

ABSTRACT
The mosquito-transmitted dengue virus (DENV) infects millions of people in tropical and subtropical regions. Maturation of DENV particles requires proper cleavage of the viral polyprotein, including processing of 8 of the 13 substrate cleavage sites by dengue virus NS2B/NS3 protease. With no available direct-acting antiviral targeting DENV, NS2/NS3 protease is a promising target for inhibitor design. Current design efforts focus on the nonprime side of the DENV protease active site, resulting in highly hydrophilic and nonspecific scaffolds. However, the prime side also significantly modulates DENV protease binding affinity, as revealed by engineering the binding loop of aprotinin, a small protein with high affinity for DENV protease. In this study, we designed a series of cyclic peptides interacting with both sides of the active site as inhibitors of dengue virus protease. The design was based on two aprotinin loops and aimed to leverage both key specific interactions of substrate sequences and the entropic advantage driving aprotinin's high affinity. By optimizing the cyclization linker, length, and amino acid sequence, the tightest cyclic peptide achieved a Ki value of 2.9 μM against DENV3 wild-type (WT) protease. These inhibitors provide proof of concept that both sides of DENV protease active site can be exploited to potentially achieve specificity and lower hydrophilicity in the design of inhibitors targeting DENV.
IMPORTANCE Viruses of the flaviviral family, including DENV and Zika virus transmitted by Aedes aegypti, continue to be a threat to global health by causing major outbreaks in tropical and subtropical regions, with no available direct-acting antivirals for treatment. A better understanding of the molecular requirements for the design of potent and specific inhibitors against flaviviral proteins will contribute to the development of targeted therapies for infections by these viruses. The cyclic peptides reported here as DENV protease inhibitors provide novel scaffolds that enable exploiting the prime side of the protease active site, with the aim of achieving better specificity and lower hydrophilicity than those of current scaffolds in the design of antiflaviviral inhibitors.
KEYWORDS: dengue fever, Zika, antimicrobial peptides, antiviral agents, drug design, protease inhibitors, receptor-ligand interaction, structural biology, substrate
INTRODUCTION
Dengue virus (DENV), the causative agent of the disease dengue fever, is endemic in more than 110 countries, with approximately 390 million people infected yearly, leading to about 20,000 deaths (1–3). Currently, no direct-acting antiviral drugs are available either in clinics or in development to combat dengue virus infections. Thus, a better understanding of the causative virus and potential viral drug targets is needed to develop effective therapies.
DENV is a member of the family Flaviviridae and is an enveloped virus with a positive single-stranded RNA genome. There are four different serotypes (DENV1 to DENV4), and each serotype shares 65 to 70% sequence identity of the genome (4). The dengue virus RNA genome encodes a single polyprotein, which needs to get processed at the cytoplasmic side of host cell rough endoplasmic reticulum membrane by dengue virus NS2B/NS3 protease and at the luminal side by the host cell peptidase (5). Dengue virus NS2B/NS3 protease is a serine protease that belongs to the chymotrypsin family with a classic Ser-His-Asp catalytic triad (6). NS2B (amino acids 1394 to 1440), which is referred to as a cofactor (cNS2B), is required for the proper function of NS3 protease (NS3pro185; amino acids 1476 to 1660) (7) and participates in substrate recognition (8). Dengue virus protease is responsible for the cleavage at 8 of the 13 polyprotein cleavage sites (9). These cleavage steps are required for maturation of the viral particle, making dengue virus NS2B/NS3 protease a promising target for drug development.
Inhibitors targeting dengue virus protease reported in the literature (10–14) have largely been based on the P side of the substrate cleavage product, since the P1 and P2 positions are rather conserved (basic amino acids), while the rest of the cleavage sequences are diverse. While most of these inhibitors bind with only micromolar affinity, a recent study found inhibitors with nanomolar Ki values (15). However, the potential challenge in targeting dengue virus protease is that this enzyme has a P side substrate sequence preference similar to those of several human serine proteases (furin RXRR, thrombin P1 R, and trypsin P1 R); hence, P side-based inhibitors are not designed to be specific to the viral protease. Moreover, these linear peptide-derived inhibitors have either low activity in enzymatic assays or substantially low potency in cellular assays, probably due to high hydrophilicity (or low lipophilicity, commonly measured by log octanol/water partition coefficient [cLogP]) values caused by charged side chain moieties at the P1 and/or P2 position. The relatively low affinity and high hydrophilicity and concerns for stability, combined with scarcity of small-molecule inhibitor-bound crystal structures, have impeded further characterization and optimization of peptide-based inhibitors.
The serine protease inhibitor aprotinin, a protein of 58 amino acids, has a high affinity for DENV protease and inhibits DENV2 protease with a Ki value of 26 nM (16). The binding loop of aprotinin is highly analogous in sequence to the native NS3 cleavage site and spans from the P3 to P4′ position at the active site of DENV protease (8). By engineering the binding loop of aprotinin, we recently identified the optimal amino acids for each of the P′ positions (17). Ensuring specificity, the P′ side of cleavage products does not share homology with human serine protease substrates and includes relatively hydrophobic amino acids. Our previous work determined that forming specific intermolecular interactions, such as hydrogen bonds contributed by P1 and P2′ residues, hydrophobic packing of P3′ and P4′ residues, and maintaining the conformation of the aprotinin's binding loop, is key for retaining binding affinity (17).
In the current study, we leveraged the potency of aprotinin and our detailed analysis of the specificity for dengue virus protease for P′ side substrates to design the scaffold for specific inhibitors. Building on the interactions identified to be key for binding, we have designed cyclic peptides (CPs) targeting DENV protease active-site pocket from the S3 to S4′ position (Fig. 1). As we identified entropy to be the major driving force for binding (17), a second aprotinin loop was incorporated into the design with the aim of maintaining binding loop structure and rigidity. The binding loop of aprotinin (Pro13 to Ile18/Ile19) and a second loop (Tyr35/Gly36 to Arg39) were linked together with or without glycine spacers between Ile18/Ile19 and Tyr35/Gly36. The disulfide bond between Cys14 and Cys38 already in aprotinin was kept to cyclize the peptide. The inhibition constants of CPs against dengue virus protease were measured by fluorescence resonance energy transfer (FRET)-based enzymatic assays, and molecular dynamics (MD) simulations were applied to investigate the molecular interactions between CPs and the protease active-site residues.
FIG 1.

Design of cyclic peptides derived from aprotinin as dengue virus NS3/2B protease inhibitor. (A) Aprotinin-DENV3 protease complex structure (PDB code 3u1j). The NS3 protease domain is in green, the NS2B cofactor is in cyan, and aprotinin is in purple. (B) The binding loop and the second loop of aprotinin on which the cyclic peptides were based are displayed as sticks. Dengue virus NS3/2B protease is shown as a gray surface, and the catalytic triad is indicated by orange. (C) Cyclic peptide derived from aprotinin is displayed as sticks. The amino acids (P3 to P3′) are numbered based on substrate residue positions. (D) Hydrogen bonds between the cyclic peptide (magenta sticks) and dengue virus NS3/2B protease (residues labeled and side chains depicted as green sticks) are displayed as yellow dashes. The catalytic triad of protease is depicted as orange sticks, and residues are labeled.
RESULTS
The measurable inhibition constants of cyclic peptides against DENV3 protease vary from 2.9 μM (CP7) to 780.3 μM (CP9) (Tables 1 and 2). To determine the optimal length of the cyclic peptide, peptides were designed with various lengths from the longest, CP1 and CP2 (residues P3 to P4′, two glycine linkers and residues 35 to 39), to the shortest, CP12 (residues P3 to P3′ and with no linker and residues 37 to 39).
TABLE 1.
Inhibition constants for cyclic peptides of different lengths in enzymatic assays against DENV3 NS3/2B proteasea
| Peptide | Amino acid |
Ki (μM) | Fold change | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P3 | P2 | P1 | P1′ | P2′ | P3′ | P4′ | 35 | 36 | 37 | 38 | 39 | |||||
| CP1 | P | C* | K | A | R | I | I | G | G | Y | G | G | C* | R | 232.3 ± 69.9 | 1.00 |
| CP2 | P | C* | K | A | R | I | I | G | G | Y | G | G | C* | A | 678.3 ± 244.1 | 2.92 |
| CP3 | P | C* | K | A | R | I | G | Y | G | G | C* | A | 376.8 ± 204.1 | 1.62 | ||
| CP4 | P | C* | R | A | R | I | G | Y | G | G | C* | A | 966.1 ± 328.8 | 4.16 | ||
| CP5 | P | C* | K | A | R | I | Y | G | G | C* | A | 14.5 ± 6.7 | 0.06 | |||
| CP6 | P | C* | R | A | R | I | Y | G | G | C* | R | 187.3 ± 43.2 | 0.81 | |||
| CP7 | P | C* | R | A | R | I | Y | G | G | C* | A | 2.9 ± 0.8 | 0.01 | |||
| CP8 | P | C* | R | A | R | I | Y | G | C* | A | 467.9 ± 68.7 | 2.01 | ||||
| CP9 | P | C* | R | A | R | I | G | G | C* | A | 780.3 ± 195.5 | 3.36 | ||||
| CP10 | P | C* | R | A | R | I | G | G | C* | R | 29.7 ± 5.8 | 0.13 | ||||
| CP11 | C* | R | A | R | I | Y | G | G | C* | R | 145.1 ± 27.9 | 0.62 | ||||
| CP12 | P | C* | R | A | R | I | G | C* | A | 101.2 ± 24.8 | 0.44 | |||||
The amino acid sequence is given based on corresponding substrate positions (P3 to P4′; aprotinin residues 12 to 18) and the linked aprotinin second-loop residue numbers (35 to 39). The peptides were cyclized through a disulfide bond between cysteine residues indicated by an asterisk. The fold changes are relative to CP1.
TABLE 2.
Inhibition constants for cyclic peptides with sequence optimization against DENV3 NS3/2B proteasea
| Peptide | Amino acid |
Ki (μM) | Fold change | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P3 | P2 | P1 | P1′ | P2′ | P3′ | 35 | 36 | 37 | 38 | 39 | |||
| CP7 | P | C* | R | A | R | I | Y | G | G | C* | A | 2.9 ± 0.8 | 1.00 |
| CP13 | Bzb | C* | R | A | R | I | Y | G | G | C* | A | 33.2 ± 6.5 | 11.45 |
| CP14 | P | C* | R | A | Q | I | Y | G | G | C* | A | NBc | NB |
| CP15 | P | C* | R | A | W | I | Y | G | G | C* | R | 19.7 ± 8.8 | 6.79 |
| CP16 | P | C* | R | A | W | I | Y | G | G | C* | A | 144.8 ± 54.4 | 49.93 |
| CP17 | P | C* | R | A | R | I | D | G | G | C* | A | 69.3 ± 24.4 | 23.90 |
| CP18 | P | C* | R | V | R | I | D | G | G | C* | A | 35.8 ± 16.4 | 12.34 |
| CP19 | P | C* | R | V | R | I | Y | G | G | C* | A | 25.6 ± 9.9 | 8.83 |
The amino acid sequence is given based on corresponding substrate positions (P3 to P4′; aprotinin residues 12 to 18) and the linked aprotinin second loop residue numbers (35 to 39). The peptides were cyclized through a disulfide bond between cysteine residues indicated by an asterisk. The fold changes are relative to CP7.
Bz denotes benzoyl capping group.
NB, no binding.
First, the linker between the binding loop and second loop was optimized for length. Comparing CP3 to CP5, or CP4 to CP7, no glycine linker is required between Ile18 and Tyr35. Without the glycine linker, the inhibition improved, with Ki values decreasing from 376.8 μM to 14.5 μM for CP3 versus CP5 and from 966.1 μM to 2.9 μM for CP4 versus CP7. Further, comparing CP2 (Ki = 14.5 μM) to CP5 (Ki = 678.3 μM), P4′ isoleucine and two glycine residues as a linker were not favorable. Thus, no additional linker residue is required to connect the two loops, and shorter cyclic peptides have better inhibition against dengue virus protease.
Next, the optimal length of the second loop was investigated by comparing CP7 to peptides with shorter second loops: For CP12, CP8, and CP9, the Ki values increased from 2.9 μM to 101.2 μM, 467.9 μM, and 780.3 μM, respectively. Therefore, the optimal number of residues for the second loop of the cyclic peptide between P3′ Ile and Cys38 is three.
Optimizing residues for P1 and P2′ positions of the peptides.
Dengue virus protease prefers basic residues lysine and arginine at the P1 position, and whether one of these residues is preferred over the other was tested. CP3, with lysine at P1, has a lower Ki (376.8 μM) than does CP4, with arginine at P1 (966.1 μM), but CP7, with arginine at P1, has a lower Ki (2.9 μM) than does CP5, with lysine at P1 (14.5 μM) (Table 1). Thus, the preference between the two basic amino acids is subtle and context dependent. While Arg can form one more hydrogen bond than can Lys, both residues can form electrostatic interactions with the negatively charged residue D129 on the protease. The charge interaction might be the dominant force in this pocket, explaining the lack of a significant difference between these two amino acids.
Arginine is favored at the P2′ position and can form both a hydrogen bond and more van der Waals (vdW) contacts than other residues, as we have previously shown (17). In this study, arginine (large basic hydrogen bonding partner), glutamine (large hydrophilic hydrogen bonding partner), and tryptophan (large aromatic hydrogen bonding partner) at the P2′ position were compared: CP7, CP14, and CP16 have Ki values of 2.9, 1,273, and 145 μM, respectively (Table 2). Glutamine of CP14, which can also form a hydrogen bond, is less favorable at this position than arginine, and tryptophan of CP16, which was designed to interact with protease through extensive vdW contacts, is also less favored than that of CP7. These results reflect that both hydrogen bonding and vdW contacts by the P2′ residue are integral for the binding interaction in this pocket.
The optimal C-terminal residue depends on the peptide length.
To decrease the flexibility at the C terminus of the cyclic peptides, the long side chain of Arg39 (present in native aprotinin sequence) was replaced with alanine. The CP1 peptide, which is longer with linkers between the binding and second loop, has an arginine at this position (Ki = 232.3 μM). The inhibition was weaker when the arginine was replaced with an alanine (CP2; Ki = 678.3 μM). In contrast, comparing CP6, with an arginine, to CP7, with an alanine, the affinity improves, with the Ki value dropping from 187.3 μM to 2.9 μM (Table 1). Thus, decreased flexibility at the peptide C terminus was beneficial for tighter binding interactions at this peptide length. When the peptide length was further decreased by removing Tyr35, the trend was reversed: CP10, with an arginine, had better inhibition (Ki = 29.7 μM) than did CP9, with an alanine (Ki = 780.3 μM). Potentially in these smaller peptides without Tyr35, the binding conformation is altered and the terminal arginine interacts with the S2′ pocket, specifically with Asp75, resulting in the observed lower Ki value. Overall, the length of the peptide likely determines the position of the terminal residue when bound to dengue virus protease active site, resulting in peptides with different lengths favoring different amino acids at this position.
N-terminal capping.
To investigate the contribution of P3 residue (N terminus), the proline at this position was replaced with a benzoyl capping group, which can mimic interactions contributed by the proline ring. CP13 with the capping group has a higher Ki value (33.2 μM) than does CP7 (2.9 μM), indicating that proline is more favorable than a benzoyl group (Table 2). Further, to investigate the requirement of a P3 residue, we designed CP11, which does not have the P3 residue. Comparing CP6 and CP11, removal of the whole proline residue did not significantly change the Ki values, which suggests, at least in this context, that P3 proline is not required (Table 1).
Cyclic peptide inhibitors are flexible.
Based on the Ki values of the cyclic peptides designed and assayed, both the sequence and length of the cyclic peptide significantly affect binding to dengue virus protease. However, the Ki values are still much higher (lower inhibition activity) than the same peptide sequence within the binding loop of aprotinin. We have shown through isothermal titration calorimetry (ITC) that entropy is the driving force for binding of aprotinin and variants, and fluctuations in molecular dynamics (MD) simulations correlate with binding affinity for aprotinin constructs (17). To investigate how the dynamics of cyclic peptide molecular interactions compare to those within the context of aprotinin constructs, MD simulations of the highest-affinity cyclic peptide bound to dengue virus protease were carried out. MD simulations started from the protease structure with the CP7 peptide modeled into the binding pocket based on the aprotinin-DENV3 protease complex structure (PDB code 3u1j). The dynamics were compared to those of the aprotinin-protease complex structure (3u1j) to examine whether interactions important for aprotinin binding were still maintained.
Throughout the simulation, the CP7 cyclic peptide remained positioned in the binding site. Snapshots of CP7, and the corresponding binding loop/second loop with the same sequence within aprotinin (the simulation included the whole aprotinin, but only these loops are displayed), are shown in Fig. 2. Both the superimposed snapshots and calculated root-mean-square fluctuation (RMSF) values of the backbone indicate that the binding loop and second loop are more flexible in CP7, with 0.7-Å- and 1.6-Å-higher RMSF values than for the corresponding loops within the context of aprotinin, respectively. The second loop of CP7 was incorporated with the aim of sustaining the binding loop structure and providing some of the rigidity observed in aprotinin; however, these results illustrate that the cyclic peptide, although still stable in the binding site, is relatively more flexible. The decreased rigidity of the cyclic peptide compared to aprotinin correlates with the loss in potency.
FIG 2.

Molecular dynamics (MD) simulations of aprotinin and the cyclic peptide CP7 bound to the dengue virus protease active site. (A) Snapshots from MD simulation trajectories of aprotinin and CP7 bound to DENV3 protease. The protease is depicted in cartoon representation in green and the aprotinin's binding/second loops or CP7 as sticks from the start (in red) to the end (in blue) of the trajectory. (The simulations were performed with full-length aprotinin, but only residues corresponding to native substrates are shown for clarity.) (B) The RMSF values during MD simulations were tabulated as the average for the binding loop and the second loop and are displayed for individual residue positions.
Cyclic peptide maintains contacts with DENV protease.
To investigate how the cyclic peptide inhibitor packs against the protease active site, both the intermolecular vdW contacts and the hydrogen bonding patterns were analyzed. The average vdW contact energy of each binding loop residue was calculated throughout the simulation time (Fig. 3). Similar to the contacts of same positions within the aprotinin binding loop (17), P1 Lys/Arg and P2′ Arg contribute the most contact energies. Intermolecular contacts are formed by P3-P3′ residues, with the only other residue beyond this region making contact with the protease being the side chain of Cys38 in the disulfide bond. Overall, the rest of the residues in CP7 P3-P3′ make similar contacts at each corresponding position as in the aprotinin binding, but the total vdW contact is slightly decreased. The intermolecular hydrogen bonds formed by the binding loop residues were also analyzed and compared to those in aprotinin (Table 3). The hydrogen bonds that are observed for aprotinin binding to the protease are conserved in the cyclic peptides; however, these hydrogen bonds are more stable in aprotinin, existing for a larger percentage of the simulation time. As these binding loops within aprotinin and within the cyclic peptide have the same sequence (except the P1 residue, lysine or arginine), the differences in vdW contacts and hydrogen bonding tightness likely reflect how stable the whole macrocyclic peptide is within the binding pocket.
FIG 3.

Average intermolecular van der Waals (vdW) contact energies during MD simulations between aprotinin or CP7 binding loop residues and DENV3 protease.
TABLE 3.
Hydrogen bonds between residues of aprotinin or cyclic peptides and DENV3 protease and the percentage of time the hydrogen bonds existed during the MD simulationsa
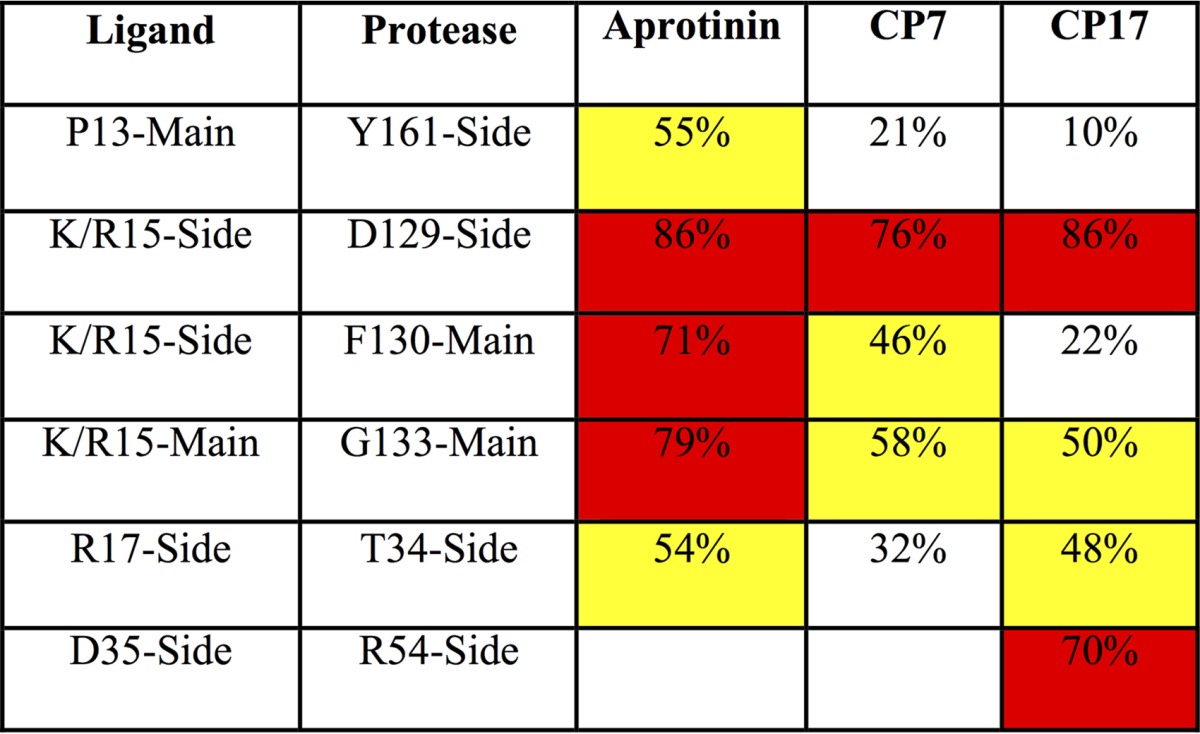
Yellow highlights values higher than 40%, and red highlights values higher than 60%.
Targeting Arg54 on the protease.
To further increase the specificity of the cyclic peptide, potential interactions of the cyclic peptide were assessed. Arg54 of dengue virus protease is conserved throughout all four serotypes of dengue virus, is a lysine in both West Nile virus (WNV) and Zika virus, but is not conserved in human furin, thrombin, or trypsin. To build interactions with this particular residue and thus potentially enhance specificity, various residues on the second loop of the cyclic peptides were replaced with acidic residues. Replacing Tyr35 with aspartic acid in both CP17 (with an alanine at P1′) and CP18 (valine at P1′) resulted in Ki values of 69.3 μM and 35.8 μM, respectively (Table 2). While P1′ alanine is more favorable over valine (comparing CP7 to CP19), there is no significant difference between alanine and valine with Asp35. Since the S1′ pocket is relatively small, alanine may fit better than valine in this pocket. However, the P1′ valine may push the second loop closer to Arg54 on the protease, stabilizing the interaction between Asp35 of cyclic peptide and Arg54 of the protease. The interactions we aimed to stabilize between these two residues would be reflected by an extra hydrogen bond between this cyclic peptide-protease pair.
To test whether this design achieved this extra hydrogen bond, CP17 was modeled and MD simulations were performed on CP17 bound to dengue virus protease to investigate the dynamics of molecular interactions between Asp35 of cyclic peptide and Arg54 of the protease. Similar to the simulations of the tightest binder CP7, MD simulations started from the protease structure with the CP17 peptide modeled into the binding pocket based on the aprotinin-DENV3 protease complex structure (PDB code 3u1j) (8). For the peptide CP17, there is one extra hydrogen bond formed between Asp35 of CP17 and Arg54 of the protease (Table 3), which is promising. As Arg54 on the protease is conserved in the flaviviral proteases, further optimization of interactions with this residue is a potential strategy to increase the specificity of these peptides against dengue virus protease.
DISCUSSION
In this study, we designed cyclic peptide inhibitors targeting both P and P′ sites of dengue virus protease and optimized the sequence and length of these peptides. The best binder (CP7) has a Ki value of 2.9 μM against DENV3 wild-type (WT) protease. These inhibitors were derived based on the binding loop of aprotinin, and as intended, interactions important for aprotinin binding were preserved, specifically the hydrogen bonds contributed by P1 and P2′ residues and vdW contacts of each residue position. However, these interactions in the cyclic peptides were not as strong as those in aprotinin upon binding to dengue virus protease. The MD simulations and the resulting RMSF values reflect that even though a second loop was included in the design to support the structure and stability of the binding loop, the cyclic peptide is still more flexible than the corresponding loops in aprotinin. This increased flexibility may account for the decreased affinity between the cyclic peptides and protease, which explains why these cyclic peptides have higher Ki values against DENV3 WT protease than aprotinin. Nevertheless, these peptides demonstrate the proof of principle and viability of targeting the primed side of the DENV3 protease active site as a potential strategy for specific inhibition. A similar strategy of inhibitor design based on aprotinin was applied to WNV protease (18), indicating that cyclic peptide derived from aprotinin can be a promising scaffold for inhibitor design targeting serine proteases.
To move these inhibitors from proof-of-principle micromolar inhibitors to potentially viable antiviral drugs, many other additional strategies are needed to further increase the affinity. The first such strategy is to increase the rigidity. The two loops of cyclic peptide inhibitor (CP7 as an example) are linked together through a peptide bond between Ile18 and Tyr35, and a disulfide bond between Cys14 and Cys38 (Fig. 1; Tables 1 and 2), which could be broken under reducing conditions, including in the cellular environment. Using a nonreducible covalent bond instead is a strategy to increase the stability of the cyclic peptide, or incorporating heterocycles at the same position is a strategy to increase both stability and rigidity. Incorporating heterocycles between the binding loop and second loop of cyclic peptides at different positions could be another strategy to rigidify the peptides, as could replacing one or more peptide bonds with more rigid nonrotatable bonds. The second strategy for increasing the affinity is to decrease the molecular weight of the cyclic peptide by cutting residues at both termini. The third strategy is to target the hydrophobic protease residues 30 and 31, which define the S4′ pocket and were proposed to interact with the membrane (19, 20). The fourth strategy is to couple the prime side inhibitors with P site inhibitors, which are generally highly charged and polar, to potentially increase their cell permeability. The fifth strategy is to replace the peptide bond between P1 and P1′ residues with a noncleavable bond, which would be beneficial to the integrity of the cyclic peptide. Replacing the carbonyl group of P1 residue, which forms a key hydrogen bond with protease's Ser135 backbone amide nitrogen (Fig. 1D and Table 3), with peptide bioisosteres such as reduced amide, ketoethylene, or hydroxyethylene, the hydrogen bonds can be maintained while making the bond noncleavable. A viable high-affinity inhibitor may be attainable by combining one or more of these strategies to form a more rigid cyclic inhibitor.
Macrocyclic inhibitors were shown to successfully inhibit hepatitis C virus (HCV) NS3/4A protease compared to their linear analogs, and these inhibitors were cyclized through links between side chains (21), unlike through-backbone bonds in dengue virus cyclic peptides. Since dengue virus protease's S2′ and S4′ pockets are solvent exposed and overlapping (Fig. 1C), similar to the S4 and S2 pockets of HCV NS3/4A protease, introducing a P2′-P4′ macrocycle to these cyclic peptides can potentially increase both peptide rigidity and the packing against residues in these two pockets.
Recently, DENV/WNV protease inhibitors targeting P site pockets with nanomolar activity in enzymatic assays have been reported (Ki = 12 nM and 39 nM) (15). Since these inhibitors are highly polar, possibly underlying the micromolar-level activity in cell-based assays, incorporating the critical hydrophobic interactions we identified at P3′ or P4′ pockets (17) is a strategy to reduce the hydrophilicity. Further, building interactions with the conserved DENV protease residue Arg54 could increase the specificity of this tighter-binding lead compound. Unnatural amino acids such as Orn at the P1 position instead of Lys to maintain critical polar interactions without distributing the charge, or halogenated amino acids to exploit halogen bonding, can be used in the future improvement of these compounds. A crystal structure of DENV protease bound to a lead compound spanning both P and P′ sides would be immensely useful for further optimization in the design of more potent and specific inhibitors. In the future, further improvement of P2 and P3 moieties, such as those in a peptide-based inhibitor with an available crystal structure (PDB code 3U1I), could leverage additional interactions with the NS2B cofactor. Lastly, as dengue virus protease shares substrate sequence similarity with other flaviviral proteases causing human infections, notably Zika virus protease, the lead compounds and strategies identified in this study can lead to the design of pan-flaviviral inhibitors or inhibitors targeting other flaviviruses.
MATERIALS AND METHODS
Protein and peptides.
The DENV protease gene (cDNA encoding NS2B cofactor [cNS2B; amino acids 1394 to 1440] and NS3 protease [NS3pro185; amino acids 1476 to 1660] with a G4SG4 linker in between) was constructed for protein expression in Escherichia coli, as described previously (8). The protease gene was constructed between BamHI and XhoI sites of plasmid pGEX6p1 (GE Healthcare), and BL21(DE3) cells were used for protein expression. Published protein expression and purification protocols were used (22). All cyclic peptides were purchased from 21st Century Biochemicals.
Enzyme inhibition assay.
A fluorescence resonance energy transfer (FRET)-based enzymatic assay was used to determine the inhibition constants of cyclic peptides against DENV3 WT protease. A 100 nM concentration of the DENV3 protease was incubated with various concentrations of cyclic peptides (5 mM to 500 nM) for 60 min in 50 mM Tris assay buffer {20% glycerol, 1 mM 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]; pH 8.5} (23). A 5 μM concentration of protease substrate [Ac-[D-EDANS]KRRSWP[K-DABCYL]-AMIDE] (21st Century Biochemicals) was added to initiate proteolysis and monitored using the EnVision plate reader (PerkinElmer) at excitation and emission wavelengths of 340 nm and 490 nm, respectively. One-phase association nonlinear fit to the whole progress curve was applied to determine the initial cleavage velocities (24). By nonlinear regression fitting to the Morrison equation of initial velocity versus inhibitor concentration using Prism 6 (GraphPad Software), apparent inhibition constants (Ki) were obtained. Data were collected in triplicate and processed independently to calculate the shared inhibition constant and standard deviation.
Molecular dynamics simulations.
All molecular dynamics simulations were performed in triplicate by following previously published protocols (25). To prepare the modeled complex structures, the aprotinin-bound DENV3 WT protease structure (PDB code 3u1j) was used as a starting point. Aprotinin residues except the binding loop (Pro13 to Ile18/Ile19) and the second loop (Tyr35/Gly36 to Arg39) were removed from the structure, and these two loops were linked together with a peptide bond using Maestro (Suite 2012: Maestro, version 9.3; Schrödinger). The modeled structures were prepared for simulation by keeping the crystallographic waters within 4.0 Å of any protein atom but removing the buffer salts from the coordinate file. Next, hydrogen atoms were added to the structure using Protein Preparation Tool from Schrödinger (26), and the optimal protonation states for the ionizable side chains were determined. The hydrogen bonding network of the initial structures was optimized, and by using the Impact refinement module with the OPLS2005 force field, the structures were minimized in vacuum (27). Using the System Builder utility, these prepared systems were then solvated with the TIP3P water model extending 10 Å beyond the protein in all directions in a truncated octahedron solvent box. The overall charge of the system was neutralized by adding counter ions (Na+ or Cl−). Desmond was used in all simulations with the OPLS2005 force field. Each simulation was carried out at 300 K and 105 Pa (= 1 atm) for 100 ns, and the coordinates and energies were recorded every 5 ps.
Structural analysis.
The hydrogen bonds were determined using VMD (28). A hydrogen bond was defined by the donor and acceptor distance less than 3 Å and a donor-hydrogen-acceptor angle of >150°. To calculate the vdW contact energies between cyclic peptides and protease, a simplified Lennard-Jones potential was applied, as described in detail before (29).
ACKNOWLEDGMENT
This work was supported by NIH grant R01 AI085051.
REFERENCES
- 1.Monath TP. 1994. Dengue: the risk to developed and developing countries. Proc Natl Acad Sci U S A 91:2395–2400. doi: 10.1073/pnas.91.7.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. 2009. Dengue: guidelines for diagnosis, treatment, prevention and control. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 3.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rico-Hesse R. 1990. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 174:479–493. doi: 10.1016/0042-6822(90)90102-W. [DOI] [PubMed] [Google Scholar]
- 5.Chambers TJ, Hahn CS, Galler R, Rice CM. 1990. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688. [DOI] [PubMed] [Google Scholar]
- 6.Bera AK, Kuhn RJ, Smith JL. 2007. Functional characterization of cis and trans activity of the flavivirus NS2B-NS3 protease. J Biol Chem 282:12883–12892. doi: 10.1074/jbc.M611318200. [DOI] [PubMed] [Google Scholar]
- 7.Yusof R, Clum S, Wetzel M, Murthy HM, Padmanabhan R. 2000. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J Biol Chem 275:9963–9969. doi: 10.1074/jbc.275.14.9963. [DOI] [PubMed] [Google Scholar]
- 8.Noble CG, Seh CC, Chao AT, Shi PY. 2012. Ligand-bound structures of the dengue virus protease reveal the active conformation. J Virol 86:438–446. doi: 10.1128/JVI.06225-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falgout B, Pethel M, Zhang YM, Lai CJ. 1991. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol 65:2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin Z, Patel SJ, Wang WL, Chan WL, Ranga Rao KR, Wang G, Ngew X, Patel V, Beer D, Knox JE, Ma NL, Ehrhardt C, Lim SP, Vasudevan SG, Keller TH. 2006. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg Med Chem Lett 16:40–43. [DOI] [PubMed] [Google Scholar]
- 11.Yin Z, Patel SJ, Wang WL, Wang G, Chan WL, Rao KR, Alam J, Jeyaraj DA, Ngew X, Patel V, Beer D, Lim SP, Vasudevan SG, Keller TH. 2006. Peptide inhibitors of dengue virus NS3 protease. Part 1: warhead. Bioorg Med Chem Lett 16:36–39. doi: 10.1016/j.bmcl.2005.09.062. [DOI] [PubMed] [Google Scholar]
- 12.Nitsche C, Steuer C, Klein CD. 2011. Arylcyanoacrylamides as inhibitors of the dengue and West Nile virus proteases. Bioorg Med Chem 19:7318–7337. doi: 10.1016/j.bmc.2011.10.061. [DOI] [PubMed] [Google Scholar]
- 13.Nitsche C, Behnam MA, Steuer C, Klein CD. 2012. Retro peptide-hybrids as selective inhibitors of the dengue virus NS2B-NS3 protease. Antiviral Res 94:72–79. doi: 10.1016/j.antiviral.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Bastos Lima A, Behnam MA, El Sherif Y, Nitsche C, Vechi SM, Klein CD. 2015. Dual inhibitors of the dengue and West Nile virus NS2B-NS3 proteases: synthesis, biological evaluation and docking studies of novel peptide-hybrids. Bioorg Med Chem 23:5748–5755. doi: 10.1016/j.bmc.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 15.Behnam MA, Graf D, Bartenschlager R, Zlotos DP, Klein CD. 2015. Discovery of nanomolar dengue and West Nile virus protease inhibitors containing a 4-benzyloxyphenylglycine residue. J Med Chem 58:9354–9370. doi: 10.1021/acs.jmedchem.5b01441. [DOI] [PubMed] [Google Scholar]
- 16.Mueller NH, Yon C, Ganesh VK, Padmanabhan R. 2007. Characterization of the West Nile virus protease substrate specificity and inhibitors. Int J Biochem Cell Biol 39:606–614. doi: 10.1016/j.biocel.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 17.Lin KH, Nalivaika EA, Prachanronarong KL, Yilmaz NK, Schiffer CA. 4 September 2016. Dengue protease substrate recognition: binding of the prime side. ACS Infect Dis doi: 10.1021/acsinfecdis.6b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ang MJ, Lim HA, Poulsen A, Wee JL, Ng FM, Joy J, Hill J, Chia CS. 31 May 2016. Miniature bovine pancreatic trypsin inhibitors (m-BPTIs) of the West Nile virus NS2B-NS3 protease. J Enzyme Inhib Med Chem doi: 10.1080/14756366.2016.1190713. [DOI] [PubMed] [Google Scholar]
- 19.Chappell KJ, Stoermer MJ, Fairlie DP, Young PR. 2008. West Nile virus NS2B/NS3 protease as an antiviral target. Curr Med Chem 15:2771–2784. doi: 10.2174/092986708786242804. [DOI] [PubMed] [Google Scholar]
- 20.Assenberg R, Mastrangelo E, Walter TS, Verma A, Milani M, Owens RJ, Stuart DI, Grimes JM, Mancini EJ. 2009. Crystal structure of a novel conformational state of the flavivirus NS3 protein: implications for polyprotein processing and viral replication. J Virol 83:12895–12906. doi: 10.1128/JVI.00942-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, Coleman PJ, Dimuzio JM, Ferrara M, Di Filippo M, Gates AT, Graham DJ, Harper S, Hazuda DJ, Huang Q, McHale C, Monteagudo E, Pucci V, Rowley M, Rudd MT, Soriano A, Stahlhut MW, Vacca JP, Olsen DB, Liverton NJ, Carroll SS. 2012. MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother 56:4161–4167. doi: 10.1128/AAC.00324-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Lim SP, Beer D, Patel V, Wen D, Tumanut C, Tully DC, Williams JA, Jiricek J, Priestle JP, Harris JL, Vasudevan SG. 2005. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J Biol Chem 280:28766–28774. doi: 10.1074/jbc.M500588200. [DOI] [PubMed] [Google Scholar]
- 23.Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. 2001. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J Biol Chem 276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- 24.Salykin A, Kuzmic P, Kyrylenko O, Musilova J, Glatz Z, Dvorak P, Kyrylenko S. 2013. Nonlinear regression models for determination of nicotinamide adenine dinucleotide content in human embryonic stem cells. Stem Cell Rev 9:786–793. doi: 10.1007/s12015-013-9454-3. [DOI] [PubMed] [Google Scholar]
- 25.Özen A, Lin KH, Kurt Yilmaz N, Schiffer CA. 2014. Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc Natl Acad Sci U S A 111:15993–15998. doi: 10.1073/pnas.1414063111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. 2013. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 27.Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. 2010. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J Chem Theory Comput 6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 28.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 29.Ozen A, Haliloglu T, Schiffer CA. 2011. Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J Mol Biol 410:726–744. doi: 10.1016/j.jmb.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]