodies (MAbs) (Fig. 3D). Both viruses failed to form plaques on noncomplementing cells (data not shown). IFA of infected BHK cells showed assembly defects that were similar for the two mutants, so only the results from vVACV-A6C are shown in Fig. 7. The defects of vVACV-A6N and vVACV-A6C were more similar to those of vΔA6 than to those of vYLDV-97, with the accumulation of larger but fewer viroplasms (GFP positive) and the distribution of A11 proteins throughout the cytoplasm (Fig. 7A). This indicates that neither A6 domain possesses the partial function in viral assembly observed for YLDV-97.
FIG 7.
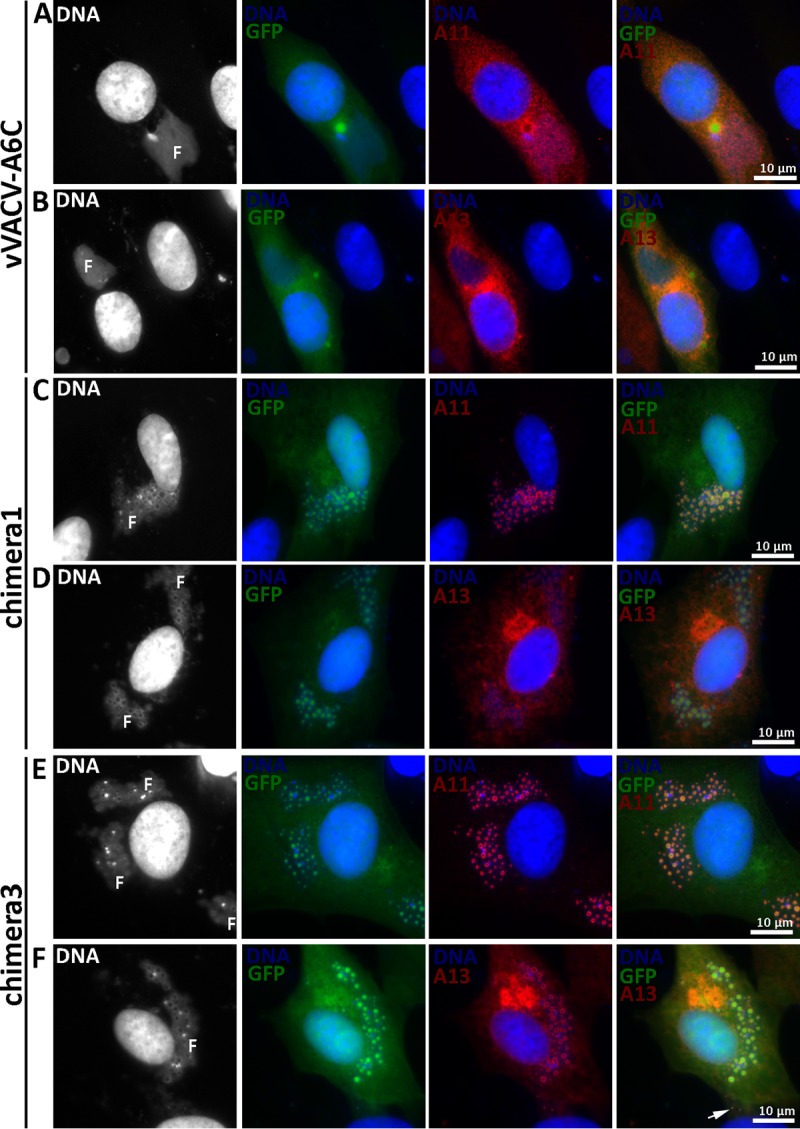
IFA of BHK cells infected with A6 mutants. BHK cells were infected with vVACV-A6C (A and B), chimera 1 (C and D), or chimera 3 (E and F) at an MOI of 0.5 PFU/cell. After 8 h, the infected cells were fixed, permeabilized, and stained with MAbs for A13 (11F7) (29) or A11 (10G11) (18), followed by DAPI and goat anti-mouse IgG coupled to Cy3. The fluorescence signal from DAPI is shown in white in the first column. Note the difference in A11 localizations between panels A and C. F, viral factory.
As stated above, we initially constructed chimeras of different halves of VACV-A6 and YLDV-97 without considering the actual A6 domain architecture (chimeras 1 and 2) (Fig. 3). VACV mutants with these chimeras expressed the chimeric proteins, as detected by Western blotting of the C-terminal epitope tag (Fig. 3E). The mutants failed to replicate in noncomplementing cells (data not shown). IFA of infected BHK cells showed similar phenotypes for the two mutants, so only the results from chimera 1 are shown in Fig. 7. The assembly defects of mutants with chimera 1 and chimera 2 were more similar to those of vYLDV-97 than to those of vΔA6, with the accumulation of many smaller viroplasms (GFP positive) and the accumulation of A11 proteins around the viroplasms in viral factories (Fig. 7C). TEM analysis confirmed that the mutant with chimera 2 had an assembly defect similar to that of vYLDV-97, with crescents surrounding the viroplasms (Fig. 4E). Interestingly, in cells infected with the mutant with chimera 1, immature virions were predominant (IVs in Fig. 4D), indicating that this mutant progressed further in virion assembly than did vYLDV-97.
As chimera 1, which contained the N-terminal half of VACV-A6, functioned better than YLDV-97 and chimera 2 in virion assembly, we next constructed a more precise chimera of the VACV-A6 N domain and the YLDV-97 C domain (chimera 3) (Fig. 3) based on the domain boundary defined by limited proteolysis analysis. The VACV mutant that encoded chimera 3 formed small plaques and increased its titer more than 10-fold in BS-C-1 cells (Fig. 8). IFA of infected BHK cells at 8 h postinfection (hpi) still showed the accumulation of many small viroplasms (GFP positive) in viral factories, but there were some A13-positive, virion-like particles at the periphery of the cells (Fig. 7F, arrow). TEM analysis revealed the presence of MVs (Fig. 4F) in infected cells, in addition to many immature virions. The difference in phenotypes between vYLDV-97, chimera 1, and chimera 3 suggest that the cognate N domain of VACV-A6 worked better than the noncognate N domain of YLDV-97 in a later step of VACV virion assembly after forming the initial crescents.
FIG 8.
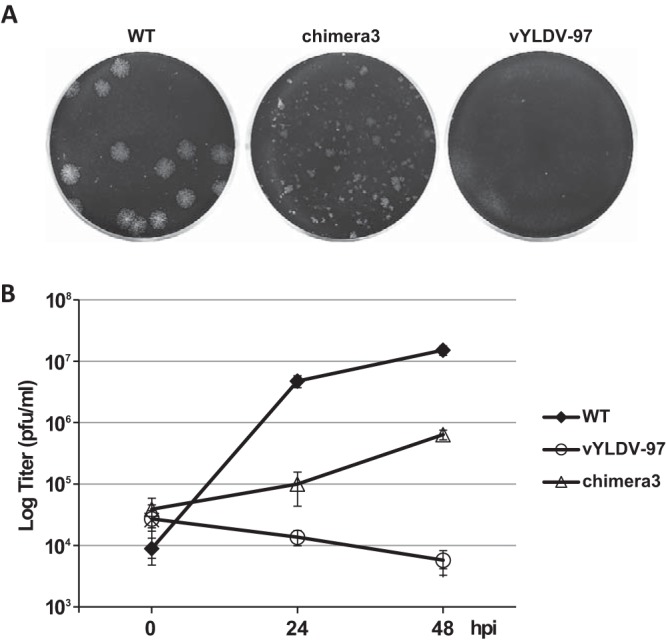
Plaque morphology and growth curves of A6 mutant VACVs. (A) BS-C-1 cells in 12-well plates were infected with the indicated VACVs in semisolid medium for 48 h. The cells were then stained with crystal violet to reveal plaques. (B) BS-C-1 cells in 12-well plates were infected with the indicated viruses at an MOI of 0.5 PFU/cell. Viral titers at 0, 24, and 48 hpi were determined by a plaque assay on HeLa-iA6 cells.
DISCUSSION
Conditional lethal VACV mutants are extremely valuable for studying the function of essential viral genes. However, they often reveal only the earliest step of viral replication that requires the product of the essential viral gene. In the case of A6, previous studies of a conditional lethal mutant that repressed A6 expression showed that A6 deficiency arrested virion assembly at the stage of crescent membrane formation (13). Whether A6 plays a role after crescent formation was unknown. In the present study, we established an experimental system that allowed us to construct VACVs with any desired A6 mutation. The identification of A6 mutants that were normal in crescent formation but defective for viral replication indicates that the A6 function is also required after the formation of crescent membranes. Furthermore, we defined the domain architecture of A6 and suggested that the function of A6 at a later step of virion assembly requires cooperation between the A6 N domain and some other VACV proteins.
The generation of deletion viruses with the aid of appropriate complementing cells has increasingly been used for defining the function of vaccinia virus genes (7, 21–23). We previously established an H7 cell line (23), which was instrumental for our structure-function analysis of H7 (24). While establishing the H7 cell line was relatively straightforward, this was not the case for making the A6 cell line. We were unable to establish cell lines that constitutively express A6, most likely due to the toxicity of A6 to cells. We overcame this problem by using the “Tet-on” system to establish a cell line (HeLa-iA6) that expresses A6 only in the presence of an inducer. Short-term induction of A6 expression did not have any perceivable negative effect on cell growth, giving us ample time to use the cell line for propagating VACVs defective in A6. Prolonged induction of A6 expression, however, changed the cell morphology and slowed the growth of HeLa-iA6 cells (data not shown). The precise effect of A6 expression on cell physiology is currently unclear. This will be an area of further investigation once a better understanding of the molecular function of A6 is achieved.
The characterization of the A6 deletion VACV mutant (vΔA6) not only confirmed many findings from previous studies of an inducible A6 mutant (iA6) but also removed some ambiguity about the A6 function from those previous studies. Similarly to iA6 under nonpermissive conditions, vΔA6 showed an assembly defect characterized by the accumulation of viroplasms and the mislocalization of A11 proteins and many viral membrane proteins. However, while short crescents around some viroplasms in iA6-infected cells were previously observed (13), they were not detected around viroplasms in vΔA6-infected cells (Fig. 4). We believe that the small number of short crescents in iA6-infected cells were due to the small and undetectable amount of A6 under nonpermissive conditions. vΔA6 lacks even a low level of the A6 protein, and its phenotypes demonstrate that A6 is required for the earliest step of crescent formation.
With the complementing cell line and the deletion virus, we were able to introduce precise A6 mutations into VACV to probe the function of A6. Without a structure or obvious motif for A6, we decided to test A6 orthologs from other poxviruses as the products of the mutagenesis experiments performed by nature. With nearly 60% amino acid sequence identities, VACV-A6 and YLDV-97 almost certainly adopt an identical protein fold and perform the same function for their respective viruses. We were therefore surprised to find that YLDV-97 failed to replace the function of A6 in VACV virion assembly. Similarly, we found that the A6 ortholog from myxoma virus also failed to replace the function of A6 in VACV replication (data not shown). This suggests that there are important interactions between A6 and other VACV proteins, which were lost by replacing A6 with the noncognate orthologs. Our previous immunoprecipitation (IP) and mass spectrometry analyses identified only A11 as a weak interacting partner for A6 (18). However, we found that the A6/YLDV-97 chimeric proteins maintain the weak interaction with A11. In addition, unlike vΔA6, vYLDV-97 and the mutants expressing the chimeric proteins had normal A11 localization and formed crescents in infected cells (Fig. 5 and 7), suggesting that YLDV-97 may be defective for a function after crescent formation due to a defect in interactions with unknown binding partners of A6. These interactions were probably too weak or transient to be detected by IP and mass spectrometry in our previous study.
Among the five VMAPs, A6 is the largest, with a molecular mass of 43 kDa. Our limited proteolysis of the purified recombinant A6 protein showed that A6 has a small N domain and a twice-larger C domain. This A6 domain architecture is supported by the successful expression and purification of the A6 N and C domains from prokaryotic cells. It is also supported by the observation that only the precise chimera of the YLDV-97 N domain and the VACV-A6 C domain (chimera 3) was functional for VACV replication, while chimeras of different halves of A6 orthologs were not functional (Fig. 3).
The deletion of either the N or C domain of A6 from VACV resulted in an assembly defect that was similar to that of vΔA6, suggesting that both domains are required for crescent formation. However, vYLDV-97 formed crescents, so the cognate N or C domain is not required for this process. It is possible that, for forming the initial crescents, YLDV-97 is able to maintain the necessary protein interactions or that cognate protein interactions may not be critical. In comparison to vYLDV-97, chimera 3 could be considered a mutant of vYLDV-97 that replaced the N domain of YLDV-97 with that of VACV-A6 (Fig. 3). While vYLDV-97 failed to replicate in noncomplementing cells, chimera 3 replicated with low efficiency, suggesting that a cognate A6 N domain is essential for a subsequent step of virion assembly after the formation of the initial crescents. Since a cognate C domain is not required, the cooperation of A6 with other VACV proteins at this step of virion assembly may be mediated mainly by the N domain. The reason why chimera 3 replicated less efficiently than WT VACV could be that the two A6 domains have to function cooperatively and that this cooperation was reduced by the noncognate C domain. A further understanding of how the two A6 domains function together in poxvirus morphogenesis may have to await a better understanding of the A6 structure. In this respect, we have just solved the crystal structure of the A6 N domain (our unpublished data), which shows a compact, independent fold. We are currently working on determining the crystal structures of the C domain and full-length A6, as we believe that more insight into the A6 molecular function should be gained by comparing the individual domain structures with the full-length structure. The experimental system that we establish here will be valuable for future structure-guided functional studies of A6.
MATERIALS AND METHODS
Cells and antibodies.
BS-C-1 cells were maintained in minimum essential medium with Earle's salts supplemented with 10% fetal bovine serum (FBS). BHK cells and HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. A murine MAb against V5 (Sigma-Aldrich) was purchased from the vendor. Anti-A6 MAb 10F1 was reported previously (18). It was developed from mice immunized with the recombinant A6 protein. For this study, additional anti-A6 MAbs generated from the same immunization were screened again for reactivity against aa 1 to 121 of A6 by Western blotting, and 3B6 was found to be positive in this screen.
Cell line construction.
HeLa cell lines inducibly expressing VACV-A6 (HeLa-iA6) were established in two steps. First, a cell line (HeLa-TetR) that stably expressed the tetracycline repressor was established by the transfection of HeLa cells with pcDNA6/TR (Invitrogen), selection with blasticidin (Invitrogen) at 7.5 μg/ml, and screening by immunoblotting with an anti-TetR antibody (MoBiTec). Subsequently, HeLa-TetR cells were transfected with a plasmid that contained VACV-A6 under the control of a tetracycline-regulated promoter. The plasmid was constructed by inserting the human codon-optimized A6 gene (synthesized commercially by GenScript) between the HindIII and BamHI sites of pcDNA4/TO (Invitrogen). The transfected cells were selected with zeocin (Invitrogen) at 100 μg/ml. Cell clones were treated with 100 ng/ml tetracycline and screened by immunoblotting with the anti-A6 10F1 MAb (18). HeLa-iA6 cells were maintained in DMEM, and Tet was added to the medium a day before the cells were used for infection with VACV.
VACV mutant construction.
VACV with a deletion in A6L (vΔA6) was constructed as follows. 293T cells were infected with the VACV WR strain at a multiplicity of infection (MOI) of 0.1 PFU/cell and transfected with pA5-GUS-A7 (13). Crude viruses from the cells were used to infect HeLa-iA6 cells grown in medium containing 100 ng/ml Tet. GUS-expressing viruses were plaque purified according to a standard protocol (25). After 4 rounds of plaque purification, the recombinant virus was confirmed to be free of WT VACV WR by PCR amplification of the A6L-flanking region and immunoblotting with the anti-A6 10F1 antibody.
A transfer plasmid (pRY57) for putting A6L back into vΔA6 was constructed by inserting the coding sequence for A6 with a C-terminal V5 epitope tag between the A7 and GFP cassettes of the pA5-GFP-A7 plasmid (13). A plasmid (pBO19) for putting the YLDV A6 ortholog into vΔA6 was constructed by replacing the A6L ORF in pRY57 with YLDV-97, which was PCR amplified from genomic DNA of YLDV (ATCC VR-937). Similarly to A6L in pRY57, YLDV-97 in pBO19 was under the control of the VACV A6L promoter and had a C-terminal V5 tag. Plasmids for making various chimeras of VACV-A6 and YLDV-97 were constructed by making chimeras of the pRY57 and pBO19 plasmids. All plasmids were confirmed by Sanger sequencing. The plasmids were transfected into HeLa-iA6 cells that were infected with the vΔA6 virus. Recombinant viruses expressing GFP were isolated after four rounds of plaque purification on HeLa-iA6 cells, which had been grown with medium containing 100 ng/ml of tetracycline.
Electron microscopy.
BS-C-1 cells in 60-mm-diameter dishes were infected with 1 PFU/cell of various VACVs. At 24 hpi, the cells were fixed with 4% formaldehyde and 1% glutaraldehyde for 1 h at 4°C. The cells were then scraped off and prepared for transmission electron microscopy by the Electron Microscopy Core Laboratory at the University of Texas Health Science Center at San Antonio. Thin sections were examined with a JEOL 1230 transmission electron microscope.
Fluorescence microscopy.
BHK or HeLa cells grown on coverslips were infected at 0.5 PFU/cell. At 8 hpi, the cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% saponin (Sigma-Aldrich) for 5 min, blocked with 10% FBS for 60 min, and stained with various antibodies for 1 h and an appropriate secondary antibody for an additional hour. The DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Coverslips were imaged with an Olympus IX-81 fluorescence microscope.
Plaque formation and growth curve analysis of VACV mutants.
BS-C-1 or HeLa-iA6 cells in 12-well plates were incubated with different A6 mutants for 2 h at room temperature. Following adsorption, the cells were washed twice with phosphate-buffered saline (PBS) and moved to a 37°C incubator. For comparison of plaque morphologies, medium containing 0.5% methyl cellulose was added, and plaques were visualized at 48 hpi by staining the cells with crystal violet. For growth curve analysis, infected cells were harvested at 0, 24, and 48 hpi. The viral titers in the cell lysates were determined by plaque assays on HeLa-iA6 cells that had been treated with tetracycline.
Western blot analysis.
Western blot analysis was performed as previously described (26). Briefly, the samples were solubilized in sodium dodecyl sulfate (SDS) sample buffer, resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto nitrocellulose membranes, and blocked with Tris-buffered saline supplemented with 5% nonfat dried milk and 0.05% Tween 20 for 1 h at room temperature. Subsequently, membranes were incubated with antibodies and analyzed with a chemiluminescence reagent (Pierce).
Protein purification.
The full-length N domain (aa 1 to 121) and C domain (aa 122 to 372) of VACV-A6 were cloned into a modified pET vector as a SUMO fusion with an N-terminal 6×His tag. Recombinant A6 was expressed in E. coli and purified by using Ni-nitrilotriacetic acid (NTA) as previously described (27). Briefly, the A6 protein was first purified from the soluble cell lysate as a His-tagged SUMO fusion by using Ni-NTA affinity column. The eluted protein was subsequently subjected to Ulp1 protease cleavage to remove the SUMO moiety and was collected as the flowthrough of a second subtracting Ni-NTA column.
ACKNOWLEDGMENT
This work was supported by a grant from the NIAID to Y.X. (AI079217).
REFERENCES
- 1.Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2946. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Downie AW, Espana C. 1972. Comparison of Tanapox virus and Yaba-like viruses causing epidemic disease in monkeys. J Hyg (Lond) 70:23–32. doi: 10.1017/S0022172400022051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol 77:7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- 5.Krijnse Locker J, Chlanda P, Sachsenheimer T, Brugger B. 2013. Poxvirus membrane biogenesis: rupture not disruption. Cell Microbiol 15:190–199. doi: 10.1111/cmi.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chlanda P, Carbajal MA, Cyrklaff M, Griffiths G, Krijnse-Locker J. 2009. Membrane rupture generates single open membrane sheets during vaccinia virus assembly. Cell Host Microbe 6:81–90. doi: 10.1016/j.chom.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 7.Maruri-Avidal L, Weisberg AS, Bisht H, Moss B. 2013. Analysis of viral membranes formed in cells infected by a vaccinia virus L2-deletion mutant suggests their origin from the endoplasmic reticulum. J Virol 87:1861–1871. doi: 10.1128/JVI.02779-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulaeto D, Grosenbach D, Hruby DE. 1995. Brefeldin A inhibits vaccinia virus envelopment but does not prevent normal processing and localization of the putative envelopment receptor P37. J Gen Virol 76(Part 1):103–111. doi: 10.1099/0022-1317-76-1-103. [DOI] [PubMed] [Google Scholar]
- 9.Husain M, Moss B. 2003. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J Virol 77:11754–11766. doi: 10.1128/JVI.77.21.11754-11766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Resch W, Weisberg AS, Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J Virol 79:6598–6609. doi: 10.1128/JVI.79.11.6598-6609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satheshkumar PS, Weisberg A, Moss B. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J Virol 83:8439–8450. doi: 10.1128/JVI.00877-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maruri-Avidal L, Domi A, Weisberg AS, Moss B. 2011. Participation of vaccinia virus l2 protein in the formation of crescent membranes and immature virions. J Virol 85:2504–2511. doi: 10.1128/JVI.02505-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng X, Embry A, Rose L, Yan B, Xu C, Xiang Y. 2012. Vaccinia virus A6 is essential for virion membrane biogenesis and localization of virion membrane proteins to sites of virion assembly. J Virol 86:5603–5613. doi: 10.1128/JVI.00330-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Direct formation of vaccinia virus membranes from the endoplasmic reticulum in the absence of the newly characterized L2-interacting protein A30.5. J Virol 87:12313–12326. doi: 10.1128/JVI.02137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moss B. 2015. Poxvirus membrane biogenesis. Virology 479–480:619–626. doi: 10.1016/j.virol.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reference deleted.
- 17.Meng X, Embry A, Sochia D, Xiang Y. 2007. Vaccinia virus A6L encodes a virion core protein required for formation of mature virion. J Virol 81:1433–1443. doi: 10.1128/JVI.02206-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, Meng X, Yan B, Rose L, Deng J, Xiang Y. 2012. Vaccinia virus virion membrane biogenesis protein A11 associates with viral membranes in a manner that requires the expression of another membrane biogenesis protein, A6. J Virol 86:11276–11286. doi: 10.1128/JVI.01502-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maruri-Avidal L, Weisberg AS, Moss B. 2013. Association of the vaccinia virus A11 protein with the endoplasmic reticulum and crescent precursors of immature virions. J Virol 87:10195–10206. doi: 10.1128/JVI.01601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong A, Xu X, Edwards AM, Midwest Center for Structural Genomics, Structural Genomics Consortium, Chang C, Chruszcz M, Cuff M, Cymborowski M, Di Leo R, Egorova O, Evdokimova E, Filippova E, Gu J, Guthrie J, Ignatchenko A, Joachimiak A, Klostermann N, Kim Y, Korniyenko Y, Minor W, Que Q, Savchenko A, Skarina T, Tan K, Yakunin A, Yee A, Yim V, Zhang R, Zheng H, Akutsu M, Arrowsmith C, Avvakumov GV, Bochkarev A, Dahlgren LG, Dhe-Paganon S, Dimov S, Dombrovski L, Finerty P Jr, Flodin S, Flores A, Graslund S, Hammerstrom M, Herman MD, Hong BS, Hui R, Johansson I, Liu Y, Nilsson M, Nedyalkova L, et al. . 2007. In situ proteolysis for protein crystallization and structure determination. Nat Methods 4:1019–1021. doi: 10.1038/nmeth1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warren RD, Cotter CA, Moss B. 2012. Reverse genetics analysis of poxvirus intermediate transcription factors. J Virol 86:9514–9519. doi: 10.1128/JVI.06902-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyle KA, Greseth MD, Traktman P. 2015. Genetic confirmation that the H5 protein is required for vaccinia virus DNA replication. J Virol 89:6312–6327. doi: 10.1128/JVI.00445-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng X, Wu X, Yan B, Deng J, Xiang Y. 2013. Analysis of the role of vaccinia virus H7 in virion membrane biogenesis with an H7-deletion mutant. J Virol 87:8247–8253. doi: 10.1128/JVI.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolli S, Meng X, Wu X, Shengjuler D, Cameron CE, Xiang Y, Deng J. 2015. Structure-function analysis of vaccinia virus H7 protein reveals a novel phosphoinositide binding fold essential for poxvirus replication. J Virol 89:2209–2219. doi: 10.1128/JVI.03073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Earl PL, Moss B, Wyatt LS, Carroll MW. 2001. Generation of recombinant vaccinia viruses. Curr Protoc Mol Biol Chapter 16:Unit 16.17. doi: 10.1002/0471142727.mb1617s43. [DOI] [PubMed] [Google Scholar]
- 26.Meng X, Xiang Y. 2006. Vaccinia virus K1L protein supports viral replication in human and rabbit cells through a cell-type-specific set of its ankyrin repeat residues that are distinct from its binding site for ACAP2. Virology 353:220–233. doi: 10.1016/j.virol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 27.Krumm B, Meng X, Li Y, Xiang Y, Deng J. 2008. Structural basis for antagonism of human interleukin 18 by poxvirus interleukin 18-binding protein. Proc Natl Acad Sci U S A 105:20711–20715. doi: 10.1073/pnas.0809086106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reference deleted.
- 29.Xu C, Meng X, Yan B, Crotty S, Deng J, Xiang Y. 2011. An epitope conserved in orthopoxvirus A13 envelope protein is the target of neutralizing and protective antibodies. Virology 418:67–73. doi: 10.1016/j.virol.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]