

ABSTRACT
Among prion infections, two scenarios of prion spread are generally observed: (i) early lymphoid tissue replication or (ii) direct neuroinvasion without substantial antecedent lymphoid amplification. In nature, cervids are infected with chronic wasting disease (CWD) prions by oral and nasal mucosal exposure, and studies of early CWD pathogenesis have implicated pharyngeal lymphoid tissue as the earliest sites of prion accumulation. However, knowledge of chronological events in prion spread during early infection remains incomplete. To investigate this knowledge gap in early CWD pathogenesis, we exposed white-tailed deer to CWD prions by mucosal routes and performed serial necropsies to assess PrPCWD tissue distribution by real-time quaking-induced conversion (RT-QuIC) and tyramide signal amplification immunohistochemistry (TSA-IHC). Although PrPCWD was not detected by either method in the initial days (1 and 3) postexposure, we observed PrPCWD seeding activity and follicular immunoreactivity in oropharyngeal lymphoid tissues at 1 and 2 months postexposure (MPE). At 3 MPE, PrPCWD replication had expanded to all systemic lymphoid tissues. By 4 MPE, the PrPCWD burden in all lymphoid tissues had increased and approached levels observed in terminal disease, yet there was no evidence of nervous system invasion. These results indicate the first site of CWD prion entry is in the oropharynx, and the initial phase of prion amplification occurs in the oropharyngeal lymphoid tissues followed by rapid dissemination to systemic lymphoid tissues. This lymphoid replication phase appears to precede neuroinvasion.
IMPORTANCE Chronic wasting disease (CWD) is a universally fatal transmissible spongiform encephalopathy affecting cervids, and natural infection occurs through oral and nasal mucosal exposure to infectious prions. Terminal disease is characterized by PrPCWD accumulation in the brain and lymphoid tissues of affected animals. However, the initial sites of prion accumulation and pathways of prion spread during early CWD infection remain unknown. To investigate the chronological events of early prion pathogenesis, we exposed deer to CWD prions and monitored the tissue distribution of PrPCWD over the first 4 months of infection. We show CWD uptake occurs in the oropharynx with initial prion replication in the draining oropharyngeal lymphoid tissues, rapidly followed by dissemination to systemic lymphoid tissues without evidence of neuroinvasion. These data highlight the two phases of CWD infection: a robust prion amplification in systemic lymphoid tissues prior to neuroinvasion and establishment of a carrier state.
KEYWORDS: chronic wasting disease, prions
INTRODUCTION
Chronic wasting disease (CWD) is a naturally occurring transmissible spongiform encephalopathy (TSE) of cervids, including deer, elk, and moose (1). CWD was first recognized in Colorado in 1967 and has since spread to captive and free-ranging cervid populations in 24 states and three additional countries, Canada, the Republic of Korea, and Norway (2, 3). Clinical signs include progressive wasting despite polyphagia, bruxism, head tremors, and ataxia (1, 4). Like other transmissible spongiform encephalopathies, clinical disease is associated with spongiform change and accumulation of the infectious prion isoform, PrPCWD, in the central nervous system, most consistently observed affecting the nucleus of the vagus nerve in the obex region of the medulla oblongata (5–7).
The majority of natural, horizontal CWD transmission occurs by mucosal contact with infectious prions, either by contact with infected animals or by indirect environmental exposure associated with foraging and rutting (8). Transmission of CWD prions by mucosal exposure has been experimentally confirmed by aerosol and oral exposure to prions, with minor mucosal lesions in the oral cavity enhancing transmission efficiency (9, 10). In infected cervids, CWD prions have been identified in body fluids and excreta, including blood, saliva, urine, and feces, all of which are potential natural sources of environmental contamination (8, 11–14). Once CWD has contaminated the environment, it has been shown to persist and remain infectious for years, potentially through binding to soil (15). Vertical transmission has also been described; however, its role in the spread of CWD remains unclear (16).
As with other TSEs, the pathogenesis of CWD is influenced by the PRNP gene sequence, and four polymorphisms have been identified thus far in white-tailed deer (WTD): Q95H, G96S, A116G, and Q226K (17). Of these polymorphisms, Q95, G96, and A116 are more common in the deer population and reported with higher frequency in natural cases of CWD (18). Studies have demonstrated at least one copy of the S96 allelic variation prolongs the CWD incubation period in deer, up to 230%, and appears to impede but not prevent PrPCWD conversion (2, 19, 20).
Investigations of early prion pathogenesis in naturally acquired disease have described two patterns of prion spread: an early lymphoid replication phase prior to accumulation in the brain (neuroinvasion), as in scrapie, and exclusive central nervous system prion replication, as reported in bovine spongiform encephalopathy (BSE) (21, 22). Previous studies suggest CWD pathogenesis shares many similarities with scrapie. A survey of mule deer from areas where CWD is endemic identified PrPCWD lymphoid replication in tonsil biopsy specimens from animals that did not display overt signs of clinical disease (23). Experimental studies of CWD transmission in deer following relatively large oral challenges with CWD-positive brain homogenate provided a glimpse of the disease at individual early time points and demonstrated PrPCWD in alimentary-associated lymphoid tissues at 42 days and in systemic lymphoid tissues at 90 days postexposure (24, 25). However, there remains relatively little information regarding the chronological progression of PrPCWD in deer tissues during the early postexposure period following a smaller, more physiologically relevant dose.
To further investigate early CWD pathogenesis, we examined the temporal distribution and accumulation of PrPCWD after mucosal exposure in white-tailed deer using immunohistochemistry and real-time conversion. Following mucosal exposure, we found the earliest evidence of prion replication in the upper alimentary tract in the lymphoid tissues draining the oropharyngeal cavity, followed by rapid dissemination to systemic lymphoid tissues without evidence of prion neuroinvasion. This study documents the earliest sites of CWD prion entry and highlights the importance of the lymphoid system in early CWD pathogenesis.
RESULTS
CWD prions reach and amplify in lymphoid tissues early in infection. (i) Twenty-four and 72 h postmucosal exposure.
In WTD collected at 24 or 72 h postoronasal CWD exposure, occasional real-time quaking-induced conversion (RT-QuIC) of a minimal number of sample replicates of the prescapular, mandibular, and hepatogastric lymph nodes (LN) displayed amyloid seeding activity; however, the average rates of amyloid seeding for these samples did not statistically differ from those of corresponding negative-control tissues (P value of >0.05 by unpaired t test) (Fig. 1). Analysis of tissues by tyramide signal amplification immunohistochemistry (TSA-IHC) from these early time points did not detect any PrPCWD immunoreactivity.
FIG 1.

PrPCWD amyloid seeding activity is not detected at 24 and 72 h following oronasal exposure. (A) RT-QuIC analysis of tissues collected 24 h postoronasal exposure. Rare tissues, such as the prescapular LN, mandibular LN, and RAMALT, displayed minor SHrPrP substrate conversion; however, rates of amyloid formation were not statistically different from those of corresponding negative-control tissues. Data from two deer (963 and 965) are displayed as means and standard errors of the means (SEM) from 8 replicates, representing 2 separate experiments. LN, lymph node; Retro LN, retropharyngeal lymph node; RAMALT, rectoanal mucosa-associated lymphoid tissue; VNO, vomeronasal organ. (B) RT-QuIC analysis of tissues collected 72 h postmucosal exposure. A few tissues, such as the prescapular LN and spleen, displayed minor SHrPrP substrate amyloid conversion; however, rates of amyloid formation were not statistically different from those of the corresponding negative-control tissues (P > 0.05, unpaired t test). Data from two deer (967 and 968) are displayed as means and SEM from 8 replicates, representing 2 separate experiments. (C) RT-QuIC analysis of negative-control tissues collected 72 h postmucosal exposure. Rare tissues displayed minimal spontaneous amyloid formation of SHrPrP substrate. Data from one deer are displayed as means and SEM from a minimum of 8 replicates and 2 separate experiments.
(ii) One month postexposure.
At 1 month postexposure (MPE), statistically significant PrPCWD amyloid seeding activity (P value of <0.01 by unpaired t test) was detected in the retropharyngeal lymph nodes from two 96GG WTD and in the parotid lymph node from one 96GG WTD (Fig. 2A). In comparison, no significant amyloid seeding was observed in tissues from 96GS WTD at 1 MPE. The negative results in the 96GS deer suggested that the amyloid seeding detected in tissues from 96GG WTD at this collection time is indicative of de novo prion generation in these tissues and not detection of inoculum.
FIG 2.

PrPCWD is detected in retropharyngeal and parotid lymph nodes in 96GG deer at 1 month postexposure. (A) RT-QuIC analysis of 96GG, 96GS, and negative-control tissues collected at 1 MPE. Three samples displayed statistically significant RT-QuIC amyloid seeding: 1219 retropharyngeal lymph node, 1202 retropharyngeal lymph node, and 1219 parotid lymph node (P < 0.05, unpaired t test). No statistically significant PrPCWD amyloid seeding formation was observed in 96GS WTD tissues. Data from three 96GG deer (1174, 1219, and 1202), one 96GS deer, and one negative-control deer are displayed as means and SEM of 12 replicates from 3 separate experiments. (B) Representative TSA-IHC from 96GG WTD collected at 1 MPE. Minimal PrPCWD immunoreactivity in germinal centers from less than five lymphoid follicles was detected in tonsils from two 96GG WTD at 1 MPE. No PrPCWD immunoreactivity was observed in retropharyngeal lymph nodes, ileocecocolic lymph nodes, or negative-control tissues. All images are at ×200 magnification; scale bar, 50 μm.
TSA-IHC evaluation of lymphoid tissues at 1 MPE demonstrated faint granular PrPCWD immunoreactivity in germinal centers of a few (1 to 8) tonsil lymphoid follicles in two of the three 96GG WTD examined (Fig. 2B). Similar faint granular PrPCWD immunoreactivity was detected in the parotid lymph node sample that displayed amyloid seeding detected by RT-QuIC. Despite statistically significant RT-QuIC amyloid seeding in retropharyngeal lymph nodes, TSA-IHC failed to demonstrate PrPCWD immunoreactivity. The variation in PrPCWD tissue detection at this early disease time point is likely due to unequal tissue distribution and sectioning differences. No PrPCWD immunoreactivity was observed in 96GS tissues.
(iii) Two months postexposure.
At 2 MPE, statistically significant PrPCWD seeding activity (P value of <0.01 by unpaired t test) was consistently detected in lymph nodes draining the head and neck, including the tonsil, retropharyngeal lymph node, and mandibular lymph node (Fig. 3A). In addition, one 96GG WTD had statistically significant seeding activity in the parotid lymph node (P value of <0.05 by unpaired t test). Compared to 1 MPE (Fig. 2A), the tonsil and retropharyngeal lymph node at 2 MPE displayed faster amyloid formation rates, consistent with a greater accumulation of PrPCWD in these tissues. As at 1 MPE, lymphoid tissues from 96GS WTD at 2 MPE contained no detectable PrPCWD amyloid seeding activity.
FIG 3.

PrPCWD is detected in oropharyngeal lymphoid tissues in 96GG deer at 2 months postexposure (2-MPE collection). (A) RT-QuIC of 96GG WTD, 96GS WTD, and negative-control tissues collected at 2 MPE. PrPCWD amyloid seeding was detected in oropharyngeal lymphoid tissues from at least 1 96GG WTD collected at 2 MPE, including the tonsil and retropharyngeal, mandibular, and parotid lymph nodes (P < 0.01, unpaired t test). Amyloid formation rates from 96GS WTD tissues were not statistically different from those of the negative control. Data from three 96GG deer (1148, 1157, and 1218), one 96GS deer, and one negative-control deer are represented as means and SEM of 12 replicates from 3 separate experiments. (B) Representative TSA-IHC from 96GG WTD collection at 2 MPE. Mild PrPCWD immunoreactivity was detected in a small number of germinal center lymphoid follicles (<10 follicles/tissue) in tonsil and retropharyngeal lymph nodes. No PrPCWD immunoreactivity was observed in corresponding negative controls. Images are at ×200 magnification; scale bar, 50 μm.
TSA-IHC evaluation of lymphoid tissues from 96GG WTD revealed greater PrPCWD immunoreactivity in tonsils and retropharyngeal lymph nodes in two 96GG WTD compared to that of 1-MPE animals (Fig. 3B) (summarized in Table 1). No PrPCWD immunoreactivity was observed in 96GS deer tissues.
TABLE 1.
Summary of PrPCWD detection in 96GG WTD by RT-QuIC and TSA-IHC following mucosal exposurea
| Tissue | Value atb: |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 MPE |
2 MPE |
3 MPE |
4 MPE |
|||||
| QuIC | IHC | QuIC | IHC | QuIC | IHC | QuIC | IHC | |
| Retro LN | 2/3 | 0/3 | 3/3 | 2/3 | 2/2 | 2/2 | 3/3 | 3/3 |
| Tonsil | 0/3 | 2/3 | 2/3 | 3/3 | 2/2 | 2/2 | 3/3 | 3/3 |
| Mandibular LN | 0/3 | 0/3 | 2/3 | 0/3 | 2/2 | 2/2 | 3/3 | 3/3 |
| Parotid LN | 1/3 | 1/3 | 1/3 | 0/3 | 1/2 | 1/2 | 3/3 | 2/3 |
| Prescapular LN | 0/3 | 0/3 | 0/3 | 0/3 | 2/2 | 1/2 | 3/3 | 2/3 |
| Mesenteric LN | 1/3 | 0/3 | 0/3 | 0/1 | 1/2 | 0/2 | 3/3 | 2/3 |
| Hepatogastric LN | 0/3 | 0/3 | 0/3 | 0/3 | 2/2 | 2/2 | 3/3 | 3/3 |
| Ileocecocolic LN | 0/3 | 0/2 | 0/3 | 0/3 | 2/2 | 1/2 | 3/3 | 3/3 |
| Spleen | 0/3 | 0/2 | 0/3 | 0/2 | 2/2 | 1/2 | 3/3 | 2/3 |
| RAMALT | 0/3 | 0/3 | 0/3 | 0/2 | 2/2 | 0/2 | 2/3 | 2/3 |
| VNO | 0/3 | NA | 0/3 | NA | 2/2 | NA | 2/3 | NA |
| Ileum | NA | 0/2 | NA | 0/2 | NA | 1/2 | NA | 2/2 |
| Obex | 0/3 | 0/3 | 0/3 | 0/3 | 0/2 | 0/2 | 0/3 | 0/3 |
RT-QuIC and TSA-IHC detected similar patterns of PrPCWD accumulation during early infection: initial detection in oropharyngeal lymph nodes prior to systemic lymphoid dissemination. Although the two assays displayed similar lymphoid distribution patterns, RT-QuIC analysis identified more positive tissues compared with TSA-IHC. Abbreviations: QuIC, RT-QuIC; IHC, TSA-IHC; Retro LN, retropharyngeal lymph node; VNO, vomeronasal organ; NA, not applicable.
Data for each tissue are displayed as the number of animals positive by the detection method out of the total number of animals examined.
(iv) Three months postexposure.
At 3 MPE, statistically significant PrPCWD seeding activity (P value of <0.05 by unpaired t test) expanded from oropharyngeal lymphoid tissues to all systemic lymphoid tissues examined and the vomeronasal organ in at least one of the two 96GG WTD (Fig. 4A). The lymphoid tissues draining the head and neck had the highest rates of amyloid seeding activity, consistent with a greater PrPCWD burden. The PrPCWD seeding activity in spleen was consistent with systemic spread of prions, either through lymphoid cell trafficking or blood.
FIG 4.
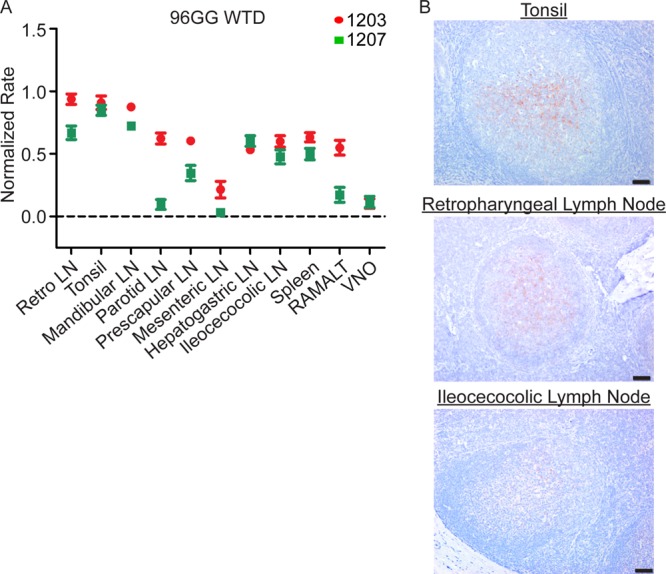
PrPCWD is detected in systemic lymphoid tissues in 96GG deer at 3 months postexposure. (A) RT-QuIC of 96GG WTD tissues collected at 3 MPE. The majority of tissues display positive rates of amyloid formation that are statistically different from those of the corresponding negative-control tissues (P < 0.05, unpaired t test). The two tissues that are not significant are 1207 mesenteric lymph node and 1207 parotid lymph node. The increase in lymphoid tissue distribution at 3 MPE is consistent with systemic prion spread. Amyloid formation rates in the oropharyngeal lymphoid tissues are higher at 3 MPE than at 2 MPE (Fig. 3A), consistent with an accumulation of PrPCWD with the longer incubation time. Data from two 96GG deer (1207 and 1203) are represented as means and SEM of 12 replicates from 3 separate experiments. 3-MPE tissues were compared with 4-MPI negative-control tissues for statistical analysis. (B) Representative TSA-IHC from 96GG WTD collected at 3 MPE. Increased PrPCWD immunoreactivity staining was identified in oropharyngeal lymphoid tissues in lymphoid follicle germinal centers. TSA-IHC additionally detected PrPCWD immunoreactivity in the majority of systemic lymphoid tissues (not pictured). IHC images are at ×200 magnification; scale bar, 50 μm.
TSA-IHC analysis of tissues at 3 MPE identified PrPCWD immunoreactivity in the majority of systemic lymphoid tissues in addition to oropharyngeal lymphoid tissues (Fig. 4B) (summarized in Table 1). Lymph nodes draining the head and neck had the greatest PrPCWD staining distribution and intensity.
(v) Four months postexposure.
Tissues from 96GG WTD sacrificed at 4 MPE had statistically significant PrPCWD amyloid seeding activity (P value of <0.01 by unpaired t test) in the majority of tissues evaluated, with only rectoanal mucosa-associated lymphoid tissue (RAMALT) and vomeronasal organ (VNO) displaying variable seeding activity (Fig. 5A). Overall, tissues had a higher rate of amyloid formation than tissues at 3 MPE, consistent with greater PrPCWD tissue accumulation. The first identification of amyloid seeding activity in a 96GS WTD was in the tonsil at this time point.
FIG 5.

Increased PrPCWD tissue burden is present in systemic lymphoid tissues in 96GG deer at 4 months postexposure. (A) RT-QuIC PrPCWD amyloid formation was detected in all tissues from at least two of the three 96GG WTD collected at 4 MPE (P < 0.05, unpaired t test). The two tissues that did not have statistically significant rates of amyloid formation were 1201 vomeronasal organ and 1201 RAMALT. Compared with 3-MPE tissues (Fig. 4), 4-MPE tissues displayed higher rates of amyloid formation, consistent with a larger PrPCWD tissue burden. Tonsil from 96GS WTD had detectable amyloid formation at this collection time (P < 0.01, unpaired t test). Data from three 96GG deer (1171, 1201, and 1215), one 96GS deer, and one negative-control deer are represented as means and SEM of 12 replicates from 3 separate experiments. (B) Representative TSA-IHC from 96GG WTD collected at 4 MPE. All lymphoid tissues displayed PrPCWD immunoreactivity in the germinal centers of follicles. The greatest staining was observed in oropharyngeal lymphoid tissues. No staining was observed in corresponding negative-control tissues. IHC images are at ×200 magnification; scale bar, 50 μm.
TSA-IHC analysis of 4-MPE tissues revealed PrPCWD immunoreactivity in all tissues from at least 2 of the 3 96GG WTD (Fig. 5B). The tonsil and retropharyngeal lymph nodes had diffuse PrPCWD immunoreactivity in lymphoid follicular germinal centers. Compared to 3-MPE tissues, all lymphoid tissues had an increased number of follicles displaying PrPCWD immunoreactivity and greater staining intensity consistent with increased prion accumulation. No PrPCWD immunoreactivity was observed in tissues from 96GS WTD.
Evaluation of PrPCWD in 96SS WTD.
PrPCWD tissue distribution was evaluated from a single WTD collected at 16 MPE. RT-QuIC detected statistically significant amyloid seeding activity (P value of <0.05 by unpaired t test) in the majority of tissues examined (retropharyngeal LN, tonsil, mandibular lymph node, parotid LN, prescapular LN, hepatogastric LN, spleen, RAMALT, and VNO) (Fig. 6A). The seeding activity in the mesenteric and ileocecocolic lymph nodes was statistically indistinguishable from negative-control tissues. No seeding activity was detected in the obex. TSA-IHC demonstrated PrPCWD immunoreactivity in all tissues except the mesenteric lymph node, spleen, RAMALT, and obex (Fig. 6B) (summarized in Table 2).
FIG 6.
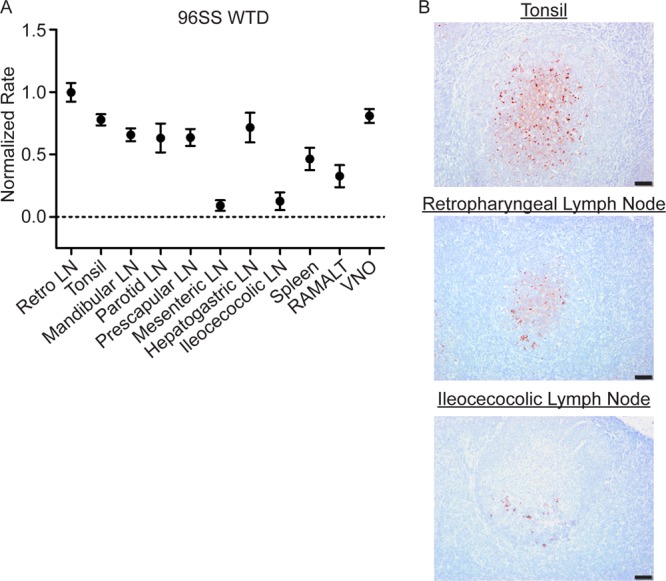
PrPCWD is detected in systemic lymphoid tissues from a subclinical 96SS deer collected at 16 MPE. (A) RT-QuIC PrPCWD seeded amyloid formation was detected in all tissues except the mesenteric LN and ileocecocolic LN (P < 0.05, unpaired t test), consistent with systemic lymphoid PrPCWD distribution. (B) Representative TSA-IHC from 96SS WTD. The tonsil and retropharyngeal lymphoid tissues displayed strong PrPCWD immunoreactivity in the germinal centers of a majority of follicles. Rare immunoreactivity limited to a few germinal centers was observed in the ileocecocolic lymph node. IHC images are at ×200 magnification; scale bar, 50 μm.
TABLE 2.
Summary of PrPCWD detection in 96SS WTD at 16 MPE by RT-QuIC and TSA-IHCa
| Tissue | Result at 16 MPE |
|
|---|---|---|
| QuIC | IHC | |
| Retro LN | 1/1 | 1/1 |
| Tonsil | 1/1 | 1/1 |
| Mandibular LN | 1/1 | 1/1 |
| Parotid LN | 1/1 | 1/1 |
| Prescapular LN | 1/1 | 1/1 |
| Mesenteric LN | 0/1 | 0/1 |
| Hepatogastric LN | 1/1 | 1/1 |
| Ileocecocolic LN | 0/1 | 1/1 |
| Spleen | 1/1 | 0/1 |
| RAMALT | 1/1 | 0/1 |
| VNO | 1/1 | NA |
| Ileum | NA | 1/1 |
| Obex | 0/1 | 0/1 |
RT-QuIC and TSA-IHC detected similar patterns of PrPCWD distribution in the majority of systemic lymphoid tissues. Data are displayed as the number of animals positive by detection method out of the total number of animals examined. Abbreviations: QuIC, RT-QuIC; IHC, TSA-IHC; Retro LN, retropharyngeal lymph node; VNO, vomeronasal organ; NA, not applicable.
Evaluation of PrPCWD in obexes.
The obex region of the medulla oblongata, the region considered to be the first site of CWD prion neuroinvasion, was evaluated for evidence of PrPCWD by RT-QuIC and TSA-IHC. Neither PrPCWD RT-QuIC seeding nor TSA-IHC immunoreactivity was detected at any collection time (Fig. 7). Thus, there was no evidence of neuroinvasion in the time period we evaluated in this study.
FIG 7.
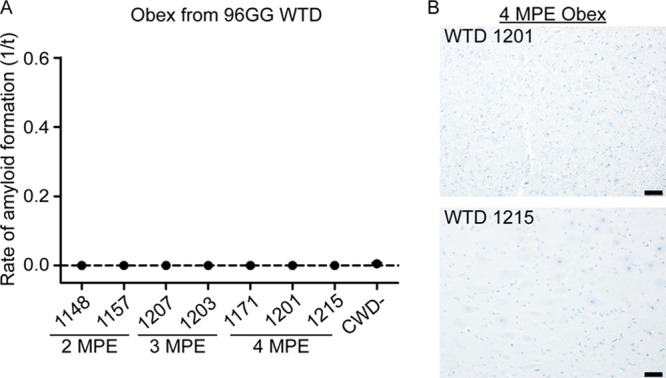
No evidence of CWD neuroinvasion in 96GG deer at 4 MPE. (A) RT-QuIC evaluation of obex samples collected from 2 MPE to 4 MPE. No evidence of PrPCWD amyloid seeding was detected in the obexes evaluated from 96GG deer collected during early infection time points. (B) Representative TSA-IHC of the obex from two 96GG WTD collected at 4 MPE. No PrPCWD immunoreactivity was observed in sections of the obex from the 4-MPE collection. IHC images are at ×200 magnification; scale bar, 50 μm.
Estimation of prion burden during early CWD infection.
We have previously reported that the prion concentration in the sample used to seed the RT-QuIC reaction directly correlates with the rate of amyloid formation (26, 27). To explore the kinetics of PrPCWD accumulation in lymphoid tissues over the course of early infection, we compared the average amyloid formation rates in 96GG WTD lymphoid tissues from the 1- to 4-MPE collections with those collected from 3 white-tailed deer with terminal disease (20 to 24 months postinfection). Evaluation of lymphoid tissues over time revealed two patterns. First, the oropharyngeal lymph nodes, including the tonsil, retropharyngeal lymph node, and mandibular lymph node, displayed a rapid accumulation of PrPCWD, approximating terminal tissue levels by 3 MPE, with no statistical difference observed between 3 MPE, 4 MPE, and the terminal disease states (3 MPE versus terminal and 4 MPE versus terminal, P value of >0.05 by unpaired t test) (Fig. 8A). The parotid lymph node displayed a lower rate of prion accumulation but reached prion levels comparable to those of terminal disease by 4 MPE (4 MPE versus terminal, P value of >0.05 by unpaired t test) (Fig. 8A). Second, distal gastrointestinal lymphoid tissues (GALT) and spleen displayed a later accumulation of PrPCWD and did not reach peak tissue burdens during the time course of our study (4-MPE tissues versus terminal tissues, P value of <0.01 by unpaired t test) (Fig. 8B). We approximated the PrPCWD amyloid formation kinetics of the oropharyngeal and gastrointestinal lymphoid patterns by linear regression analysis. We found no statistical difference between the slopes of the best-fit linear regression lines (P value of 0.4229 by linear regression analysis). These data indicate that the lymphoid tissues examined have similar prion replication kinetics and that the difference in lymphoid tissue prion levels at 4 MPE reflects the sequence of onset of PrPCWD accumulation in tissues (i.e., the oropharyngeal tissues were infected earlier than systemic lymphoid tissues) and not due to a difference in prion replication kinetics among tissues.
FIG 8.
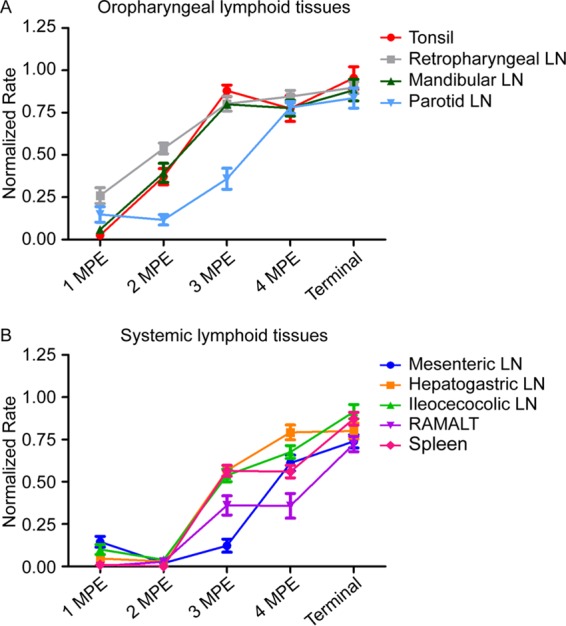
PrPCWD tissue burden in early infection approximates terminal disease tissue levels in oropharyngeal lymphoid tissues. The prion concentration in oropharyngeal tissues was estimated by averaging the amyloid formation rate from all deer at each collection time point. (A) Estimation of prion replication kinetics in oropharyngeal lymphoid tissues during early CWD infection. In oropharyngeal lymph nodes, the tonsil and retropharyngeal and mandibular lymph nodes displayed a rapid PrPCWD accumulation and reached tissue PrPCWD levels comparable to that for terminal disease burden by 3 MPE (3 MPE versus terminal, P > 0.05, unpaired t test). The parotid lymph node displayed a slightly slower prion accumulation but reached prion levels comparable to that of terminal disease by 4 MPE (4 MPE versus terminal, P > 0.05, unpaired t test). (B) Estimation of prion replication kinetics in gastrointestine-associated lymphoid tissues (GALT) and systemic lymphoid tissues during early CWD infection. PrPCWD seeding activity was not detected in distal GALT and the spleen until 3 MPE. At 4 MPE, GALT and spleen had statistically significantly lower rates of amyloid formation than corresponding terminal tissues (4 MPE versus terminal, P < 0.01, unpaired t test), indicating these lymphoid tissues did not reach maximum prion burdens during the time course of our study.
DISCUSSION
We analyzed PrPCWD distribution in WTD at early time points following oral exposure to CWD prions. We demonstrated early PrPCWD seeding in oropharyngeal lymph nodes at 1 MPE and rapid systemic accumulation of PrPCWD in lymphoid tissues by 4 MPE with tissue prion levels comparable to those for terminal disease. These findings are consistent with previous investigations of early CWD pathogenesis (24, 25), even though the exposure dose in the present study was 10-fold lower. This suggests the dose of CWD prions, as long as it is greater than the infectious dose, does not impact infection kinetics and that most experimental doses far surpass the natural infectious dose and thereby produce the same disease course. Interestingly, a recent report determined the minimal effective oral dose of sheep-adapted BSE to be 0.05 g, and that higher inoculation doses did not alter the disease course (28). Investigations into the minimum infectious oral dose required for CWD in the natural host are ongoing.
The pattern of CWD lymphoid tropism observed in our study is consistent with an early and sustained lymphoid prion replication phase prior to neuroinvasion, and in that respect it is similar to several other orally transmitted prion diseases, such as scrapie and variant Creutzfeldt-Jakob disease (22, 29). Temporal investigations of scrapie identified alimentary-associated lymphoid tissues, including tonsil, retropharyngeal lymph node, Peyer's patches, and ileocecocolic lymph nodes, as the first sites of scrapie detection (30, 31). The oropharyngeal lymphoid tissues draining the head and neck displayed PrPCWD amyloid seeding activity and IHC immunoreactivity, consistent with either a larger initial PrPCWD dose arriving at these tissues due to direct drainage from the oral mucosa or to faster prion replication kinetics in these tissues. The initial oropharyngeal lymphoid replication phase was followed by systemic lymphoid PrPCWD replication. PrPCWD may be trafficked systemically from initial lymphoid replication sites via either blood or lymphoid cells and/or secondary mucosal entry sites in the intestinal mucosa. Blood has been shown previously to harbor CWD infectivity, particularly associated with B cells and platelets (12, 32). In contrast, in PRNP knockout mice orally inoculated with the Fukuoka-1 strain of CJD, macrophages were identified as the cells responsible for transporting prions from intestinal mucosa to mesenteric lymph nodes and spleen (33). Further studies are needed to illuminate which lymphoid cell or cells are responsible for systemic prion trafficking in subclinical CWD and other prion diseases. We did not observe evidence of neuroinvasion in the obex region of the medulla oblongata, the first site of PrPCWD accumulation in the brain, during the time course of our study (1). However, we cannot exclude PrPCWD accumulation in peripheral nerves during early disease prior to transport to the brainstem.
A surprising finding was the early detection of PrPCWD seeding activity in the vomeronasal organ at 3 MPE. The vomeronasal organ is a collection of accessory olfactory cells located in the anterior nasal mucosa that functions in pheromone detection in cervids (34). RT-QuIC analysis of nasal brushings have recently been shown to be a useful preclinical prion diagnostic tool for CJD in humans and CWD in cervids (35–37). The early PrPCWD accumulation in the nasal cavity, not only in 96GG deer but also during the prolonged subclinical phase in 96SS deer, may provide an additional pathway contributing to indirect environmental transmission in cervids.
In order for oral TSE transmission to occur, prions must cross alimentary mucosal barriers. Studies with radioactively labeled scrapie prions in mice demonstrated prions can rapidly cross the intestinal mucosa in minutes, followed by distribution to systemic organs via the vascular system (38). Investigations of specific routes of intestinal uptake of transmissible mink encephalopathy and scrapie prion have identified M cells of the follicle-associated epithelium of Peyer's patches as a site of prion entry (39, 40). Additionally, gastrointestinal-associated lymphoid tissue (GALT) uptake appears to be required for neuroinvasion, as depletion of M cells or decreased numbers of Peyer's patches, but not lymphocytes, blocked the development of prion disease in mice (33, 41–43). Other imaging studies have suggested prions can use a paracellular route to cross the epithelial mucosal epithelium (40). Our study strongly supports oronasal mucosal uptake as the most important pathway of prion entry in early CWD pathogenesis. This does not exclude that additional intestinal mucosal uptake may also contribute to total CWD prion uptake.
During early CWD disease stages, the low prion tissue burden limits PrPCWD detection by traditional immunoblotting techniques. Here, we demonstrate that both RT-QuIC and TSA-IHC can enhance PrPCWD detection, with RT-QuIC being more sensitive during early infection. This finding is consistent with recent studies reporting RT-QuIC has sensitivity at least equal to that of IHC in detecting PrPCWD in RAMALT biopsy specimens from WTD and elk (35, 36); however, a larger sample size is necessary to evaluate RT-QuIC as a diagnostic tool for subclinical infection.
The present study provides further support for the influence of the PRNP genotype, specifically at codon 96, on CWD pathogenesis in deer (19). Lymphoid tissues from 96GS WTD displayed slower disease progression, with RT-QuIC amyloid seeding activity only detected in tonsil at 4 MPI and in some systemic lymphoid tissues in the 96SS WTD despite a 4× longer incubation course. The slower disease course in 96GS and 96SS WTD may enable distinction of the exact chronological sequence of PrPCWD lymphoid replication that is masked in the faster 96GG WTD disease course. Thus, the initial identification of amyloid seeding in 96GS WTD tonsil suggests this tissue is the first location of PrPCWD entry following oral exposure.
In summary, we used RT-QuIC and TSA-IHC to explore the early pathogenesis of CWD infection in white-tailed deer. Our results indicate oropharyngeal prion entry and early prion amplification in draining oropharyngeal lymphoid tissues followed by dissemination to systemic lymphoid tissues, with tissue burdens approaching terminal disease levels by 4 months postexposure prior to neuroinvasion. The detection methods used, particularly RT-QuIC, make possible whole-body profiling of CWD prion infection throughout the course of disease.
MATERIALS AND METHODS
Deer CWD inoculum.
CWD-positive (CWD+) or CWD-negative (CWD−) 10% (wt/vol) brain homogenates in phosphate-buffered saline were used as inocula for deer studies. The CWD+ inoculum was produced by pooling eight random CWD+ brain sections, confirmed positive by Western blotting, from six experimentally inoculated, white-tailed deer with terminal disease. The titer of this inoculum was calculated to be 3.33 × 106 50% lethal doses/g of brain tissue by cervid-PrPC transgenic mouse bioassay (44). The CWD− inoculum was produced by pooling eight random CWD− brain sections, confirmed negative by Western blotting, from three uninoculated, captive, indoor-housed white-tailed deer (WTD). The CWD status of the pooled inoculum as positive or negative was confirmed by Western blotting.
Animal care and study design.
Indoor-housed, hand-raised WTD (Odocoileus virginianus) were maintained in strict accordance with Colorado State University Animal Care and Use Committee-approved protocols. Deer in the very early harvest cohort, identified as 900 series, were inoculated oronasally with 0.55 g of CWD+ or CWD− pooled cervid brain homogenate. Oronasal inoculation was performed by atomizing the equivalent of 0.05 g of brain homogenate directly into the nasal cavity with a Nasonex-style sprayer and separately instilling the equivalent of 0.5 g of brain homogenate directly into the animal's mouth using a syringe. These 900 series WTD were sacrificed at either 24 h or 72 h postinoculation. Deer in the early pathogenesis cohort, identified by an 1100 or 1200 series, were inoculated with either 0.5 g of CWD+ or CWD− brain homogenate per os (p.o.), by instilling the equivalent of 0.5 g brain homogenate directly into the animal's mouth with a syringe, and sacrificed at either 1, 2, 3, or 4 months postexposure (MPE). A single WTD (number 1205) homozygous for serine at PRNP codon 96 (96SS) was inoculated p.o. with 0.5 g of CWD-positive brain homogenate and sacrificed at 16 months postexposure. No signs of clinical disease were detected in any deer throughout the course of study.
PRNP genotype at codon 96 was determined as previously described through collaborations with Katherine O'Rourke at Washington State University and Wilfred Goldmann at the Roslin Institute, University of Edinburgh (16, 45–47). Deer were determined to be homozygous G96G (96GG), heterozygous G96S (96GS), or homozygous S96S (96SS), and each genotype was evenly distributed throughout the collection time points in an attempt to equalize genetic representation (Fig. 9).
FIG 9.
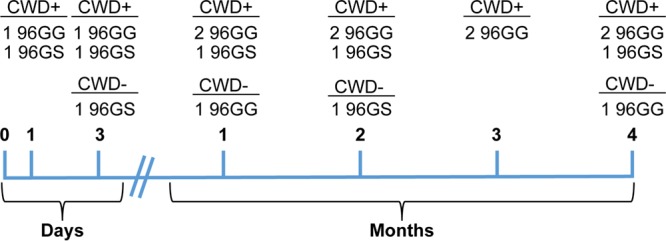
Overview of the PRNP genotype distribution of white-tailed deer at each collection time point. White-tailed deer were collected at 24 and 72 h or 1, 2, 3, and 4 months postexposure. PRNP codon 96 genotypes, homozygous G96G (96GG) and heterozygous G96S (96GS), were distributed throughout the collections to ensure adequate genetic representation. An additional deer (not shown), homozygous S96S (96SS), was collected at 16 months postexposure.
WTD collected at terminal disease were experimentally inoculated p.o. by instilling the equivalent of 0.01 g of pooled CWD+ brain homogenate directly into the animal's mouth with a syringe. Deer were monitored for clinical signs using a previously established scoring system and necropsied when animals reach a terminal disease state between 20 and 26 months postinoculation.
Tissue collection and processing.
A full necropsy and tissue collection was performed of each WTD at the time of sacrifice. For this study, tissue analysis focused on lymphoid tissues, brain (obex), ileum, RAMALT, and VNO. Each tissue was collected with an individual, prion-free instrument to avoid cross-contamination. Tissues were divided into two halves; one-half was frozen and stored at −80°C, and the other half was fixed in periodate-lysine-paraformaldehyde (PLP) for 4 days before transferring to sterile 1× phosphate-buffered saline (PBS) until trimming into histology cassettes. Tissue cassettes were processed into paraffin-embedded blocks by following routine histologic techniques. Frozen tissues were processed into 10% (wt/vol) tissue homogenates using 1× PBS (Life Technologies) and a Bullet Blender tissue homogenizer (Next Advance).
IHC for PrPCWD.
Tissue PrPCWD was detected with a two-step immunostaining procedure using TSA enhancement, with modifications to a previously described protocol (48). Briefly, 5-μm tissue sections were mounted on positively charged glass slides. Following routine deparaffinization with xylene and rehydration with graded alcohols, tissues were subjected to 1 μg/ml proteinase K digestion at 37°C to remove native PrPC. Epitope exposure was performed using hydrated autoclaving antigen retrieval in 10 mM EDTA, pH 6.0. Tissues were exposed to 88% formic acid for 5 min prior to quenching endogenous peroxide activity with 3.0% hydrogen peroxide in methanol. Tissue sections were blocked with a proprietary protein block, TNB (Perkin-Elmer), followed by sequential application of primary anti-prion protein antibody BAR224 (Cayman Chemical) at 0.5 μg/ml and secondary anti-mouse antibody conjugated to horseradish peroxidase (Envision+; Dako). Antibody signal was enhanced with tyramide signal amplification (PerkinElmer) by following the manufacturer's suggested protocol. Immunoreactivity was detected using AEC substrate-chromogen (Dako). Coverslips were mounted using aqueous mounting medium, and staining was visualized by light microscopy.
RT-QuIC substrate protein purification.
Real-time quaking-induced conversion (RT-QuIC) assays were performed with recombinant truncated Syrian hamster PrPC (SHrPrP) encompassing residues 90 to 231, which was expressed and purified as previously described (26, 27). Briefly, SHrPrP was expressed in BL21 Rosetta Escherichia coli (Novagen). Bacteria were cultured at 37°C in lysogeny broth (LB) medium in the presence of the selection antibiotics, kanamycin and chloramphenicol, until the final optical density at 600 nm (OD600) was at least 2.5. Cell lysis was performed using BugBuster reagent supplemented with Lysonase (EMD Biosciences), and inclusion bodies were harvested by centrifugation at 212,000× relative centrifugal force (RCF). The inclusion body pellet was solubilized overnight (8 M guanidine hydrochloride, 100 mM Na2PO4) prior to application to nickel-nitrilotriacetic acid (Ni-NTA) Superflow resin (Qiagen) that had been previously equilibrated with denaturation buffer (6 M guanidine hydrochloride, 100 mM Na2PO4, 10 mM Tris). The resin-SHrPrP was applied to an XK16-60 column (GE Healthcare) and purified using a Bio-Rad Duoflow fast-performance liquid chromatograph (FPLC), with a gradient from denaturation buffer to refolding buffer (100 mM Na2HPO4, 10 mM Tris) to induce protein refolding. Refolding was followed by a gradient from refolding to elution buffer (100 mM NaH2PO4, 10 mM Tris, 0.5 M imidazole) with all fractions being collected. Fractions from the elution peak were combined and dialyzed against 4 liters of buffer (20 mM NaH2PO4) overnight. Final protein concentration was determined using the A280 and a coefficient of extinction of 25,900 by Beer's law.
RT-QuIC assay. (i) Lymphoid and parenchymal tissues.
Tissue homogenates were subjected to sodium phosphotungstic acid (NaPTA) precipitation to enhance detection by RT-QuIC. Briefly, 100 μl of a 1% tissue homogenate solution was incubated and shaken with 7 μl NaPTA solution (0.5 g sodium phosphotungstic acid, 0.43 g MgCl2-6-hydrate, pH 4.0) for 1 h at 37°C and 1,400 rpm. The solution was pelleted by centrifugation at 21,130× RCF for 30 min. The supernatant was removed, and remaining pellets were resuspended in 10 μl of RT-QuIC dilution buffer (0.1% SDS–1× PBS). Two microliters of resuspended NaPTA-precipitated tissue sample was used to seed each RT-QuIC reaction.
(ii) Nervous system tissues.
Brain tissue was alcohol precipitated prior to RT-QuIC assay as previously described (49). Briefly, 10 μl of a 10% brain homogenate was incubated at room temperature with 100% ethanol for 5 min. Proteins were pelleted by centrifugation at 21,130× RCF for 5 min and the supernatant discarded. The procedure was repeated and the final pellet was resuspended in 10 μl of RT-QuIC dilution buffer. Two microliters of a 10−1 dilution of the precipitated nervous tissue proteins were used to seed each RT-QuIC reaction.
(iii) Assay conditions.
RT-QuIC was performed as previously described (26, 27). Briefly, RT-QuIC experiments were carried out in black, optical-bottom 96-well plates (Nunc), with each well containing 0.1 mg/ml SHrPrP substrate and RT-QuIC reaction buffer (20 mM NaH2PO4, 320 mM NaCl, 1.0 mM EDTA, 1 mM thioflavin T). RT-QuIC experiments were carried out in a BMG Labtech Polarstar fluorometer/plate reader, with each cycle consisting of 1 min of shaking at 700 rpm followed by 1 min of rest, repeated for 15 min. Fluorescence readings with an excitation of 450 nm, emission of 480 nm, and gain of 1,700 were taken at the completion of each 15-min shake/rest cycle by measuring each well with 20 flashes and an orbital average of 4. Each RT-QuIC experiment consisted of 250 fluorescence readings, for a total of 62.5 h.
RT-QuIC data analysis.
Data for each sample were collected from a minimum of two separate RT-QuIC experiments with 4 replicates each, for a minimum of 8 replicates per tissue. Individual replicates were considered to have positive amyloid formation if their fluorescence rose above a threshold of 5 standard deviations above the average of the baseline fluorescence. RT-QuIC amyloid formation rates were calculated as previously described (26, 50). Rates of amyloid formation were normalized to a plate positive control to minimize any individual experiment variation by dividing the rate of amyloid formation for each sample by the highest rate of conversion for the positive control. Sample data were statistically analyzed using GraphPad software. RT-QuIC replicate distributions were determined to be normally distributed using the D'Agostino test for normality. Amyloid formation rates from tissue homogenates from PrPCWD-exposed animals were compared to corresponding negative controls by an unpaired t test and considered statistically different if the P value was at least <0.05.
ACKNOWLEDGMENTS
White-tailed deer were kindly provided through long collaboration with wildlife biologists David Osborn, Karl Miller, and Robert Warren at the Warnell School of Forestry, University of Georgia, and hand raised by Sally Dahmes (founder of WASCO, Inc.). We thank laboratory members Amy Nalls and Erin McNulty for their assistance with deer necropsies and sample handling. We thank students Nikki Buhrdorf and Sarah Accardi for assisting in sample preparation and performing immunohistochemistry. We thank Jeanette Hayes-Klug and Kelly Anderson for their excellent care and observation of the deer.
This work was supported by NIH grants R01-NS061902 (E.A.H.), R01-NS078745 (C.S.), and R01-NS076894 (C.K.M.). C.E.H. was supported by NIH training grant T32–OD010437-14.
REFERENCES
- 1.Williams ES. 2005. Chronic wasting disease. Vet Pathol 42:530–549. doi: 10.1354/vp.42-5-530. [DOI] [PubMed] [Google Scholar]
- 2.Haley NJ, Hoover EA. 2015. Chronic wasting disease of cervids: current knowledge and future perspectives. Annu Rev Anim Biosci 3:305–325. doi: 10.1146/annurev-animal-022114-111001. [DOI] [PubMed] [Google Scholar]
- 3.ProMED-mail. 4 April 2016. Chronic wasting disease, Cervid–Europe: Norway. ProMed-mail archive no. 20160410.4149651 http://www.promedmail.org.
- 4.Williams ES, Young S. 1980. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 5.Spraker TR, Miller MW, Williams ES, Getzy DM, Adrian WJ, Schoonveld GG, Spowart RA, O'Rourke KI, Miller JM, Merz PA. 1997. Spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus) and Rocky Mountain elk (Cervus elaphus nelsoni) in northcentral Colorado. J Wildl Dis 33:1–6. doi: 10.7589/0090-3558-33.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Williams ES, Young S. 1993. Neuropathology of chronic wasting disease of mule deer (Odocoileus hemionus) and elk (Cervus elaphus nelsoni). Vet Pathol 30:36–45. doi: 10.1177/030098589303000105. [DOI] [PubMed] [Google Scholar]
- 7.Spraker TR, Zink RR, Cummings BA, Wild MA, Miller MW, O'Rourke KI. 2002. Comparison of histological lesions and immunohistochemical staining of proteinase-resistant prion protein in a naturally occurring spongiform encephalopathy of free-ranging mule deer (Odocoileus hemionus) with those of chronic wasting disease of captive mule deer. Vet Pathol 39:110–119. doi: 10.1354/vp.39-1-110. [DOI] [PubMed] [Google Scholar]
- 8.Miller MW, Williams ES, Hobbs NT, Wolfe LL. 2004. Environmental sources of prion transmission in mule deer. Emerg Infect Dis 10:1003–1006. doi: 10.3201/eid1006.040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denkers ND, Hayes-Klug J, Anderson KR, Seelig DM, Haley NJ, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mathiason CK, Hoover EA. 2013. Aerosol transmission of chronic wasting disease in white-tailed deer. J Virol 87:1890–1892. doi: 10.1128/JVI.02852-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denkers ND, Telling GC, Hoover EA. 2011. Minor oral lesions facilitate transmission of chronic wasting disease. J Virol 85:1396–1399. doi: 10.1128/JVI.01655-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathiason CK, Hays SA, Powers J, Hayes-Klug J, Langenberg J, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hoover EA. 2009. Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS One 4:e5916. doi: 10.1371/journal.pone.0005916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, Wild MA, Wolfe LL, Spraker TR, Miller MW, Sigurdson CJ, Telling GC, Hoover EA. 2006. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 314:133–136. doi: 10.1126/science.1132661. [DOI] [PubMed] [Google Scholar]
- 13.Tamguney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, Lemus A, DeArmond SJ, Prusiner SB. 2009. Asymptomatic deer excrete infectious prions in faeces. Nature 461:529–532. doi: 10.1038/nature08289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA. 2009. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS One 4:e4848. doi: 10.1371/journal.pone.0004848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson CJ, Phillips KE, Schramm PT, McKenzie D, Aiken JM, Pedersen JA. 2006. Prions adhere to soil minerals and remain infectious. PLoS Pathog 2:e32. doi: 10.1371/journal.ppat.0020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nalls AV, McNulty E, Powers J, Seelig DM, Hoover C, Haley NJ, Hayes-Klug J, Anderson K, Stewart P, Goldmann W, Hoover EA, Mathiason CK. 2013. Mother to offspring transmission of chronic wasting disease in reeves' muntjac deer. PLoS One 8:e71844. doi: 10.1371/journal.pone.0071844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson SJ, Samuel MD, O'Rourke KI, Johnson CJ. 2012. The role of genetics in chronic wasting disease of North American cervids. Prion 6:153–162. doi: 10.4161/pri.19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson C, Johnson J, Clayton M, McKenzie D, Aiken J. 2003. Prion protein gene heterogeneity in free-ranging white-tailed deer within the chronic wasting disease affected region of Wisconsin. J Wildl Dis 39:576–581. doi: 10.7589/0090-3558-39.3.576. [DOI] [PubMed] [Google Scholar]
- 19.Johnson CJ, Herbst A, Duque-Velasquez C, Vanderloo JP, Bochsler P, Chappell R, McKenzie D. 2011. Prion protein polymorphisms affect chronic wasting disease progression. PLoS One 6:e17450. doi: 10.1371/journal.pone.0017450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Race B, Meade-White K, Miller MW, Fox KA, Chesebro B. 2011. In vivo comparison of chronic wasting disease infectivity from deer with variation at prion protein residue 96. J Virol 85:9235–9238. doi: 10.1128/JVI.00790-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Keulen LJ, Vromans ME, van Zijderveld FG. 2002. Early and late pathogenesis of natural scrapie infection in sheep. APMIS 110:23–32. doi: 10.1034/j.1600-0463.2002.100104.x. [DOI] [PubMed] [Google Scholar]
- 22.Beekes M, McBride PA. 2007. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS J 274:588–605. doi: 10.1111/j.1742-4658.2007.05631.x. [DOI] [PubMed] [Google Scholar]
- 23.O'Rourke KI, Zhuang D, Lyda A, Gomez G, Williams ES, Tuo W, Miller MW. 2003. Abundant PrP(CWD) in tonsil from mule deer with preclinical chronic wasting disease. J Vet Diagn Investig 15:320–323. doi: 10.1177/104063870301500403. [DOI] [PubMed] [Google Scholar]
- 24.Fox KA, Jewell JE, Williams ES, Miller MW. 2006. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J Gen Virol 87:3451–3461. doi: 10.1099/vir.0.81999-0. [DOI] [PubMed] [Google Scholar]
- 25.Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O'Rourke KI, Hoover EA. 1999. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J Gen Virol 80(Part 10):2757–2764. doi: 10.1099/0022-1317-80-10-2757. [DOI] [PubMed] [Google Scholar]
- 26.Henderson DM, Davenport KA, Haley NJ, Denkers ND, Mathiason CK, Hoover EA. 2015. Quantitative assessment of prion infectivity in tissues and body fluids by real-time quaking-induced conversion. J Gen Virol 96:210–219. doi: 10.1099/vir.0.069906-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henderson DM, Denkers ND, Hoover CE, Garbino N, Mathiason CK, Hoover EA. 2015. Longitudinal detection of prion shedding in saliva and urine by chronic wasting disease-infected deer by real-time quaking-induced conversion. J Virol 89:9338–9347. doi: 10.1128/JVI.01118-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGovern G, Martin S, Jeffrey M, Dexter G, Hawkins SA, Bellworthy SJ, Thurston L, Algar L, Gonzalez L. 2016. Minimum effective dose of cattle and sheep BSE for oral sheep infection. PLoS One 11:e0151440. doi: 10.1371/journal.pone.0151440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mabbott NA, MacPherson GG. 2006. Prions and their lethal journey to the brain. Nat Rev Microbiol 4:201–211. doi: 10.1038/nrmicro1346. [DOI] [PubMed] [Google Scholar]
- 30.van Keulen LJ, Schreuder BE, Vromans ME, Langeveld JP, Smits MA. 2000. Pathogenesis of natural scrapie in sheep. Arch Virol Suppl 2000:57–71. [DOI] [PubMed] [Google Scholar]
- 31.Hadlow WJ, Kennedy RC, Race RE. 1982. Natural infection of Suffolk sheep with scrapie virus. J Infect Dis 146:657–664. doi: 10.1093/infdis/146.5.657. [DOI] [PubMed] [Google Scholar]
- 32.Mathiason CK, Hayes-Klug J, Hays SA, Powers J, Osborn DA, Dahmes SJ, Miller KV, Warren RJ, Mason GL, Telling GC, Young AJ, Hoover EA. 2010. B cells and platelets harbor prion infectivity in the blood of deer infected with chronic wasting disease. J Virol 84:5097–5107. doi: 10.1128/JVI.02169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takakura I, Miyazawa K, Kanaya T, Itani W, Watanabe K, Ohwada S, Watanabe H, Hondo T, Rose MT, Mori T, Sakaguchi S, Nishida N, Katamine S, Yamaguchi T, Aso H. 2011. Orally administered prion protein is incorporated by m cells and spreads into lymphoid tissues with macrophages in prion protein knockout mice. Am J Pathol 179:1301–1309. doi: 10.1016/j.ajpath.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doving KB, Trotier D. 1998. Structure and function of the vomeronasal organ. J Exp Biol 201:2913–2925. [DOI] [PubMed] [Google Scholar]
- 35.Haley NJ, Siepker C, Hoon-Hanks LL, Mitchell G, Walter WD, Manca M, Monello RJ, Powers JG, Wild MA, Hoover EA, Caughey B, Richt JA. 2016. Seeded amplification of chronic wasting disease prions in nasal brushings and recto-anal mucosa associated lymphoid tissues from elk by real time quaking-induced conversion. J Clin Microbiol 54:1117–1126. doi: 10.1128/JCM.02700-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haley NJ, Siepker C, Walter WD, Thomsen BV, Greenlee JJ, Lehmkuhl AD, Richt JA. 2016. Antemortem detection of chronic wasting disease prions in nasal brush collections and rectal biopsies from white-tailed deer by real time quaking-induced conversion. J Clin Microbiol 54:1108–1116. doi: 10.1128/JCM.02699-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orru CD, Bongianni M, Tonoli G, Ferrari S, Hughson AG, Groveman BR, Fiorini M, Pocchiari M, Monaco S, Caughey B, Zanusso G. 2014. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med 371:519–529. doi: 10.1056/NEJMoa1315200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urayama A, Concha-Marambio L, Khan U, Bravo-Alegria J, Kharat V, Soto C. 2016. Prions efficiently cross the intestinal barrier after oral administration: study of the bioavailability, and cellular and tissue distribution in vivo. Sci Rep 6:32338. doi: 10.1038/srep32338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kujala P, Raymond CR, Romeijn M, Godsave SF, van Kasteren SI, Wille H, Prusiner SB, Mabbott NA, Peters PJ. 2011. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog 7:e1002449. doi: 10.1371/journal.ppat.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kincaid AE, Hudson KF, Richey MW, Bartz JC. 2012. Rapid transepithelial transport of prions following inhalation. J Virol 86:12731–12740. doi: 10.1128/JVI.01930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donaldson DS, Kobayashi A, Ohno H, Yagita H, Williams IR, Mabbott NA. 2012. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol 5:216–225. doi: 10.1038/mi.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prinz M, Huber G, Macpherson AJ, Heppner FL, Glatzel M, Eugster HP, Wagner N, Aguzzi A. 2003. Oral prion infection requires normal numbers of Peyer's patches but not of enteric lymphocytes. Am J Pathol 162:1103–1111. doi: 10.1016/S0002-9440(10)63907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glaysher BR, Mabbott NA. 2007. Role of the GALT in scrapie agent neuroinvasion from the intestine. J Immunol 178:3757–3766. doi: 10.4049/jimmunol.178.6.3757. [DOI] [PubMed] [Google Scholar]
- 44.Hoover CE, Davenport KA, Henderson DM, Pulscher LA, Mathiason CK, Zabel MD, Hoover EA. 2016. Detection and quantification of CWD prions in fixed paraffin embedded tissues by real-time quaking-induced conversion. Sci Rep 6:25098. doi: 10.1038/srep25098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunter N, Houston F, Foster J, Goldmann W, Drummond D, Parnham D, Kennedy I, Green A, Stewart P, Chong A. 2012. Susceptibility of young sheep to oral infection with bovine spongiform encephalopathy decreases significantly after weaning. J Virol 86:11856–11862. doi: 10.1128/JVI.01573-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stewart P, Campbell L, Skogtvedt S, Griffin KA, Arnemo JM, Tryland M, Girling S, Miller MW, Tranulis MA, Goldmann W. 2012. Genetic predictions of prion disease susceptibility in carnivore species based on variability of the prion gene coding region. PLoS One 7:e50623. doi: 10.1371/journal.pone.0050623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O'Rourke KI, Spraker TR, Hamburg LK, Besser TE, Brayton KA, Knowles DP. 2004. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J Gen Virol 85:1339–1346. doi: 10.1099/vir.0.79785-0. [DOI] [PubMed] [Google Scholar]
- 48.Seelig DM, Nalls AV, Flasik M, Frank V, Eaton S, Mathiason CK, Hoover EA. 2015. Lesion profiling and subcellular prion localization of cervid chronic wasting disease in domestic cats. Vet Pathol 52:107–119. doi: 10.1177/0300985814524798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoover CE, Davenport KA, Henderson DM, Zabel MD, Hoover EA. 15 February 2017. Endogenous brain lipids inhibit prion amyloid formation in vitro. J Virol doi: 10.1128/JVI.02162-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davenport KA, Henderson DM, Bian J, Telling GC, Mathiason CK, Hoover EA. 2015. Insights into chronic wasting disease and bovine spongiform encephalopathy species barriers by use of real-time conversion. J Virol 89:9524–9531. doi: 10.1128/JVI.01439-15. [DOI] [PMC free article] [PubMed] [Google Scholar]