

ABSTRACT
Measles virus (MeV) is a member of the family Paramixoviridae that causes a highly contagious respiratory disease but has emerged as a promising oncolytic platform. Previous studies of MeV entry focused on the identification of cellular receptors. However, the endocytic and trafficking pathways utilized during MeV entry remain poorly described. The contribution of each endocytic pathway has been examined in cells that express the MeV receptors SLAM (signaling lymphocyte-activating molecule) and PVRL4 (poliovirus receptor-like 4) (nectin-4). Recombinant MeVs expressing either firefly luciferase or green fluorescent protein together with a variety of inhibitors were used. The results showed that MeV uptake was dynamin independent in the Vero.hPVRL4, Vero.hSLAM, and PVRL4-positive MCF7 breast cancer cell lines. However, MeV infection was blocked by 5-(N-ethyl-N-propyl)amiloride (EIPA), the hallmark inhibitor of macropinocytosis, as well as inhibitors of actin polymerization. By using phalloidin staining, MeV entry was shown to induce actin rearrangements and the formation of membrane ruffles accompanied by transient elevated fluid uptake. Small interfering RNA (siRNA) knockdown of p21-activated kinase 1 (PAK1) demonstrated that MeV enters both Vero.hPVRL4 and Vero.hSLAM cells in a PAK1-independent manner using a macropinocytosis-like pathway. In contrast, MeV entry into MCF7 human breast cancer cells relied upon Rac1 and its effector PAK1 through a PVRL4-mediated macropinocytosis pathway. MeV entry into DLD-1 colon and HTB-20 breast cancer cells also appeared to use the same pathway. Overall, these findings provide new insight into the life cycle of MeV, which could lead to therapies that block virus entry or methods that improve the uptake of MeV by cancer cells during oncolytic therapy.
IMPORTANCE In the past decades, measles virus (MeV) has emerged as a promising oncolytic platform. Previous studies concerning MeV entry focused mainly on the identification of putative receptors for MeV. Nectin-4 (PVRL4) was recently identified as the epithelial cell receptor for MeV. However, the specific endocytic and trafficking pathways utilized during MeV infections are poorly documented. In this study, we demonstrated that MeV enters host cells via a dynamin-independent and actin-dependent endocytic pathway. Moreover, we show that MeV gains entry into MCF7, DLD-1, and HTB-20 cancer cells through a PVRL4-mediated macropinocytosis pathway and identified the typical cellular GTPase and kinase involved. Our findings provide new insight into the life cycle of MeV, which may lead to the development of therapies that block the entry of the virus into the host cell or alternatively promote the uptake of oncolytic MeV into cancer cells.
KEYWORDS: MCF7 cell, nectin-4, PAK1, PVRL4, Rac1, breast cancer, endocytosis, macropinocytosis, measles virus, virus entry, morbillivirus, receptor, SLAM, dynamin, p21-activated kinase
INTRODUCTION
Measles virus (MeV) is a member of the family Paramixoviridae of the genus Morbillivirus. It causes a highly contagious disease associated with respiratory symptoms, fever, gastrointestinal distress, and a characteristic rash (1, 2). Measles is often complicated by secondary infections due to MeV-induced immunosuppression in humans (1, 3–7). Neurological complications associated with MeV infection are rare but can arise at late stages of the disease (8–12). Over the past decade, numerous measles outbreaks have occurred throughout the world, causing thousands of deaths despite success in controlling the disease with a safe and effective vaccine (13–18). Currently, no specific treatment for measles is available, although vitamin A is recommended by the World Health Organization (WHO) for populations where the infant mortality rate due to measles is over 1% (19, 20). Retinoic acid is believed to provide resistance against MeV infection by enhancing innate immunity (21). Alternatively, effective measles virus inhibitors have been explored, which target virus-mediated membrane fusion (22, 23) and viral polymerase activity (24), but have yet to be developed for clinical use. However, natural recovery from measles in healthy patients takes about 7 to 10 days following the appearance of the rash, and the individual usually acquires lifelong immunity to the disease.
Measles virus particles are polymorphic, with an average diameter ranging between 50 and 1,000 nm (25). Virions are surrounded by a host cell-derived envelope that contains the viral attachment protein hemagglutinin (H) and the fusion protein (F). The 15-kb negative-sense genomic and positive-sense template RNAs are encapsidated by the nucleocapsid (N) protein. The viral phosphoprotein (P) and large polymerase proteins (L) associate with these viral ribonucleoproteins to form the viral polymerase complex (1, 2).
Entry into the host cell is the first step of the virus life cycle and represents an attractive target for antiviral treatments. In the context of measles infection, MeV was previously believed to enter the host cell through the attachment of MeV H to its cellular receptor, followed by fusion between viral and cellular membranes mediated by MeV F (26–31). However, viruses that fuse with the plasma membrane can often enter the cell by endocytosis as well, depending upon the cell type (32). Interestingly, MeV is able to use different receptors depending on the target cell type. Three major cellular receptors have been identified for measles virus. Signaling lymphocyte activation molecule (SLAM) serves as an immune cell receptor (33–37), and the tumor-associated marker nectin-4 (poliovirus receptor-like 4 [PVRL4]) is an epithelial receptor for morbilliviruses (38–43). Although vaccine and laboratory cell-adapted strains of MeV can use the human membrane cofactor protein (CD46) as a receptor (44, 45), other morbilliviruses, including wild-type MeV (wtMeV), do not use CD46 to enter the host cell (46–49).
Many enveloped viruses hijack the process of endocytosis to infect and enter cells. The normal function of endocytosis ranges from the ingestion of fluid and solutes via pinocytosis to the ingestion of large particles by phagocytosis (50–53). Different viruses enter the cell by a variety of endocytotic mechanisms, including clathrin-mediated endocytosis (CME), macropinocytosis, caveolin-associated uptake (caveola-mediated endocytosis [CavME]), interaction with lipid rafts, direct fusion with the plasma membrane, or other poorly defined pathways (54, 55). Macropinocytosis is a transient, growth factor-induced, actin-dependent endocytotic process that causes rearrangements in the plasma membrane resulting in the internalization of fluids and membranes through the formation of large vacuoles and is used by many viruses to enter the host cell (56, 57). This process is normally growth factor induced and transient and can occur in almost all cell types. Four types of membrane rearrangements or ruffles have been identified, as summarized by Mercer and Helenius (57), including cup-shaped membrane extensions (circular), flat sheet-like folds (lamellipodium-like), large irregular membrane extrusions (blebs), and thin finger-like extensions (filopodial protrusions). Ruffles form in the extracellular space and fold back upon the cell, enclosing particles and fluid near the cell surface to form large, irregularly shaped vacuoles known as macropinosomes (56). Unlike CME and CavME, macropinocytosis is dynamin independent and characterized by vigorous actin reorganization induced by the growth factor-associated activation of kinases and Rho GTPases. p21-activated kinase 1 (PAK1) plays a key role in the process of macropinocytosis by regulating the actin-myosin cytoskeleton and has been identified as a target for the small activated GTPases Rac1 and cdc42 (58). In turn, C-terminal binding protein 1 (CtBP1) is phosphorylated by PAK1, which initiates membrane fission and the stabilization of the emerging macropinosomal vesicle (59, 60). The formation of macropinosomes is blocked by inhibitors of sodium/proton exchangers like 5-(N-ethyl-N-propyl)amiloride (EIPA), which decrease the cytosolic pH and inhibit the activation of Rac1/Cdc42 (61).
Macropinocytosis has been reported to be involved in host cell entry by vaccinia virus mature virions (62), adenovirus 3 (63), adenovirus 35 (64), echovirus 1 (65), Ebola virus (66), influenza A virus (67), Nipah virus (68), respiratory syncytial virus (69), and HIV-1 (70). Although this pathway characteristically involves the nonspecific receptor-independent uptake of fluid, solute, and particles, it has been demonstrated that some viruses can use a receptor-dependent macropinocytosis-like pathway to gain entry into host cells (56, 57, 68, 70). Some preliminary reports suggest that MeV might also use a macropinocytosis-like pathway to gain entry into the host cell. However, the mechanism by which this is achieved still remains poorly described (71, 72).
The present study was aimed at characterizing wtMeV entry into cells that express endogenous or genetically introduced SLAM and PVRL4. MeV was previously thought to enter cells solely by direct fusion with the plasma membrane. The contribution of a variety of endocytotic pathways to MeV entry was quantitatively examined by measuring the impact of various inhibitory drugs, small interfering RNA (siRNA), and the expression of well-characterized dominant negative (DN) mutants of cell trafficking proteins that affect virus entry and infection. Combined with the use of a recombinant MeV expressing firefly luciferase (Luc) or enhanced green fluorescent protein (eGFP), these approaches provide a better understanding of receptor-mediated endocytosis involved in MeV uptake. Overall, our data indicate that MeV enters Vero cells that express SLAM and PVRL4 in a Rac1-PAK1-independent manner using a receptor-mediated macropinocytosis-like pathway. However, MeV entry into the PVRL4-positive MCF7, DLD-1, and HTB-20 human cancer cell lines relied upon Rac1 and its effector PAK1, using a PVRL4-mediated macropinocytosis pathway.
RESULTS
Generation and analysis of recombinant wtMeV expressing the firefly luciferase reporter gene (wtMeV-Luc).
To investigate the early steps of MeV entry, recombinant wild-type MeV expressing the firefly luciferase reporter gene (wtMeV-Luc) was generated by reverse genetics (Fig. 1). The transcriptional unit encoding eGFP in wtMeV-eGFP was replaced by the firefly luciferase open reading frame (ORF) upstream of the N gene (Fig. 1A). The virus stock was propagated in B95a cells and titrated by the 50% tissue culture infective dose (TCID50) using Vero.hSLAM cells. Three cell lines (Vero, Vero.hSLAM, and Vero.hPVRL4 cells) were used to compare multicycle growth kinetics of wtMeV-Luc and wtMeV-eGFP. Each cell line was infected with viruses at a multiplicity of infection (MOI) of 0.01, and syncytium formation and virus titers were monitored for 2 days postinfection. It appeared that both recombinant viruses were indistinguishable in their spread and replication in Vero.hSLAM and Vero.hPVRL4 cells, as demonstrated by syncytium formation and the kinetics of MeV production (Fig. 1B and C). In contrast, the replication and spread of both wtMeV-eGFP and wtMeV-Luc were significantly reduced in Vero cells that do not express MeV receptors (Fig. 1B and C). Together, these data indicate that the replacement of the transcriptional unit encoding eGFP in the wtMeV-eGFP genome with the firefly luciferase open reading frame had no effect on virus replication and spread in Vero cell lines.
FIG 1.

Levels of growth of wtMeV-Luc and wtMeV-eGFP are similar in Vero, Vero.hSLAM, and Vero.hPVRL4 cells. (A) wtMeV-Luc was engineered from the wtMeV-eGFP genome by replacing the transcription unit encoding eGFP located upstream of the MeV H ORF with the firefly luciferase (Luc) ORF. (B) Vero cell lines were infected with wtMeV-Luc and wtMeV-eGFP at an MOI of 0.01. The replication of recombinant viruses was monitored by using fluorescence and phase-contrast microscopy on the indicated days postinfection. Syncytia are delimited (black lines) in the phase-contrast image. (C) At 1 and 2 days postinfection, cell-associated virus titers were quantified and expressed as TCID50 per milliliter. (D) Vero.hSLAM and Vero.hPVRL4 cells were infected with wtMeV-Luc at an MOI of 1. At 4 and 8 hpi, luciferase activity was measured and is displayed as fold induction relative to the luciferase activity measured at 4 hpi. The means of data from three independent experiments are shown, and error bars indicate standard deviations. The asterisks indicate a statistically significant difference (P < 0.05 by ANOVA) in comparisons of data from day 1 and day 2, as well as 4 and 8 hpi, for each virus.
Next, the infectivity of wtMeV-Luc was assessed at the early steps of virus infection by using a luciferase reporter assay. Vero, Vero.hSLAM, and Vero.hPVRL4 cells were infected with wtMeV-Luc at an MOI of 1, and the accumulation of reporter activity was quantified at 4 and 8 h postinfection (hpi) (Fig. 1D). Luciferase activity was detected from 4 hpi in Vero cells expressing SLAM and PVRL4, and a 4-fold increase was observed at 4 to 8 h postinfection (Fig. 1D). In contrast, luciferase accumulation was not observed in Vero cells at 4 and 8 hpi (data not shown). Together, these data indicated that wtMeV-Luc receptor-dependent entry could be assessed at 8 hpi by quantifying luciferase activity in Vero cells expressing SLAM and PVRL4.
Wild-type MeV induces both actin rearrangement and fluid-phase uptake in Vero.hSLAM and Vero.hPVRL4 cells.
To test whether wtMeV uses macropinocytosis as a route of entry into target cells, actin rearrangement was monitored following wtMeV-Luc infection of serum-starved Vero, Vero.hSLAM, and Vero.hPVRL4 cells at an MOI of 1 (Fig. 2A). Macropinocytosis is normally induced by growth factors present in serum, which produces changes in the actin distribution within the cell (67). Thirty minutes after MeV infection, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain and Alexa Fluor 546-conjugated phalloidin, which binds specifically to the polymerized form of actin (F-actin). Interestingly, infection with wtMeV-Luc led to depolymerization and changes in the actin distribution (Fig. 2A). The number of F-actin filaments decreased, and actin-driven membrane protrusions were observed on the cell surface of Vero.hSLAM and Vero.hPVRL4 cells but were absent in mock-infected control cells (Fig. 2A). A higher MOI increased the frequency of the formation of actin protrusions (data not shown). In contrast, the actin filaments were not rearranged in Vero cells that were exposed to wtMeV-Luc (Fig. 2A). Disruption of actin filaments with cytochalasin D and latrunculin A was found to inhibit both MeV entry, measured with a luciferase reporter virus at 8 hpi (Fig. 2B), and infectivity, assessed with wtMeV-eGFP at 20 h hpi (Fig. 2C), in Vero.hSLAM and Vero.hPVRL4 cell lines. Cell viability assays confirmed that these effects were due to impaired virus entry and not to the cytotoxicity of cytochalasin D and latrunculin A (Fig. 2D). The endosomal acidification inhibitor bafilomycin A1 (Baf), which raises the pH of late endosomes, did not affect MeV entry (Fig. 2B and C). This may have been expected since membrane fusion that is mediated by MeV occurs at neutral pH, and these experiments rule out CME as a mechanism for entry into the host cell. Using a virus binding assay based upon flow cytometry (fluorescence-activated cell sorter [FACS]) analysis, cytochalasin D treatment had no effect on wtMeV binding to its cellular receptor (Fig. 2E), although wtMeV binding may be slightly reduced following latrunculin A treatment (Fig. 2F). Together, these results indicate that actin plays a critical role during wtMeV entry and infection of Vero cells expressing SLAM and PVRL4, which are associated with increased actin depolymerization and rearrangement, a hallmark of macropinocytosis (56, 57).
FIG 2.

Actin cytoskeleton dynamics are involved in wtMeV-Luc entry. (A) Serum-starved Vero, Vero.hSLAM, and Vero.hPVRL4 cells were exposed to wtMeV-Luc (MOI of 1) or PBS for 30 min and fixed with formaldehyde. Actin filaments were labeled with Alexa Fluor 546-conjugated phalloidin (red), and the nucleus was labeled with DAPI (blue). Images were captured with a 100× oil immersion objective. A higher magnification of the boxed area reveals the formation of actin protrusions at the cell surface membrane (arrows). Experiments were repeated 3 times, with similar results. (B and C) Vero.hSLAM and Vero.hPVRL4 cells in serum-free medium were pretreated with 500 nM bafilomycin A1 (Baf.), 40 μM cytochalasin D (Cyto. D), 1 μM latrunculin A (Lat. A), and DMSO for 30 min at 37°C. Cells were then infected with wtMeV-Luc and wtMeV-eGFP at an MOI of 1. (B) As a measure of cell entry by wtMeV-Luc, bioluminescence was measured at 8 hpi and is displayed as a percentage of luciferase activity relative to that under untreated conditions (DMSO). Both cytochalasin D and latrunculin A treatments inhibited MeV-Luc entry into Vero hSLAM and Vero hPVRL4 cells. (C) After wtMeV-eGFP infection, the inoculum was removed and replaced with fresh medium containing 200 μM FIP to prevent secondary infection. Twenty hours later, virus infectivity was measured by FACS analysis and is displayed as a percentage of eGFP-positive cells relative to that under untreated conditions (DMSO). Bafilomycin, cytochalasin D, and latrunculin A treatments had no effect upon MeV-eGFP infection. (D) To test for potential drug toxicity, Vero.hSLAM and Vero.hPVRL4 cells in serum-free medium were pretreated with 40 μM cytochalasin D, 1 μM latrunculin A, and DMSO for 30 min at 37°C. Toxicity assays, using the MTS-based assay described in Materials and Methods, were performed, and results are displayed as a percentage of viable cells relative to those under untreated conditions (DMSO). No toxicity due to cytochalasin D or latrunculin A was observed at the concentrations tested. (E and F) To determine effects on virus binding, Vero.hSLAM and Vero.hPVRL4 cells were pretreated with DMSO, cytochalasin D (E), and latrunculin A (F) as indicated above and then incubated with wtMeV-Luc at an MOI of 10 for 1.5 h. Cells were incubated with an MeV anti-H primary antibody followed by an Alexa Fluor 647-conjugated goat anti-mouse secondary antibody to detect MeV-bound cells by FACS analysis. Cells incubated in the absence of virus (no virus) (filled histograms) were stained with anti-MeV hemagglutinin antibody. Experiments were repeated 3 times, with similar results. Data from one representative experiment are shown in panels E and F. The studies showed that cytochalasin D had no effect and that latrunculin A had a minimal effect on cell binding to MeV-Luc. The results shown in panels B to D are expressed as the means of data from 3 independent experiments, and the error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks.
Another property of macropinocytosis activation is fluid-phase uptake. Various viruses that enter host cells via macropinocytosis have been shown to transiently enhance fluid-phase uptake (62–64, 73). To quantitatively assess fluid-phase uptake following wtMeV infection, Vero cell lines were preincubated with wtMeV-Luc (MOI of 5) at 4°C for 1 h, followed by a temperature switch to 37°C for 15 min or 30 min. The fluid-phase marker fluorescein isothiocyanate (FITC)-dextran was added for 5 min at the end of each incubation period, and the uptake of FITC-dextran was measured by FACS analysis (Fig. 3). As might be expected, FITC-dextran uptake was significantly increased by ∼1.5-fold in wtMeV-infected cells expressing SLAM and PVRL4, compared to constitutive baseline levels in mock-infected cells. The effect was observed 15 min after the administration of wtMeV in cells expressing SLAM and PVRL4 and returned to baseline levels within 30 min (Fig. 3A and B). No increase in FITC-dextran uptake was observed in Vero cells following exposure to wtMeV (Fig. 3A and B). These observations indicate that fluid-phase uptake is rapidly and transiently induced over the first 15 min at 37°C following wtMeV infection of Vero.hSLAM and Vero.hPVRL4 cells. Again, these data suggest that MeV might enter host cells through a SLAM- and PVRL4-mediated macropinocytosis pathway.
FIG 3.
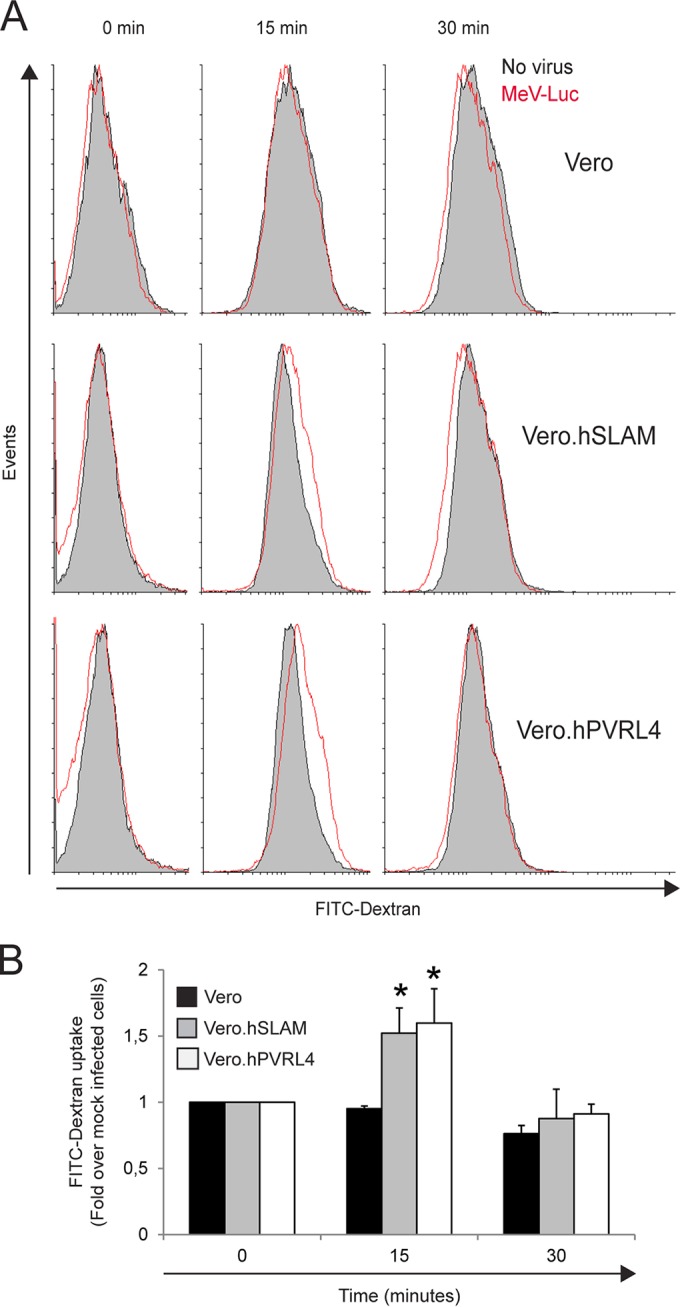
Wild-type measles virus stimulates FITC-dextran uptake in Vero.hSLAM and Vero.hPVRL4 cells. Wild-type MeV-Luc and PBS (no virus) were incubated with Vero, Vero.hSLAM, and Vero.hPVRL4 cells on ice for 1 h, shifted to 37°C for the indicated time periods, exposed to 1 mg/ml FITC-dextran at a molecular weight of 70,000 during the last 5 min, and analyzed by flow cytometry. (A) Histogram overlays illustrating the uptake of FITC-dextran molecules by cells incubated with PBS (filled histograms) and virus 0, 15, and 30 min after incubation of Vero, Vero.hSLAM, and Vero.hPVRL4 cells. Experiments were repeated 3 times, with similar results. Data from one representative experiment are shown. (B) FITC-dextran endocytosis in the presence of virus compared to that in the absence of virus. The uptake of FITC-dextran was quantified as a percentage of FITC-dextran-positive cells, which was calculated by subtracting the value for nonspecific FITC-dextran uptake (no virus) from the value obtained in the presence of the virus. Results are displayed as fold induction normalized to values under untreated conditions at time zero. The means of data from three independent experiments are shown, and error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks.
Dynamin is not required for wtMeV entry in Vero.hSLAM and Vero.hPVRL4 cells.
The GTPase dynamin is a signaling enzyme that plays a key role in actin cytoskeleton dynamics, mitogen-activated protein kinase signaling, and apoptosis (74). Importantly, dynamin function is also essential for the scission of newly formed vesicles from the plasma membrane in various endocytosis pathways such as CME, CavME, and some of the non-clathrin/non-caveolin-dependent pathways (54). However, dynamin is not required for the process of macropinocytosis (56, 57). To determine whether wtMeV entry requires dynamin, the effect of Dynasore, a GTPase inhibitor that targets dynamin-1 and -2 (75), was tested by using a wtMeV-Luc entry assay (Fig. 4). As a control to verify that Dynasore inhibits dynamin-mediated endocytosis, its effect on the internalization of transferrin (CME marker) was determined in Vero.hSLAM cells (Fig. 4A). FACS analysis revealed that Dynasore treatment reduced the internalization of transferrin in SLAM-expressing Vero cells (Fig. 4A). In addition, vesicular stomatitis virus (VSV) expressing firefly luciferase (VSV-Luc), which enters cells by CME (76), is sensitive to 100 μM Dynasore in Vero.hSLAM cells (Fig. 4C). Viability assays with Vero.hSLAM and Vero.hPVRL4 cells confirmed that Dynasore had no cytotoxic effects at 25 μM, 50 μM, and 100 μM concentrations (Fig. 4C). Subsequently, Vero cell lines were pretreated with increasing concentrations of Dynasore for 1 h in serum-free medium prior to infection with wtMeV-Luc at an MOI of 1. Luciferase activity was measured at 8 hpi, which revealed that Dynasore treatment failed to have a significant effect on wtMeV entry into Vero.hPVRL4 cells, even at the highest concentration tested (Fig. 4D). In Vero.hSLAM cells, Dynasore also did not decrease virus entry but in fact caused a slight increase (Fig. 4D). It is possible that Dynasore treatment might increase MeV entry in Vero.hSLAM cells (Fig. 4D) and subsequently redirect the virus to a nonproductive pathway, as previously observed for Ebola virus (77). To further assess the efficiency of virus entry following Dynasore treatment, an infectivity assay was performed by using wtMeV-eGFP instead of wtMeV-Luc (Fig. 4E). After infection, the inoculum was removed and replaced with fresh medium containing 200 μM fusion-inhibitory peptide (FIP) (Z-fFG; carbobenzoxyl-d-phenylalanine-l-phenylalanine-glycine) to prevent secondary infection and syncytium formation in cells (22, 78, 79). Twenty hours later, numbers of GFP-positive MeV-infected cells were quantified by FACS analysis (Fig. 4E). As expected, the results revealed that Dynasore treatment of Vero.hPVRL4 cells failed to have a significant effect on infection by wtMeV-eGFP. Dynasore also failed to inhibit infection by wtMeV-eGFP in Vero.hSLAM cells (Fig. 4E). To further examine and validate the dynamin independence of wtMeV infections, Vero cell lines were transfected with a plasmid expressing either wild-type dynamin-2 (DYN WT) or a DN form of dynamin-2 (K44A) (DYN DN) (Fig. 4F), and both wtMeV entry and infection assays were again performed (Fig. 4G and H). As expected, both the entry and infectivity of wtMeV were not affected by DYN WT or DYN DN overexpression in Vero.hSLAM and Vero.hPVRL4 cells (Fig. 4G and H).
FIG 4.

Wild-type MeV entry is dynamin independent. (A) As a positive control to demonstrate the effect of dynamin inhibition on CME, Vero.hSLAM cells in serum-free medium were pretreated with 100 μM Dynasore and DMSO for 1 h at 37°C, washed in PBS, and incubated with 50 μg/ml Texas Red-conjugated human transferrin for 30 min at 37°C. Cells were then washed three times with cold PBS and once with low-pH buffer for 10 min and processed for FACS analysis. The samples pretreated with DMSO and Dynasore are represented. The filled histogram (mock) represents cells that have not been incubated with Texas Red-conjugated human transferrin. Experiments were repeated 3 times, with similar results, and data from one representative experiment are shown. As expected, Dynasore treatment inhibited the uptake of Texas Red-conjugated transferrin. (B) As another control, Vero.hSLAM cells were pretreated with 100 μM Dynasore and infected with VSV-Luc at an MOI of 1. Dynasore treatment inhibited the CME-mediated uptake of VSV-Luc, as expected. (C) Toxicity assays were performed with Dynasore (25 μM, 50 μM, and 100 μM)-pretreated cells, and viability is displayed as a percentage relative to values under untreated conditions (DMSO). Viability was determined by measuring the absorbance at 490 nm in the MTS substrate-based assay as described in the text. (D and E) Vero.hSLAM and Vero.hPVRL4 cells in serum-free medium were pretreated with 25, 50, and 100 μM Dynasore or DMSO for 1 h at 37°C. The cells were infected with either MeV-Luc or MeV-GFP. Luciferase activity was measured at 8 hpi. After wtMeV-eGFP adsorption, the inoculum was removed and replaced with fresh medium containing 200 μM FIP to prevent secondary infection and syncytium formation. Twenty hours later, virus infectivity was measured by FACS analysis, and data are displayed as a percentage of eGFP-positive cells relative to the values under untreated conditions (DMSO). The inhibitor had little effect on MeV-Luc uptake or MeV-GFP infection. (F to H) Vero.hSLAM and Vero.hPVRL4 cells were transfected with plasmids expressing wild-type dynamin 2 (DYN WT) or the dominant negative form of dynamin 2 (DYN DN) for 2 days. (F) The expression plasmids for DYN WT and DYN DN also contained a red fluorescent protein reporter gene, which confirmed the expression of the genes in Vero.hSLAM and Vero.hPVRL4 cells when viewed by fluorescence microscopy. (G and H) Vero.hSLAM and Vero.hPVRL4 cells were transfected with plasmids expressing DYN WT and DYN DN. At 2 days posttransfection, the cells were infected with wtMeV-Luc (G) and wtMeV-eGFP (H) at an MOI of 2. Luciferase activity was measured at 8 hpi and is reported as relative light units compared to values for the DYN WT control. (H) After wtMeV-eGFP adsorption, the inoculum was removed and replaced with fresh medium containing 200 μM FIP to prevent secondary infection and syncytium formation. Twenty hours later, virus infectivity was measured by FACS analysis, and data are displayed as a percentage of eGFP-positive cells relative to the DYN WT control. DYN DN had no effect on MeV-Luc uptake or MeV-eGFP infection in either Vero hSLAM or Vero hPVRL4 cells. In panels B to E, G, and H, the means of data from three independent experiments are shown, and error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks.
To verify whether CavME was involved in virus uptake, the effect of the inhibition of this endocytosis pathway was measured by the overexpression of a dominant negative form of caveolin (CAV DN) (Fig. 5). As expected, both MeV entry and infectivity were not affected in PVRL4- and SLAM-expressing Vero cells following CAV DN overexpression (Fig. 5A to C). Altogether, these findings indicate that the cell entry of wtMeV is independent of caveolin and dynamin in Vero.hSLAM and Vero.hPVRL4 cells.
FIG 5.

Wild-type MeV entry is caveola independent. (A) Vero.hSLAM and Vero.hPVRL4 cells were transfected with plasmids expressing the wild-type (CAV WT) and negative (CAV DN) forms of caveolin 1. At 2 days posttransfection, eGFP reporter gene expression was monitored by fluorescence microscopy. (B and C) Vero.hSLAM and Vero.hPVRL4 cells were transfected with CAV WT or CAV DN expression plasmids and incubated for 2 days. Cells were then infected with wtMeV-Luc and wtMeV-eGFP at an MOI of 2. (B) Luciferase activity was measured at 8 hpi, and values are reported as a percentage of luciferase activity relative to that of the wild-type control. (C) After wtMeV-eGFP infection, the inoculum was removed and replaced with fresh medium containing 200 μM FIP to prevent secondary infection and syncytium formation. Twenty hours later, cells were incubated with an MeV anti-H primary antibody followed by an Alexa Fluor 647-conjugated goat anti-mouse secondary antibody to detect MeV-infected cells. Virus infectivity was measured by FACS analysis, and values are displayed as a percentage of Alexa Fluor-positive cells relative to the CAV WT control. The means of data from three independent experiments are shown, and error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks. Again, the CAV DN expression plasmid did not prevent MeV-Luc entry and MeV-eGFP infections.
Wild-type MeV entry and infection are sensitive to amiloride (EIPA) treatment.
As indicated above, wtMeV uptake occurs through an actin-dependent, dynamin-independent endocytic mechanism, suggesting that wtMeV might use a receptor-mediated macropinocytosis pathway in Vero.hSLAM and Vero.hPVRL4 cells. To assess the involvement of macropinocytosis, the effect of EIPA on wtMeV entry and infection was studied. EIPA is a potent and specific inhibitor of Na+/H+ exchanger activity important for macropinosome formation (61). This inhibitor, by itself, did not affect cell viability or produce cytolytic effects on Vero.hSLAM or Vero.hPVRL4 cells (Fig. 6A). The entry and infection of Vero.hSLAM cells by VSV-Luc were unaffected by 100 μM EIPA treatment, which is consistent with the fact that VSV does not enter the cells by macropinocytosis (Fig. 6B). However, treatment of both Vero.hSLAM and Vero.hPVRL4 cells with 100 μM EIPA inhibited wtMeV-Luc (Fig. 6C) and wtMeV-eGFP (Fig. 6D) entry and infection of host cells. Luciferase activity was measured at 8 hpi and was expressed as a percentage of the luciferase activity in the untreated dimethyl sulfoxide (DMSO) control (Fig. 6C). FACS analysis of wtMeV-eGFP-infected cells that were pretreated with 100 μM EIPA showed a marked drop (∼40%) in the frequency of eGFP-positive cells compared to that of control DMSO-treated cells (Fig. 6D). To rule out the possibility that EIPA may have an impact on virus attachment to Vero.hPVRL4 cells, virus binding assays were performed (Fig. 6E). Cells were pretreated with EIPA or DMSO for 1 h, dissociated from the well, and then incubated with wtMeV-Luc for 90 min on ice. Virus binding was detected by using a monoclonal antibody directed against MeV H and an Alexa Fluor 647-conjugated goat anti-mouse secondary antibody. In Vero.hPVRL4 cells, there was a shift in the histogram peak for DMSO plus wtMeV-Luc treatment, indicating that wtMeV had bound to these cells (Fig. 6E). In comparison to DMSO-treated control cells, an identical peak was observed in Vero.hPVRL4 cells pretreated with EIPA (Fig. 6E). These data suggest that EIPA treatment has no effect on wtMeV binding to Vero.hPVRL4 cells. Overall, these results suggest that wtMeV entry may occur through an EIPA-sensitive pathway in Vero cells expressing SLAM and PVRL4, which is consistent with entry by macropinocytosis.
FIG 6.

(A to D) Wild-type MeV entry and infection are sensitive to EIPA treatment. Vero.hSLAM and Vero.hPVRL4 cells were pretreated in serum-free medium with 50 μM and 100 μM EIPA and DMSO for 1 h at 37°C. (A) Toxicity assays with 50 μM and 100 μM EIPA were performed by using the MTS cell viability assay, and data are reported as a percentage relative to the value for the untreated control (DMSO). (B) Pretreatment of Vero.hSLAM cells with 100 μM EIPA appears not to affect the uptake VSV-Luc at an MOI of 1, since this virus enters cells by CME and not macropinocytosis. Luciferase activity was measured at 8 hpi, and values are reported as a percentage relative to the value for the DMSO control. (C) Vero.hSLAM and Vero.hPVRL4 cells were pretreated with 50 μM and 100 μM EIPA and infected with wtMeV-Luc at an MOI of 1. Luciferase activity was again measured at 8 hpi, and data are reported as a percentage relative to the value for the untreated control (DMSO). (D) Both cell lines were pretreated with 50 μM EIPA, 100 μM EIPA, or DMSO and incubated with wtMeV-eGFP for 1 h. The virus inoculum was removed and replaced with fresh medium containing 200 μM FIP to prevent secondary infection and syncytium formation. Twenty hours later, virus infectivity was measured by FACS analysis, and data are displayed as a percentage of eGFP-positive cells relative to the value for the untreated control (DMSO). EIPA inhibited the uptake of MeV-Luc and MeV-eGFP in both cell lines. In all experiments in panels A to D, the means of data from three independent experiments are shown, and error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks. (E) Vero.hPVRL4 cells were pretreated with 100 μM EIPA and DMSO as indicated above, and cells were dissociated from the culture plate and then incubated with wtMeV-Luc at an MOI of 10 for 1.5 h. Vero.hPVRL4 cells were incubated with an MeV anti-H primary antibody followed by an Alexa Fluor 647-conjugated goat anti-mouse secondary antibody, and bound MeV was detected by FACS analysis. Cells incubated in the absence of virus (no virus) were stained with anti-MeV hemagglutinin antibody. Data for the samples containing WtMeV-Luc plus DMSO and wtMeV-Luc plus EIPA are represented. Experiments were repeated 3 times, with similar results. Data from one representative experiment are shown. EIPA appeared to have no effect on the binding of MeV-Luc to Vero.hPVRL4 cells.
MeV enters Vero.hSLAM and Vero.hPVRL4 cells through a Rac1- and PAK1-independent pathway.
Another characteristic property of macropinocytosis is its dependence on the activity of PAK1 (58). During the process of macropinocytosis, Rac1-dependent activation of PAK1 regulates actin-myosin cytoskeletal dynamics that lead to the formation of membrane protrusions (58). Initially, we studied the role of Rac1 in MeV entry in Vero cells that had been previously transfected with either wild-type GFP-Rac1 (Rac1 WT) or dominant negative mutant pGFP-Rac1-N17 (Rac1 DN) expression plasmids (Fig. 7A). Cells were subsequently infected with wtMV-Luc, but the expression of Rac1 DN had no effect on MeV entry in both Vero.hSLAM and Vero.hPVRL4 cells (Fig. 7B). Similarly, siRNA-mediated knockdown of PAK1 did not decrease MeV entry in either cell line but seemed to increase it (Fig. 7C and D). However, the siRNA-mediated knockdown of PAK1 did not enhance virus infection in these cells lines, although MeV entry was increased (Fig. 7E). Because there is no correlation between virus entry and infectivity following the siRNA knockdown of PAK1, we further examined MeV entry by overexpressing either Myc-PAK1 WT or glutathione S-transferase (GST)-PAK1 DN in Vero.hSLAM and Vero.hPVRL4 cells (Fig. 7F to H). A nonsignificant decrease in MeV entry was observed in both cell lines (Fig. 7G), which was correlated with a nonsignificant decrease in MeV infectivity in these Vero cells (Fig. 7H). As a consequence, our data indicate that MeV entry/infection is PAK1 independent.
FIG 7.

MeV enters Vero.hSLAM and Vero.hPVRL4 cells through a Rac1- and PAK1-independent pathway. Vero.hSLAM and Vero.hPVRL4 cells were transfected with plasmids expressing wild-type Rac1 (Rac1 WT), the dominant negative form of Rac1 (Rac1 DN), wild-type PAK1 (Myc-PAK1 WT), the dominant negative form of PAK1 (GST-PAK1 DN), or siRNA targeting human PAK1. (A) Expression plasmids for Rac1 or its dominant negative form contained an eGFP reporter gene, and the synthesis of Rac1 WT or Rac1 DN was confirmed by fluorescence microscopy. (B) Vero.hSLAM and Vero.hPVRL4 cells were transfected with Rac1 WT and Rac1 DN expression plasmids and incubated for 2 days. The transfected cells were infected with MeV-Luc at an MOI of 2 for 8 h, and luciferase activity is reported as a percentage relative to the value for the Rac1 WT control. Rac1 DN appeared to have no effect on MeV-Luc entry. (C) Vero.hSLAM and Vero.hPVRL4 cells were transfected with PAK1 siRNA or scrambled control siRNA and incubated for 2 days. Cell lysates were subjected to SDS-PAGE and analyzed by Western immunoblot analysis using antibodies directed against PAK1. Knockdown of PAK1 by PAK1-targeting siRNA was confirmed in both cell lines. (D) Vero.hSLAM and Vero.hPVRL4 cells were transfected with PAK1 siRNA or scrambled control siRNA and incubated for 2 days. The transfected cells were infected with MeV-Luc at an MOI of 2 for 8 h, and luciferase activity is reported as a percentage relative to the values for scrambled control siRNA. PAK1 siRNA failed to inhibit the entry of MeV-Luc in both cell lines. (E) Vero.hSLAM and Vero.hPVRL4 cells were transfected with PAK1 siRNA or scrambled control siRNA and incubated for 2 days. The transfected cells were infected with MeV-eGFP at an MOI of 0.1 and incubated for 24 h. Syncytia were observed by using fluorescence microscopy, and PAK1 siRNA did not appear to affect MeV-eGFP infections. Experiments were repeated 3 times, with similar results. Data from one representative experiment are shown. (F) Vero.hSLAM and Vero.hPVRL4 cells were transfected with expression plasmids for Myc-PAK1 and GST-PAK1 DN and incubated for 2 days. Expression in cell lysates from these cells was confirmed by using Western immunoblot analysis and antibodies directed against the Myc and GST tags. (G) Vero.hSLAM and Vero.hPVRL4 cells were transfected with PAK1 WT and PAK1 DN expression plasmids and incubated for 2 days. These cells were infected with wtMeV-Luc for 8 h, and luciferase activity is reported as a percentage relative to the value for PAK1 WT-transfected cells. The PAK1 DN form had little effect on MeV-Luc entry into Vero.hSLAM and Vero.hPVRL4 cells. (H) Vero.hSLAM and Vero.hPVRL4 cells were transfected with PAK1 WT and PAK1 DN expression plasmids and incubated for 2 days. These cells were incubated with wtMeV-eGFP at an MOI of 2. After 1 h of adsorption, the virus inoculum was removed and replaced with fresh medium containing 200 μM FIP to prevent secondary infection and syncytium formation. Twenty-four hours later, fluorescence due to viral infection was measured by FACS analysis, and data are displayed as a percentage of eGFP-positive cells relative to the value for the PAK1 WT control. All experiments are the means of data from three independent experiments, and error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks.
Overall, these findings indicate that the route of entry for MeV in Vero.hSLAM and Vero.hPVRL4 cells differs from the classical macropinocytosis pathway with regard to the requirement for Rac1 and PAK1. Since hSLAM and hPVRL4 are not normally expressed in Vero cells and were introduced through the use of expression vectors, the possibility of an adaptation to use redundant kinase pathways exists. Our results are consistent with another recent paper (80) that shows that the receptor hSLAM can mediate the uptake of measles virus particles via a macropinocytosis-like pathway in A549 cells that had been engineered to express hSLAM (SLAMF1). Similar to our results, this pathway was inhibited by EIPA; was dependent on actin rearrangement; and was independent of Rac1, Cdc42, and PAK1 expression. Those investigators also used siRNA and DN mutants to interfere with this pathway. They proposed that this form of macropinocytosis is dependent on the RhoA–ROCK–myosin-II signal pathway, which can be inhibited by the myosin inhibitor blebbistatin. The remaining experiments in our paper deal with cancer cell lines that express endogenous PVRL4 and contain a functional PAK1 signal pathway.
MeV enters the MCF7 human breast cancer cell line through a PVRL4-mediated macropinocytosis pathway.
Although the expression of PVRL4 in Vero.hPVRL4 cells mediates MeV entry, the signaling pathways that are triggered following MeV binding to its receptor are likely different from those activated in cells that endogenously express PVRL4. To determine whether MeV can enter host cells through a classical macropinocytosis pathway, the PVRL4-positive MCF7 human breast cancer cell line was studied (Fig. 8). MeV entry into MCF7 cells was previously shown to rely entirely upon PVRL4 cell surface expression (39). Actin rearrangement was first monitored following wtMeV-Luc infection of serum-starved MCF7 cells at an MOI of 10 (Fig. 8A). As expected, infection with wtMeV-Luc led to depolymerization and changes in the actin distribution (Fig. 8A). The number of F-actin filaments decreased, and actin-driven membrane protrusions were observed on the cell surface of MCF7 cells but were absent in mock-infected control cells (Fig. 8A). The same agents as those used to study macropinocytosis were employed in the following studies. Each drug used to assess endocytosis produced no cytotoxicity at the concentrations tested on MCF7 cells (Fig. 8B). As another control, VSV-Luc entry into MCF7 cells was shown to be blocked by the dynamin inhibitor Dynasore (100 μM) (Fig. 8C). To determine whether MeV entry was dynamin independent in MCF7 cells, the cells were pretreated with 100 μM Dynasore for 1 h in serum-free medium prior to infection with wtMeV-Luc at an MOI of 10. As expected, luciferase activity measured at 8 hpi revealed that Dynasore treatment failed to have any significant effect on wtMeV entry into the MCF7 cell line (Fig. 8D). To confirm a role for actin during the initial stages of MeV infection of MCF7 cells, actin filaments were disrupted with cytochalasin D and found to inhibit the entry of the MeV-Luc reporter virus (Fig. 8E). To determine whether macropinocytosis was again involved in MeV entry of these cells, the effect of EIPA on wtMeV entry into MCF7 cells was measured. Inhibition of wtMeV-Luc entry in a dose-dependent manner was observed (Fig. 8F). As a control, VSV-Luc, which enters cells by CME, was not affected by 100 μM EIPA treatment (Fig. 8C). To further assess the effect of EIPA on wtMeV infection, MCF7 cells were pretreated with DMSO or different EIPA concentrations. They were subsequently incubated with the wtMeV-eGFP reporter virus. FACS analysis revealed that there were marked decreases of ∼40 and ∼75% eGFP-positive cells in wtMeV-eGFP-infected MCF7 cells pretreated with 50 and 100 μM EIPA, respectively, compared to those in DMSO-treated cells (Fig. 8G).
FIG 8.

MeV enters the MCF7 human breast cancer cell line through a PVRL4-mediated macropinocytosis pathway. (A) Serum-starved MCF7 cells were exposed to wtMeV-Luc (MOI of 10) or PBS for 30 min and fixed with formaldehyde. Actin filaments were labeled with Alexa Fluor 546-conjugated phalloidin (white). Images were captured with a 100× oil immersion objective. A higher magnification of the boxed area reveals the formation of actin protrusions at the cell surface membrane (arrows). Experiments were repeated 3 times, with similar results. (B) Toxicity assays of MCF7 cells treated with 100 μM Dynasore, 50 and 100 μM EIPA, and 40 μM cytochalasin D were performed by using the MTS cell viability assay, and data are reported as a percentage relative to value for the untreated control (DMSO). (C) As a control, MCF7 cells were pretreated with 100 μM Dynasore or 100 μM EIPA and were infected with VSV-Luc at an MOI of 1. Luciferase activity was measured at 8 hpi, and data are reported as a percentage relative to the value for the DMSO control. Dynasore treatment inhibited the uptake of VSV-Luc, which occurs via the process of CME. However, EIPA treatment had little effect on VSV-Luc entry. (D) MCF7 cells were pretreated with 100 μM Dynasore and infected with wtMeV-Luc. Luciferase activity was measured at 8 hpi, and values are reported as a percentage of luciferase activity relative to that of the untreated control (DMSO). Dynasore did not prevent the entry of MeV-Luc into MCF7 cells. (E) MCF7 cells were pretreated with 40 μM cytochalasin D and infected with wtMeV-Luc. Luciferase activity was measured at 8 hpi, and data are reported as a percentage of luciferase activity relative to that of the untreated control (DMSO). Cytochalasin D greatly reduced the entry of MeV-Luc into MCF7 cells. (F) MCF7 cells were pretreated with 50 μM and 100 μM EIPA and infected with wtMeV-Luc. Luciferase activity was measured at 8 hpi, and values are reported as a percentage of luciferase activity relative to that of the untreated control (DMSO). EIPA inhibited the entry of MeV-Luc into MCF7 cells. (G) MCF7 cells were pretreated with 50 μM EIPA, 100 μM EIPA, or DMSO and incubated with wtMeV-eGFP (MOI of 10) for 1 h. The virus inoculum was removed and replaced with fresh medium containing 200 μM FIP to prevent secondary infection and syncytium formation. Twenty hours later, virus infectivity was measured by FACS analysis, and values are displayed as a percentage of eGFP-positive cells relative to the value for the untreated control (DMSO). EIPA inhibits MeV-eGFP infection of MCF7 cells. (H) MCF7 cells were transfected with siRNA directed against PAK1 or scrambled control siRNA and incubated for 2 days. Cell lysates were subjected to SDS-PAGE and analyzed by Western immunoblot analysis using antibodies directed against PAK1. The knockdown of PAK1 by PAK1-targeting siRNA was confirmed in MCF7 cells. (I) MCF7 cells were transfected with PAK1 siRNA or scrambled control siRNA and incubated for 2 days. The transfected cells were infected with MeV-Luc at an MOI of 2 for 8 h, and luciferase activity is reported as a percentage relative to that of control siRNA. PAK1 siRNA inhibited the entry of MeV-Luc into MCF7 cells. (J) MCF7 cells were transfected with expression plasmids encoding either Rac1 WT or Rac1 DN. The expression plasmids also contained an eGFP reporter gene. Cells were incubated for 2 days, and the synthesis of Rac1 WT or Rac1 DN was confirmed by fluorescence microscopy. (K) MCF7 cells were transfected with Rac1 WT and Rac1 DN expression plasmids and incubated for 2 days. Transfected cells were infected with MeV-Luc at an MOI of 2 for 8 h, and luciferase activity is reported as a percentage relative to that of the Rac1 WT control. Rac1 DN appeared to inhibit MeV-Luc entry. (L) MCF7 cells were transfected with siRNA directed against PAK1 or scrambled control siRNA. After 2 days, cells were infected with wtMeV-eGFP at an MOI of 10. Syncytium formation was observed at 2 days postinfection by using fluorescence microscopy. PAK1 siRNA clearly inhibited MeV-GFP infection. Experiments were repeated 3 times, with similar results. Data from one representative experiment are shown. In the preceding experiments (B to G, I, and K) means of data from three independent experiments are shown, and error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks.
To specifically inhibit components of the macropinocytosis pathway, the knockdown of PAK1 as well as expression of Rac1 DN were performed in MCF7 cells (Fig. 8H to L). The knockdown of PAK1 dramatically reduced MeV entry into MCF7 cells (Fig. 8H and I). In addition, MeV uptake was reduced by approximately 50% in cells expressing Rac1 DN compared to those expressing the wild-type form of Rac1 (Fig. 8J and K). To determine whether macropinocytosis might play a role in virus cell-to-cell spread, MCF7 cells previously transfected with control (Ctrl) and PAK1 siRNAs were infected with wtMeV-eGFP (MOI of 10) at 3 days posttransfection, and syncytium formation was monitored at 2 days postinfection (Fig. 8L). The knockdown of PAK1 led to a reduction in the number of syncytia (Fig. 8L). Also, the sizes of the syncytia were similar among the different siRNA-transfected MCF7 cells, suggesting that the inhibitors did not directly affect syncytium formation or cell-to-cell spread (Fig. 8L). Altogether, these data strongly indicate that MeV triggers the Rac1-PAK1 signaling axis during virus entry through a PVRL4-mediated macropinocytosis pathway in the MCF7 human breast cancer cell line.
MeV also enters DLD-1 colon and HTB-20 breast cancer cell lines through a PVRL4-mediated macropinocytosis pathway.
Finally, we extended our analyses to other PVRL4-positive cancer cell lines: DLD-1 (a colorectal adenocarcinoma cell line) and HTB-20 (a breast tumor cell line). Both cell lines have been shown to express PVRL4 (39, 81). As shown in Fig. 9A, actin rearrangement was monitored with phalloidin staining following wtMeV-Luc infection of serum-starved DLD-1 and HTB-20 cells. As expected, actin-driven membrane protrusions were observed on the cell surfaces of DLD-1 and HTB-20 cells but were absent in mock-infected control cells (Fig. 9A). Dynasore treatment had no effect on MeV infection of MCF7, DLD-1, and HTB-20 cells, while increasing concentrations of the macropinocytosis inhibitor EIPA decreased MeV infectivity (Fig. 8B). To determine whether PAK1 may play a role in MeV entry, DLD-1 cells previously transfected with Ctrl and PAK1 siRNAs were infected with wtMeV-Luc (MOI of 10) at 2 days posttransfection (Fig. 9C and D). Knockdown of PAK1 (Fig. 9C) led to a significant decrease in MeV entry (Fig. 9D). The poor transfection efficiency of plasmid DNA in HTB-20 cells limited further study of components of the macropinocytosis pathway. However, microscopic observations and treatment with EIPA, the classical inhibitor, clearly indicated that MeV enters PVRL4-positive cancer cells through the process of macropinocytosis.
FIG 9.

MeV enters the DLD-1 colorectal and HTB-20 breast cancer cell lines through a PVRL4-mediated macropinocytosis pathway. (A) Serum-starved DLD-1 and HTB-20 cells were exposed to wtMeV-Luc (MOI of 10) or PBS for 30 min and fixed with formaldehyde. Actin filaments were labeled with Alexa Fluor 546-conjugated phalloidin (red). Images were captured with a 100× oil immersion objective. A higher magnification of the boxed area reveals the formation of actin protrusions at the cell surface membrane (arrows). Experiments were repeated 3 times, with similar results. (B) MCF7, HTB-20, or DLD-1 cells were pretreated with either 50 μM EIPA, 100 μM EIPA, or 100 μM Dynasore. Cells were infected with wtMeV-eGFP (MOI of 10) and viewed by fluorescence microscopy at 2 days postinfection. Dynasore had no effect on infections by wtMeV-eGFP, but EIPA clearly inhibited infections in all 3 cell types. (C) DLD-1 cells were transfected with siRNA directed against PAK1. At 2 days posttransfection, knockdown of PAK1 expression was confirmed by a Western immunoblot assay using antibodies directed against PAK1 and actin control proteins. (D) DLD-1 cells were transfected with PAK1 siRNA and scrambled control siRNA and incubated for 2 days. Cells were infected with wtMeV-Luc, luciferase activity was measured at 8 hpi, and data are reported as a percentage of the luciferase activity relative to that of the scrambled control siRNA. PAK1 siRNA inhibited the entry of MeV-Luc into DLD-1 cells. The means of data from three independent experiments are shown, and error bars indicate standard deviations. Statistically significant differences (P < 0.05 by ANOVA) are indicated by asterisks.
DISCUSSION
Viruses have not only evolved to counteract cellular antiviral mechanisms to favor viral replication but have also adapted to exploit cellular functions that promote virus entry (54–56, 82). Endocytosis plays a key role in many cellular functions, which include nutrient uptake, cell adhesion and migration, receptor signaling, cell polarity, and pathogen entry (82). Interestingly, many viruses have been shown to hijack endocytotic machinery to promote virus entry into host cells (83). Some viruses can actually use one or more endocytic pathways to infect cells since the regulation of endocytosis is pleiotropic and cell type dependent (82). A better understanding of viral entry is fundamental, as it may lead to the development of novel antiviral therapies that block virus internalization into the cell cytoplasm.
Previous studies to elucidate MeV entry have focused mainly on the identification of receptors for MeV (33, 34, 38, 39, 44, 45). However, the specific endocytotic and trafficking pathways utilized for cell entry during MeV infections remain poorly studied. Recent publications suggested that macropinocytosis might be involved in MeV infection, but the results are preliminary, and the mechanisms involved and the cell types affected need to be validated. One early study based on electron microscopy analysis revealed that a vaccine strain of MeV induces pseudopodia in HeLa or CHO-K1 cells transfected with a plasmid expressing CD46, suggesting that CD46 internalization might be mediated by macropinocytosis (71). Another study relying on the use of an MeV glycoprotein-pseudotyped lentiviral vector (H/F-LV) together with the macropinocytosis inhibitor EIPA indicated that H/F-LVs might enter quiescent T cells through a SLAM/CD46-dependent entry mechanism that strongly resembles macropinocytosis (72). Interestingly, CD46 has also been reported to be involved in the macropinocytosis of adenovirus 35 (64). While multiple approaches suggested an entry mechanism similar to macropinocytosis in MeV infection, key data from Frecha et al. were obtained by using lentivirus-based retroviral pseudotypes together with a high concentration of a pharmacological inhibitor, and drug specificity was not examined, making the interpretation of results difficult (72). Until very recently, previous work using MeV did not use specific macropinocytosis inhibitors (71). After we submitted this paper, another research group demonstrated that hSLAM (SLAMF1) can mediate the cellular entry of MeV via a macropinocytosis-like pathway in A549 cells that had been engineered to express hSLAM (SLAMF1) (80). Similar to our results, this pathway was inhibited by EIPA; was dependent on actin rearrangement; and was independent of Rac1, Cdc42, and PAK1 expression. Those investigators also used siRNA and DN mutants to interfere with this pathway. They proposed that this form of macropinocytosis is dependent on the RhoA–ROCK–myosin-II signal pathway, which can be inhibited by the myosin inhibitor blebbistatin. Our initial work with Vero.hSLAM cells supported that group's findings, but our results regarding the lack of Rac1-PAK1 involvement in a macropinocytosis-like pathway in these cells were even more striking. This work from our laboratory focuses on the role of PVRL4 (nectin-4) in the macropinocytosis of MeV in cells that naturally express this receptor.
In this study, a detailed analysis of each major endocytic pathway was performed by combining distinct and independent approaches. Drugs were used to inhibit pathways, but issues of specificity were assessed by using highly specific dominant negative mutants and siRNA knockdown to corroborate the data obtained by using pharmacological inhibitors. Our findings demonstrate that MeV enters Vero cells through a SLAM- and PVRL4-mediated macropinocytosis-like pathway in a Rac1- and PAK1-independent manner, while MeV entry into the MCF7 human breast cancer cell line relied upon Rac1 and its effector PAK1 through a PVRL4-mediated macropinocytosis pathway. That being said, we cannot say that MeV entry occurs exclusively by this pathway but that its disruption blocks the majority of infection and virus uptake.
As discussed below, the expression of SLAM and PVRL4 in Vero cells is sufficient to induce MeV entry through a receptor-mediated macropinocytosis-like pathway. However, measles virus uptake occurs in a Rac1- and PAK1-independent manner, suggesting that signaling pathways triggered following MeV binding to its receptor in Vero cells are likely different from those activated in cells that endogenously express PVRL4. Interestingly, the Src family of nonreceptor tyrosine kinases has been shown to be involved in signaling pathways triggered by SLAM and nectin molecules (84–87). Therefore, there is increasing evidence that c-Src functions as a key signaling intermediate in the induction of macropinocytosis via a mechanism independent of Rac1, cdc42, and PAK1 (88–90). Consequently, further investigations will be needed to determine whether Src family members play a key role in MeV entry into Vero cells through a receptor-mediated macropinocytosis-like pathway.
In the past decades, measles virus vaccine strains have emerged as a promising oncolytic platform for cancer therapy (91–95). Given that PVRL4 is upregulated on the tumor surface of breast (96), lung (97), ovary (98), and colon (99) cancers, this tumor-associated marker is a promising target for oncolytic measles therapies. The data presented here establish for the first time that MeV entry into MCF7 human breast cancer cells occurs through a PVRL4-mediated macropinocytosis pathway that requires the Rac1-PAK1 signaling axis. This is consistent with data from our previous study showing that MeV infection of MCF7 cells relies entirely on PVRL4 cell surface expression (39). Of particular importance, Mycobacterium bovis bacillus Calmette-Guerin (BCG) was also shown to gain entry into bladder cancer cells by macropinocytosis rather than phagocytosis (100). In fact, many studies have reported that the macropinocytosis pathway is selectively upregulated by cancer cells (100–103) and was shown to support cancer cell proliferation by supplying nutrient uptake in Ras-transformed cancer cells (101). Extracellular ATP is also internalized by macropinocytosis and induces increased intracellular ATP levels and drug resistance in cancer cells (104). Since the difference in susceptibility between BCG-permissive and BCG-resistant bladder cancer cells results from the oncogenic activation of signaling pathways that activate macropinocytosis (100, 105, 106), a better understanding of signaling pathways involved in the PVRL4-mediated macropinocytosis of MeV will benefit the development of novel oncolytic measles therapies based on selecting PVRL4-positive cancer cells with enhanced macropinocytosis.
In conclusion, the evidence presented here indicates that MeV utilizes a PVRL4-mediated macropinocytosis pathway as a primary means of entry into the MCF7, HTB-20, and DLD-1 cancer cell lines (Fig. 10). Once MeV binds to PVRL4, Rac1/PAK1-dependent signaling pathways are activated, which trigger vigorous actin-mediated membrane protrusions. This is followed by MeV endocytosis. Although the intracellular trafficking of the macropinosome is poorly documented, the ribonucleocapsid of MeV, which enters cells in a pH-independent manner, is likely released prior to the fusion of macropinosomes with more acidic compartments of the endolysosomal pathway.
FIG 10.

Schematic model for MeV entry into MCF7 cancer cells. Once MeV binds to PVRL4 (1), the Rac1/PAK1-dependent signaling pathway is activated (2), which triggers vigorous actin-mediated membrane protrusions (3). This is followed by MeV endocytosis (4). Various inhibitors of the macropinocytosis pathway and their sites of action are shown in red.
MATERIALS AND METHODS
Antibodies and probes.
PAK1 (catalog number 2602S) rabbit polyclonal antibody was purchased from Cell Signaling Technology (Danvers, MA). Monoclonal anti-MeV H antibody produced in mouse (catalog number MAB8905) was obtained from Merck Millipore (Etobicoke, ON, Canada). Actin mouse monoclonal antibody (catalog number A3853), Alexa Fluor 647-conjugated goat anti-mouse antibody (catalog number SAB4600353), and 70-kDa FITC-dextran (catalog number 46945) were obtained from Sigma-Aldrich Canada (Oakville, ON, Canada). Peroxidase-conjugated goat anti-mouse antibody (catalog number 115-035-146) was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Peroxidase-conjugated goat anti-rabbit antibody (catalog number G21234), Texas Red-conjugated human transferrin (catalog number T2875), and Alexa Fluor 546-conjugated phalloidin (catalog number A22283) were obtained from Life Technologies (Burlington, ON, Canada). GST and Myc rabbit polyclonal antibodies were purchased from Abcam (Cambridge, MA).
Cell lines and viruses.
MCF7 human breast cancer cells were acquired from David Hoskin (Dalhousie University, Halifax, Canada). Vero cells were purchased from the American Type Culture Collection (Manassas, VA). Vero cells that express human SLAM (Vero.hSLAM) and PVRL4 (Vero.hPVRL4) were used in previous studies (40, 107, 108). The recombinant Ichinose-B 323 (IC323) (109) wild-type measles virus isolate expressing the eGFP reporter gene (wtMeV-eGFP) was obtained from Roberto Cattaneo (Mayo Clinic, Rochester, MN). To generate a recombinant IC323 wild-type measles virus expressing firefly luciferase (wtMeV-Luc), the transcription unit encoding eGFP located upstream of the MeV hemagglutinin ORF within the wtMeV-eGFP genome was replaced by the firefly luciferase ORF. Thereafter, wtMeV-Luc was rescued by reverse genetics (110). Viruses were propagated in B95a cells and titrated in Vero.hSLAM cells. Recombinant VSV-Luc was obtained from Brent Johnston (Dalhousie University, Canada).
Virus infection.
Cells were infected with viruses at various MOIs in TCID50 per cell in phosphate-buffered saline (PBS) for 1 h at 37°C. Cell monolayers were washed twice with PBS, incubated with Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal calf serum (FCS), and then harvested at the indicated times postinfection. TCID50 titers for measles virus were determined by 50% endpoint titration on Vero.hSLAM cells according to the Spearman-Kärber method (119).
Drug treatment and virus entry and infectivity assays.
Vero cell lines and MCF7 cells in serum-free medium were pretreated with 25 to 100 μM Dynasore, 50 to 100 μM EIPA, 40 μM cytochalasin D, 500 nM bafilomycin A1, and 1 μM latrunculin A for 30 to 60 min. Cells were washed twice in PBS prior infection. All drugs were purchased from Sigma-Aldrich Canada (Oakville, ON, Canada). To measure viral entry, wtMeV-Luc and VSV-Luc were adsorbed to cells for 60 min, followed by replacement with 5% FCS-containing medium. At 8 h postinfection, luciferase activity was read on a Glomax luminometer (Promega Corp., Madison, WI) by using the luciferase assay system kit purchased from Promega Corp. (Madison, WI). To measure infectivity, wtMeV-eGFP was used. After MeV infection, the inoculum was removed and replaced with fresh medium containing 200 μM FIP (catalog number C9405), purchased from Sigma-Aldrich Canada (Oakville, ON, Canada). Twenty hours later, samples were processed for FACS analysis.
Cytotoxicity assay.
To measure viable cells in proliferation (cytotoxicity assay) following drug treatment, a CellTiter 96 AQ One Solution cell proliferation assay kit was purchased from Promega Corp. (Madison, WI). Assays were performed according to the manufacturer's protocol by using the tetrazolium substrate 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and measuring the A490 of the formazan product that is produced in viable cells.
Plasmids, site-directed mutagenesis, and transfection.
Plasmids expressing WT and DN forms of Rac1 (111, 112), caveolin 1 (CAV) (113), PAK1 (114, 115), and dynamin 2 (DYN) (116) were obtained from Addgene (Cambridge, MA). To generate the dominant negative form of dynamin 2, site-directed mutagenesis (QuikChange II; Agilent Technologies) was performed on plasmid Dyn2-pmCherryN1 (Addgene, Cambridge, MA) to introduce the K44A mutation. This mutation imparts a defect in the GTPase activity of dynamin-2 (117, 118). Expression plasmids were transfected into cells by using Lipofectamine 2000 (Life Technologies, Burlington, ON, Canada) according to the manufacturer's protocol.
siRNA transfections.
siRNA duplexes targeting human PAK1 (catalog number 6361S) and negative-control siRNA (catalog number 6568S) were purchased from Cell Signaling Technology (Danvers, MA). Vero.hSLAM, Vero.hPVRL4, and MCF7 cells were plated at 30 to 40% confluence into each well of a 12-well plate the day before transfection. They were subsequently transfected with siRNA at concentrations of 100 nM using Lipofectamine 2000 (Life Technologies, Burlington, ON, Canada). Protein knockdowns were evaluated 72 h after siRNA transfection by Western blotting.
Western blot analysis.
Cell monolayers were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris HCl [pH 7.4], 1% NP-40, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA, 1 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 2 mM dithiothreitol, 1× protease inhibitor cocktail [Roche Diagnostics, Laval, QC, Canada]) for 15 min on ice. Cell lysates were clarified by centrifugation at 14,000 × g for 15 min at +4°C. SDS-PAGE and Western immunoblotting were carried out by using antibodies against PAK1 and actin.
MeV binding assay.
Cells were washed with PBS and dissociated by using a nonenzymatic cell dissociation solution (catalog number C5789) purchased from Sigma-Aldrich Canada (Oakville, ON, Canada). To assess the capacity for binding of MeV to PVRL4 and SLAM, Vero.hSLAM and Vero.hPVRL4 cells were incubated with wtMeV-Luc at an MOI of 10. Cells were washed three times with PBS containing 1% bovine serum albumin, 5 mM EDTA, and 0.1% sodium azide and incubated with an anti-MeV hemagglutinin antibody on ice for 60 min. The cells were washed prior to incubation with an Alexa Fluor 647-conjugated goat anti-mouse antibody for 45 min on ice. Cells were again washed to remove any unbound antibodies, fixed in 1% paraformaldehyde, and processed for FACS analysis.
Immunofluorescence.
Serum-starved Vero cell lines and MCF7 cells were exposed to wtMeV-Luc (MOIs of 1 and 10) at 4°C and incubated for 1 h. The temperature shift was performed after washing with PBS by using prewarmed (37°C) serum-free medium for 30 min. Afterwards, cells were fixed with 4% paraformaldehyde and permeabilized with PBS–0.2% Triton X-100 at room temperature. Alexa Fluor 546-conjugated phalloidin and DAPI were used to stain actin filaments and nuclei of cells, respectively.
Fluid-phase uptake assay.
Cells were washed with PBS and dissociated by using a nonenzymatic cell dissociation solution (catalog number C5789) purchased from Sigma-Aldrich Canada (Oakville, ON, Canada). wtMeV-Luc (MOI of 5) or PBS (internal experiment control) was then exposed to cells on ice for 1 h, and cells were shifted to 37°C by using prewarmed serum-free medium for the indicated time periods and then exposed to FITC-dextran (1 mg/ml) during the last 5 min. Dextran uptake was stopped by placing the cells on ice and washing the cells three times with cold PBS and once with low-pH buffer (0.1 M sodium acetate, 0.05 M NaCl [pH 5.5]) for 10 min. The cells were then processed for FACS analysis. The uptake of FITC-dextran was quantified as a percentage of FITC-dextran-positive cells, which was calculated by subtracting the value of the nonspecific FITC-dextran uptake (no virus) from the value obtained in the presence of the virus. Results are displayed as fold induction normalized to values under untreated conditions at time zero.
Internalization assay for transferrin.
Cells pretreated with 100 μM Dynasore were exposed to 50 μg/ml Texas Red-conjugated human transferrin for 30 min at 37°C. Cells were then washed three times with cold PBS and once with low-pH buffer (0.1 M sodium acetate, 0.05 M NaCl [pH 5.5]) for 10 min and further prepared for FACS analyses.
Flow cytometry analysis.
Samples were run on a Cyan ADP flow cytometer (Beckman Coulter, Mississauga, ON, Canada). Data were processed by using FCS Express (De Novo Software, Glendale, CA) and analyzed by using Flowing software (http://www.flowingsoftware.com/).
Microscopy.
Cells were viewed by phase-contrast and fluorescence microscopy using a Leica DMI4000B inverted microscope (Leica Microsystems, Concord, ON, Canada). A Zeiss LSM 510 microscope (Carl Zeiss Canada) was used to acquire images by oil immersion.
Data analysis.
Data were expressed as means ± standard errors of the means. Analysis of variance (ANOVA) was performed to identify statistically significant differences. P values of <0.05 were considered statistically significant. Data were processed by using GraphPad Instat software (GraphPad, La Jolla, CA).
Ethics statement.
The experiments in this article were performed at a biological safety level 2 facility in accordance with the regulations set forth by the Public Health Agency of Canada and the Canada Food and Drug Inspection Agency. This work did not involve experimentation with animals or human beings.
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institute for Health Research (CIHR) (MOP-114949 and MOP-142775) and the Canadian Breast Cancer Foundation-Atlantic Region. C.D.R. is a Canada Research Chair in Viral Vaccinology and Therapeutics (tier 1) and received an equipment grant from the Canadian Foundation for Innovation. S.D. was supported by a trainee award from the Beatrice Hunter Cancer Research Institute with funds provided by Cancer Care Nova Scotia as part of the Terry Fox Strategic Health Research Training Program in Cancer Research at the CIHR.
S.D. and C.D.R. conceived of and designed the experiments. S.D., G.S., and K.M.R. performed the experiments. S.D. and C.D.R. analyzed the data. S.D. and C.D.R. wrote the paper.
REFERENCES
- 1.Griffin DE. 2013. Measles virus, p 1042–1069. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Moss WJ, Griffin DE. 2012. Measles. Lancet 379:153–164. doi: 10.1016/S0140-6736(10)62352-5. [DOI] [PubMed] [Google Scholar]
- 3.Avota E, Gassert E, Schneider-Schaulies S. 2010. Measles virus-induced immunosuppression: from effectors to mechanisms. Med Microbiol Immunol 199:227–237. doi: 10.1007/s00430-010-0152-3. [DOI] [PubMed] [Google Scholar]
- 4.Griffin DE. 2010. Measles virus-induced suppression of immune responses. Immunol Rev 236:176–189. doi: 10.1111/j.1600-065X.2010.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Vries RD, McQuaid S, van Amerongen G, Yuksel S, Verburgh RJ, Osterhaus AD, Duprex WP, de Swart RL. 2012. Measles immune suppression: lessons from the macaque model. PLoS Pathog 8:e1002885. doi: 10.1371/journal.ppat.1002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Vries RD, de Swart RL. 2014. Measles immune suppression: functional impairment or numbers game? PLoS Pathog 10:e1004482. doi: 10.1371/journal.ppat.1004482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mueller N, Avota E, Collenburg L, Grassme H, Schneider-Schaulies S. 2014. Neutral sphingomyelinase in physiological and measles virus induced T cell suppression. PLoS Pathog 10:e1004574. doi: 10.1371/journal.ppat.1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosby SL, Duprex WP, Hamill LA, Ludlow M, McQuaid S. 2002. Approaches in the understanding of morbillivirus neurovirulence. J Neurovirol 8(Suppl 2):85–90. doi: 10.1080/13550280290167975. [DOI] [PubMed] [Google Scholar]
- 9.Schneider-Schaulies J, Meulen V, Schneider-Schaulies S. 2003. Measles infection of the central nervous system. J Neurovirol 9:247–252. doi: 10.1080/13550280390193993. [DOI] [PubMed] [Google Scholar]
- 10.Buchanan R, Bonthius DJ. 2012. Measles virus and associated central nervous system sequelae. Semin Pediatr Neurol 19:107–114. doi: 10.1016/j.spen.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe S, Ohno S, Shirogane Y, Suzuki SO, Koga R, Yanagi Y. 2015. Measles virus mutants possessing the fusion protein with enhanced fusion activity spread effectively in neuronal cells, but not in other cells, without causing strong cytopathology. J Virol 89:2710–2717. doi: 10.1128/JVI.03346-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin DE. 2014. Measles virus and the nervous system. Handb Clin Neurol 123:577–590. doi: 10.1016/B978-0-444-53488-0.00027-4. [DOI] [PubMed] [Google Scholar]
- 13.Dabbagh A, Gacic-Dobo M, Simons E, Featherstone D, Strebel P, Okwo-Bele JM, Hoekstra E, Chopra M, Uvicanin A, Cochi S. 2009. Global measles mortality, 2000-2008. MMWR Morb Mortal Wkly Rep 58:1321–1326.19959985 [Google Scholar]
- 14.Gastanaduy PA, Redd SB, Fiebelkorn AP, Rota JS, Rota PA, Bellini WJ, Seward JF, Wallace GS, Division of Viral Disease, National Center for Immunization and Respiratory Diseases, CDC. 2014. Measles—United States, January 1-May 23, 2014. MMWR Morb Mortal Wkly Rep 63:496–499. [PMC free article] [PubMed] [Google Scholar]
- 15.Perry RT, Dabbagh AJ, Gacic-Dobo M, Liu JL, Simons EA, Featherstone DA, Strebel PM, Okwo-Bele JM, Goodson JL. 2012. Progress in global measles control, 2000-2010. MMWR Morb Mortal Wkly Rep 61:73–78.22298303 [Google Scholar]
- 16.Perry RT, Gacic-Dobo M, Dabbagh A, Mulders MN, Strebel PM, Okwo-Bele JM, Rota PA, Goodson JL, Centers for Disease Control and Prevention. 2014. Global control and regional elimination of measles, 2000-2012. MMWR Morb Mortal Wkly Rep 63:103–107. [PMC free article] [PubMed] [Google Scholar]
- 17.Tanne JH. 2014. Rise in US measles cases is blamed on unimmunized travelers. BMJ 348:g3478. doi: 10.1136/bmj.g3478. [DOI] [PubMed] [Google Scholar]
- 18.Centers for Disease Control and Prevention. 2017. Measles cases and outbreaks. https://www.cdc.gov/measles/cases-outbreaks.html.
- 19.D'Souza RM, D'Souza R. 2002. Vitamin A for the treatment of children with measles—a systematic review. J Trop Pediatr 48:323–327. doi: 10.1093/tropej/48.6.323. [DOI] [PubMed] [Google Scholar]
- 20.D'Souza RM, D'Souza R. 2002. Vitamin A for preventing secondary infections in children with measles—a systematic review. J Trop Pediatr 48:72–77. doi: 10.1093/tropej/48.2.72. [DOI] [PubMed] [Google Scholar]
- 21.Trottier C, Colombo M, Mann KK, Miller WH Jr, Ward BJ. 2009. Retinoids inhibit measles virus through a type I IFN-dependent bystander effect. FASEB J 23:3203–3212. doi: 10.1096/fj.09-129288. [DOI] [PubMed] [Google Scholar]
- 22.Richardson CD, Scheid A, Choppin PW. 1980. Specific inhibition of paramyxovirus and myxovirus replication by oligopeptides with amino acid sequences similar to those at the N-termini of the F1 or HA2 viral polypeptides. Virology 105:205–222. doi: 10.1016/0042-6822(80)90168-3. [DOI] [PubMed] [Google Scholar]
- 23.Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. 2004. A target site for template-based design of measles virus entry inhibitors. Proc Natl Acad Sci U S A 101:5628–5633. doi: 10.1073/pnas.0308520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White LK, Yoon JJ, Lee JK, Sun A, Du Y, Fu H, Snyder JP, Plemper RK. 2007. Nonnucleoside inhibitor of measles virus RNA-dependent RNA polymerase complex activity. Antimicrob Agents Chemother 51:2293–2303. doi: 10.1128/AAC.00289-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraser KB, Martin SJ. 1978. Measles virus and its biology (experimental virology). Academic Press, London, United Kingdom. [Google Scholar]
- 26.Richardson C, Hull D, Greer P, Hasel K, Berkovich A, Englund G, Bellini W, Rima B, Lazzarini R. 1986. The nucleotide sequence of the mRNA encoding the fusion protein of measles virus (Edmonston strain): a comparison of fusion proteins from several different paramyxoviruses. Virology 155:508–523. doi: 10.1016/0042-6822(86)90212-6. [DOI] [PubMed] [Google Scholar]
- 27.Baker KA, Dutch RE, Lamb RA, Jardetzky TS. 1999. Structural basis for paramyxovirus-mediated membrane fusion. Mol Cell 3:309–319. doi: 10.1016/S1097-2765(00)80458-X. [DOI] [PubMed] [Google Scholar]
- 28.Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol 4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 29.Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. 1996. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol 12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 30.Navaratnarajah CK, Oezguen N, Rupp L, Kay L, Leonard VH, Braun W, Cattaneo R. 2011. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat Struct Mol Biol 18:128–134. doi: 10.1038/nsmb.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navaratnarajah CK, Negi S, Braun W, Cattaneo R. 2012. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J Biol Chem 287:38543–38551. doi: 10.1074/jbc.M112.410563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melikyan GB. 2014. HIV entry: a game of hide-and-fuse? Curr Opin Virol 4:1–7. doi: 10.1016/j.coviro.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 34.Hsu EC, Iorio C, Sarangi F, Khine AA, Richardson CD. 2001. CDw150(SLAM) is a receptor for a lymphotropic strain of measles virus and may account for the immunosuppressive properties of this virus. Virology 279:9–21. doi: 10.1006/viro.2000.0711. [DOI] [PubMed] [Google Scholar]
- 35.Wang N, Satoskar A, Faubion W, Howie D, Okamoto S, Feske S, Gullo C, Clarke K, Sosa MR, Sharpe AH, Terhorst C. 2004. The cell surface receptor SLAM controls T cell and macrophage functions. J Exp Med 199:1255–1264. doi: 10.1084/jem.20031835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baron MD. 2005. Wild-type Rinderpest virus uses SLAM (CD150) as its receptor. J Gen Virol 86:1753–1757. doi: 10.1099/vir.0.80836-0. [DOI] [PubMed] [Google Scholar]
- 37.Seki F, Ono N, Yamaguchi R, Yanagi Y. 2003. Efficient isolation of wild strains of canine distemper virus in Vero cells expressing canine SLAM (CD150) and their adaptability to marmoset B95a cells. J Virol 77:9943–9950. doi: 10.1128/JVI.77.18.9943-9950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noyce RS, Delpeut S, Richardson CD. 2013. Dog nectin-4 is an epithelial cell receptor for canine distemper virus that facilitates virus entry and syncytia formation. Virology 436:210–220. doi: 10.1016/j.virol.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 41.Pratakpiriya W, Seki F, Otsuki N, Sakai K, Fukuhara H, Katamoto H, Hirai T, Maenaka K, Techangamsuwan S, Lan NT, Takeda M, Yamaguchi R. 2012. Nectin4 is an epithelial cell receptor for canine distemper virus and involved in neurovirulence. J Virol 86:10207–10210. doi: 10.1128/JVI.00824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birch J, Juleff N, Heaton MP, Kalbfleisch T, Kijas J, Bailey D. 2013. Characterization of ovine nectin-4, a novel peste des petits ruminants virus receptor. J Virol 87:4756–4761. doi: 10.1128/JVI.02792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delpeut S, Noyce RS, Richardson CD. 2014. The tumor-associated marker, PVRL4 (nectin-4), is the epithelial receptor for morbilliviruses. Viruses 6:2268–2286. doi: 10.3390/v6062268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dorig RE, Marcil A, Chopra A, Richardson CD. 1993. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75:295–305. doi: 10.1016/0092-8674(93)80071-L. [DOI] [PubMed] [Google Scholar]
- 45.Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D. 1993. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol 67:6025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buckland R, Wild TF. 1997. Is CD46 the cellular receptor for measles virus? Virus Res 48:1–9. doi: 10.1016/S0168-1702(96)01421-9. [DOI] [PubMed] [Google Scholar]
- 47.Hsu EC, Sarangi F, Iorio C, Sidhu MS, Udem SA, Dillehay DL, Xu W, Rota PA, Bellini WJ, Richardson CD. 1998. A single amino acid change in the hemagglutinin protein of measles virus determines its ability to bind CD46 and reveals another receptor on marmoset B cells. J Virol 72:2905–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanaka K, Xie M, Yanagi Y. 1998. The hemagglutinin of recent measles virus isolates induces cell fusion in a marmoset cell line, but not in other CD46-positive human and monkey cell lines, when expressed together with the F protein. Arch Virol 143:213–225. doi: 10.1007/s007050050281. [DOI] [PubMed] [Google Scholar]
- 49.Nielsen L, Blixenkrone-Moller M, Thylstrup M, Hansen NJ, Bolt G. 2001. Adaptation of wild-type measles virus to CD46 receptor usage. Arch Virol 146:197–208. doi: 10.1007/s007050170169. [DOI] [PubMed] [Google Scholar]
- 50.Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu Rev Biochem 78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 51.Hansen CG, Nichols BJ. 2009. Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci 122:1713–1721. doi: 10.1242/jcs.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumari S, Mg S, Mayor S. 2010. Endocytosis unplugged: multiple ways to enter the cell. Cell Res 20:256–275. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. 1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 54.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu Rev Biochem 79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 55.Yamauchi Y, Helenius A. 2013. Virus entry at a glance. J Cell Sci 126:1289–1295. doi: 10.1242/jcs.119685. [DOI] [PubMed] [Google Scholar]
- 56.Mercer J, Helenius A. 2009. Virus entry by macropinocytosis. Nat Cell Biol 11:510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- 57.Mercer J, Helenius A. 2012. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol 15:490–499. doi: 10.1016/j.mib.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 58.Dharmawardhane S, Schurmann A, Sells MA, Chernoff J, Schmid SL, Bokoch GM. 2000. Regulation of macropinocytosis by p21-activated kinase-1. Mol Biol Cell 11:3341–3352. doi: 10.1091/mbc.11.10.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liberali P, Kakkonen E, Turacchio G, Valente C, Spaar A, Perinetti G, Bockmann RA, Corda D, Colanzi A, Marjomaki V, Luini A. 2008. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J 27:970–981. doi: 10.1038/emboj.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haga Y, Miwa N, Jahangeer S, Okada T, Nakamura S. 2009. CtBP1/BARS is an activator of phospholipase D1 necessary for agonist-induced macropinocytosis. EMBO J 28:1197–1207. doi: 10.1038/emboj.2009.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. 2010. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol 188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 63.Amstutz B, Gastaldelli M, Kalin S, Imelli N, Boucke K, Wandeler E, Mercer J, Hemmi S, Greber UF. 2008. Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J 27:956–969. doi: 10.1038/emboj.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalin S, Amstutz B, Gastaldelli M, Wolfrum N, Boucke K, Havenga M, DiGennaro F, Liska N, Hemmi S, Greber UF. 2010. Macropinocytotic uptake and infection of human epithelial cells with species B2 adenovirus type 35. J Virol 84:5336–5350. doi: 10.1128/JVI.02494-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krieger SE, Kim C, Zhang L, Marjomaki V, Bergelson JM. 2013. Echovirus 1 entry into polarized Caco-2 cells depends on dynamin, cholesterol, and cellular factors associated with macropinocytosis. J Virol 87:8884–8895. doi: 10.1128/JVI.03415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. 2010. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog 6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pernet O, Pohl C, Ainouze M, Kweder H, Buckland R. 2009. Nipah virus entry can occur by macropinocytosis. Virology 395:298–311. doi: 10.1016/j.virol.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 69.Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. 2013. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9:e1003309. doi: 10.1371/journal.ppat.1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gobeil LA, Lodge R, Tremblay MJ. 2013. Macropinocytosis-like HIV-1 internalization in macrophages is CCR5 dependent and leads to efficient but delayed degradation in endosomal compartments. J Virol 87:735–745. doi: 10.1128/JVI.01802-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crimeen-Irwin B, Ellis S, Christiansen D, Ludford-Menting MJ, Milland J, Lanteri M, Loveland BE, Gerlier D, Russell SM. 2003. Ligand binding determines whether CD46 is internalized by clathrin-coated pits or macropinocytosis. J Biol Chem 278:46927–46937. doi: 10.1074/jbc.M308261200. [DOI] [PubMed] [Google Scholar]
- 72.Frecha C, Levy C, Costa C, Negre D, Amirache F, Buckland R, Russell SJ, Cosset FL, Verhoeyen E. 2011. Measles virus glycoprotein-pseudotyped lentiviral vector-mediated gene transfer into quiescent lymphocytes requires binding to both SLAM and CD46 entry receptors. J Virol 85:5975–5985. doi: 10.1128/JVI.00324-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karjalainen M, Kakkonen E, Upla P, Paloranta H, Kankaanpaa P, Liberali P, Renkema GH, Hyypia T, Heino J, Marjomaki V. 2008. A raft-derived, Pak1-regulated entry participates in alpha2beta1 integrin-dependent sorting to caveosomes. Mol Biol Cell 19:2857–2869. doi: 10.1091/mbc.E07-10-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sever S. 2002. Dynamin and endocytosis. Curr Opin Cell Biol 14:463–467. doi: 10.1016/S0955-0674(02)00347-2. [DOI] [PubMed] [Google Scholar]
- 75.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 76.Regan AD, Whittaker GR. 2013. Entry of rhabdoviruses into animal cells. Adv Exp Med Biol 790:167–177. doi: 10.1007/978-1-4614-7651-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog 6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Norrby E. 1971. The effect of a carbobenzoxy tripeptide on the biological activities of measles virus. Virology 44:599–608. doi: 10.1016/0042-6822(71)90374-6. [DOI] [PubMed] [Google Scholar]
- 79.Norrby E, Sievertsson H. 1974. Effect of certain polypeptides on the biological activities of measles virus. Antimicrob Agents Chemother 5:426–430. doi: 10.1128/AAC.5.4.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goncalves-Carneiro D, McKeating JA, Bailey D. 18 January 2017. The measles virus receptor SLAMF1 can mediate particle endocytosis. J Virol doi: 10.1128/JVI.02255-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fabre-Lafay S, Monville F, Garrido-Urbani S, Berruyer-Pouyet C, Ginestier C, Reymond N, Finetti P, Sauvan R, Adelaide J, Geneix J, Lecocq E, Popovici C, Dubreuil P, Viens P, Goncalves A, Charafe-Jauffret E, Jacquemier J, Birnbaum D, Lopez M. 2007. Nectin-4 is a new histological and serological tumor associated marker for breast cancer. BMC Cancer 7:73. doi: 10.1186/1471-2407-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cossart P, Helenius A. 2014. Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol 6:a016972. doi: 10.1101/cshperspect.a016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mercer J, Greber UF. 2013. Virus interactions with endocytic pathways in macrophages and dendritic cells. Trends Microbiol 21:380–388. doi: 10.1016/j.tim.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 84.Kawakatsu T, Ogita H, Fukuhara T, Fukuyama T, Minami Y, Shimizu K, Takai Y. 2005. Vav2 as a Rac-GDP/GTP exchange factor responsible for the nectin-induced, c-Src- and Cdc42-mediated activation of Rac. J Biol Chem 280:4940–4947. doi: 10.1074/jbc.M408710200. [DOI] [PubMed] [Google Scholar]
- 85.Fukuyama T, Ogita H, Kawakatsu T, Fukuhara T, Yamada T, Sato T, Shimizu K, Nakamura T, Matsuda M, Takai Y. 2005. Involvement of the c-Src-Crk-C3G-Rap1 signaling in the nectin-induced activation of Cdc42 and formation of adherens junctions. J Biol Chem 280:815–825. doi: 10.1074/jbc.M411099200. [DOI] [PubMed] [Google Scholar]
- 86.Detre C, Keszei M, Romero X, Tsokos GC, Terhorst C. 2010. SLAM family receptors and the SLAM-associated protein (SAP) modulate T cell functions. Semin Immunopathol 32:157–171. doi: 10.1007/s00281-009-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pavlova NN, Pallasch C, Elia AE, Braun CJ, Westbrook TF, Hemann M, Elledge SJ. 2013. A role for PVRL4-driven cell-cell interactions in tumorigenesis. eLife 2:e00358. doi: 10.7554/eLife.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kasahara K, Nakayama Y, Sato I, Ikeda K, Hoshino M, Endo T, Yamaguchi N. 2007. Role of Src-family kinases in formation and trafficking of macropinosomes. J Cell Physiol 211:220–232. doi: 10.1002/jcp.20931. [DOI] [PubMed] [Google Scholar]
- 89.Mettlen M, Platek A, Van Der Smissen P, Carpentier S, Amyere M, Lanzetti L, de Diesbach P, Tyteca D, Courtoy PJ. 2006. Src triggers circular ruffling and macropinocytosis at the apical surface of polarized MDCK cells. Traffic 7:589–603. doi: 10.1111/j.1600-0854.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 90.Donepudi M, Resh MD. 2008. c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell Signal 20:1359–1367. doi: 10.1016/j.cellsig.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Knuchel MC, Marty RR, Morin TN, Ilter O, Zuniga A, Naim HY. 2013. Relevance of a pre-existing measles immunity prior immunization with a recombinant measles virus vector. Hum Vaccin Immunother 9:599–606. doi: 10.4161/hv.23241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lorin C, Mollet L, Delebecque F, Combredet C, Hurtrel B, Charneau P, Brahic M, Tangy F. 2004. A single injection of recombinant measles virus vaccines expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J Virol 78:146–157. doi: 10.1128/JVI.78.1.146-157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Msaouel P, Opyrchal M, Domingo Musibay E, Galanis E. 2013. Oncolytic measles virus strains as novel anticancer agents. Expert Opin Biol Ther 13:483–502. doi: 10.1517/14712598.2013.749851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rager-Zisman B, Bazarsky E, Skibin A, Chamney S, Belmaker I, Shai I, Kordysh E, Griffin DE. 2003. The effect of measles-mumps-rubella (MMR) immunization on the immune responses of previously immunized primary school children. Vaccine 21:2580–2588. doi: 10.1016/S0264-410X(03)00053-7. [DOI] [PubMed] [Google Scholar]
- 95.Wong-Chew RM, Beeler JA, Audet S, Santos JI. 2003. Cellular and humoral immune responses to measles in immune adults re-immunized with measles vaccine. J Med Virol 70:276–280. doi: 10.1002/jmv.10390. [DOI] [PubMed] [Google Scholar]
- 96.Fabre-Lafay S, Garrido-Urbani S, Reymond N, Goncalves A, Dubreuil P, Lopez M. 2005. Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-alpha-converting enzyme (TACE)/ADAM-17. J Biol Chem 280:19543–19550. doi: 10.1074/jbc.M410943200. [DOI] [PubMed] [Google Scholar]
- 97.Takano A, Ishikawa N, Nishino R, Masuda K, Yasui W, Inai K, Nishimura H, Ito H, Nakayama H, Miyagi Y, Tsuchiya E, Kohno N, Nakamura Y, Daigo Y. 2009. Identification of nectin-4 oncoprotein as a diagnostic and therapeutic target for lung cancer. Cancer Res 69:6694–6703. doi: 10.1158/0008-5472.CAN-09-0016. [DOI] [PubMed] [Google Scholar]
- 98.Derycke MS, Pambuccian SE, Gilks CB, Kalloger SE, Ghidouche A, Lopez M, Bliss RL, Geller MA, Argenta PA, Harrington KM, Skubitz AP. 2010. Nectin 4 overexpression in ovarian cancer tissues and serum: potential role as a serum biomarker. Am J Clin Pathol 134:835–845. doi: 10.1309/AJCPGXK0FR4MHIHB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raveh S, Gavert N, Spiegel I, Ben-Ze'ev A. 2009. The cell adhesion nectin-like molecules (Necl) 1 and 4 suppress the growth and tumorigenic ability of colon cancer cells. J Cell Biochem 108:326–336. doi: 10.1002/jcb.22258. [DOI] [PubMed] [Google Scholar]
- 100.Redelman-Sidi G, Iyer G, Solit DB, Glickman MS. 2013. Oncogenic activation of Pak1-dependent pathway of macropinocytosis determines BCG entry into bladder cancer cells. Cancer Res 73:1156–1167. doi: 10.1158/0008-5472.CAN-12-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, Bar-Sagi D. 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Amyere M, Payrastre B, Krause U, Van Der Smissen P, Veithen A, Courtoy PJ. 2000. Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol Biol Cell 11:3453–3467. doi: 10.1091/mbc.11.10.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ha KD, Bidlingmaier SM, Zhang Y, Su Y, Liu B. 2014. High-content analysis of antibody phage-display library selection outputs identifies tumor selective macropinocytosis-dependent rapidly internalizing antibodies. Mol Cell Proteomics 13:3320–3331. doi: 10.1074/mcp.M114.039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qian Y, Wang X, Liu Y, Li Y, Colvin RA, Tong L, Wu S, Chen X. 2014. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett 351:242–251. doi: 10.1016/j.canlet.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 105.Rosenbaum RS, Park MC, Fleischmann J. 1996. Intravesical bacille Calmette-Guerin therapy for muscle invasive bladder cancer. Urology 47:208–211. doi: 10.1016/S0090-4295(99)80418-X. [DOI] [PubMed] [Google Scholar]
- 106.Gontero P, Bohle A, Malmstrom PU, O'Donnell MA, Oderda M, Sylvester R, Witjes F. 2010. The role of bacillus Calmette-Guerin in the treatment of non-muscle-invasive bladder cancer. Eur Urol 57:410–429. doi: 10.1016/j.eururo.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 107.Welstead GG, Iorio C, Draker R, Bayani J, Squire J, Vongpunsawad S, Cattaneo R, Richardson CD. 2005. Measles virus replication in lymphatic cells and organs of CD150 (SLAM) transgenic mice. Proc Natl Acad Sci U S A 102:16415–16420. doi: 10.1073/pnas.0505945102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Welstead GG, Hsu EC, Iorio C, Bolotin S, Richardson CD. 2004. Mechanism of CD150 (SLAM) down regulation from the host cell surface by measles virus hemagglutinin protein. J Virol 78:9666–9674. doi: 10.1128/JVI.78.18.9666-9674.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J Virol 76:6743–6749. doi: 10.1128/JVI.76.13.6743-6749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Christiansen G, Billeter MA. 1995. Rescue of measles viruses from cloned DNA. EMBO J 14:5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. 2000. Localized Rac activation dynamics visualized in living cells. Science 290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- 112.Subauste MC, Von Herrath M, Benard V, Chamberlain CE, Chuang TH, Chu K, Bokoch GM, Hahn KM. 2000. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem 275:9725–9733. doi: 10.1074/jbc.275.13.9725. [DOI] [PubMed] [Google Scholar]
- 113.Hayer A, Stoeber M, Bissig C, Helenius A. 2010. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic 11:361–382. doi: 10.1111/j.1600-0854.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- 114.Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. 1997. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol 7:202–210. doi: 10.1016/S0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- 115.Xiao GH, Beeser A, Chernoff J, Testa JR. 2002. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem 277:883–886. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- 116.Taylor MJ, Perrais D, Merrifield CJ. 2011. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol 9:e1000604. doi: 10.1371/journal.pbio.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. 2007. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci 120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 118.Ochoa GC, Slepnev VI, Neff L, Ringstad N, Takei K, Daniell L, Kim W, Cao H, McNiven M, Baron R, De Camilli P. 2000. A functional link between dynamin and the actin cytoskeleton at podosomes. J Cell Biol 150:377–389. doi: 10.1083/jcb.150.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ramakrishnan MA. 2016. Determination of 50% endpoint titer using a simple formula. World J Virol 5:85–86. doi: 10.5501/wjv.v5.i2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]