

Abstract
Carbonic anhydrase II (CAII) deficiency is an autosomal recessive disorder characterized by renal tubular acidosis, osteopetrosis, recurrent bone fractures, renal stones, growth failure, and mental retardation. Several cases have been reported in Saudi Arabia with homozygous mutations in CA2 consistent with a high degree of consanguinity. We report a case of carbonic anhydrase II deficiency with short stature, mixed renal tubular acidosis, recurrent bone fractures due to trivial trauma, recurrent renal stones and cerebral calcification. This patient was compound heterozygous for a novel CA2 mutation and a previously reported mutation in Arabs.
Keywords: Carbonic anhydrase II deficiency, Osteopetrosis, Renal tubular acidosis, Marble brain disease, Novel mutation, Arab
Introduction
Carbonic anhydrase is an important enzyme for brain, kidney and bone physiology. This enzyme catalyzes the formation of carbonic acid from water and carbon dioxide. Carbonic acid dissociates spontaneously to release protons, which are essential for creating the acidic environment required for the dissolution of bone minerals in the resorption lacunae. Carbonic anhydrase II deficiency is an autosomal recessive inborn error of metabolism that results in cerebral calcification, renal tubular acidosis and osteopetrosis [1].
We report a case of carbonic anhydrase deficiency that presented with recurrent bone fractures, renal stones and hypokalemia and was originally diagnosed as osteomalacia. Later, she was found to have mixed-type renal tubular acidosis with other features of carbonic anhydrase deficiency. By screening the patient and her family for CA2 mutations, we found a novel mutation that has not been previously reported.
Case presentation
A 19-year-old Saudi girl was healthy until the age of 12 years when she suffered a bone fracture after a trivial trauma. She was observed to have a short stature for her age, which was investigated thoroughly; she was diagnosed with osteomalacia based on vitamin D deficiency and a bone scan. A renal stone was found at that time, and she underwent lithotripsy. Concurrent distal renal tubular acidosis was also found and resulted in hypokalemia and normal anion gap metabolic acidosis. Her menarche and secondary sexual characteristics started at 15 years of age. Mentally, she had good performance at the high school level, with the exception of some delay due to recurrent hospitalization. She had a 25-year-old brother with a history of recurrent bone fractures, renal stones, short stature and mental retardation. The mother and father were from the same tribe.
The patient was referred to our hospital as a case with recurrent bone fractures due to minimal traumas and bilateral renal stones. She had the characteristic facial features of a wide forehead, a prominent narrow nose, a small mouth with crowded teeth and prominent ears. Her height was 126 cm, and she weighed 40 kg; both were below the 5th percentile. The rest of the physical examination was unremarkable.
Her labs revealed the following: hypokalemia, 3.1 mmol/L; chloride, 112 mmol/L; corrected calcium, 2.08 mmol/L; inorganic phosphorus, 0.57 mmol/L; alkaline phosphatase, 62 µ/L; and uric acid, 43 µmol/L. A urine analysis showed a pH of 5.5. Her venous blood gas pH was 7.21, PCO2 was 35.2 mmHg, HCO3 was 13.8 mmol/L, and her anion gap was 16.3. Hormonal testing was normal except for a 25-OH vitamin D level of 15.2 nmol/L (6 ng/mL) with 16.49 pmol/L (4.49 pmol/mL) of parathyroid hormone (Table 1).
Table 1.
Laboratory parameters
| 25-OH vitamin D | 6 ng/mL |
| Serum intact parathyroid hormone | 4.49 pmol/mL |
| Potassium | 3. 1 mmol/L |
| Chloride | 112 mmol/L |
| Corrected calcium | 2.08 mmol/L |
| Inorganic phosphorous | 0.57 mmol/L |
| Uric acid | 43 µmol/L |
| Alkaline phosphatase | 62 µ/L |
| Urine pH | 5.5 |
| Urine sodium | 85 mmol/L |
| Urine potassium | 50 mmol/L |
| Urine calcium | 4.4 mmol/L |
| Urine magnesium | 1.5 mmol/L |
| Complete blood count | |
| White blood cells | 9.5 (×10.e9/L) |
| Red blood cells | 3.97 (×10.e12/L) |
| Hemoglobin | 98 g/L |
| Platelets | 290 (×10.e9/L) |
Radiologically, she had intracranial calcifications of the basal ganglia (Figs. 1, 2) and osteopetrosis. A kidney ultrasound and CT scan showed small kidneys, bilateral cortical cysts, increased echogenicity and bilateral renal stones. A bone scan showed a generalized increased uptake with pseudofractures of the femurs and right tibia (Fig. 3).
Fig. 1.

Skull X-ray and CT brain showing intracranial calcifications of the basal ganglia
Fig. 2.
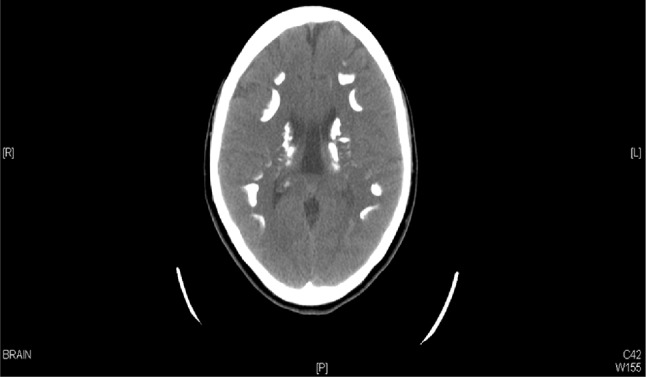
Skull X-ray and CT brain showing intracranial calcifications of the basal ganglia
Fig. 3.

Whole body bone scan performed with 20 mCi of Tc99 m MDP (methylenediphosphonic acid): showing increased tracer uptake throughout whole the skeleton with focal area of uptake in mid-shaft of both femur and mid-shaft of right tibia
DNA was extracted from the peripheral blood samples of the family members. Genomic DNA of the affected patients was then amplified by PCR for all seven exons of the CA2 gene (NM_000067.2) (Table 2). The coding regions of the gene were then screened by direct sequencing (ABI) of the PCR products, and the sequencing results were analyzed by SeqMan II (DNASTAR, Madison, WI, USA). Three family members, including our patient, were compound heterozygous for two lethal mutations. One mutation was a common Arab splice site mutation (c.232+1G>A) in intron 2 [2]. A novel mutation (c.484delG) was detected in exon 5 in the other allele. This novel mutation predicts a frame shift at amino acid 162 and premature truncation 35 residues downstream (p.V162CfsX35). Screening of the parents and all other family members revealed a heterozygosity for the c.232+1G>A mutation in the father and three daughters. The mother and one daughter were heterozygous for the c.484delG mutation. Two sons had wild-type alleles (Figs. 4, 5).
Table 2.
Forward and reverse primer sequences and exon analyzed by with real-time PCR
| Exon | Forward primer | Reverse primer |
|---|---|---|
| Exon-1 | Fw-GGGAGCCTATAAAAGCTGGTG | Rv-CGG TAA ACA GCA TGT GCG |
| Exon-2 | Fw-CAGATTGGCTCCGGAATG | Rv-TAA GAG GGT TCC TGC CAC TG |
| Exon-3 | Fw-AGGC ATG TGT TTC ATG TGT G | Rv-TTC ACT GGG CGT GAG TTG |
| Exon-4 | Fw-TGCTGTTATACCA AGTACTGTGTGG | Rv-AAC ATT TCA GGG ACA AAG GC |
| Exon-5 | Fw-AGTG GAG AAT TTG GGC TCA C | Rv- AAC TGC TCA TCC AAA CAC CAC |
| Exon-6 | Fw-GTG TCT GCT GCTCTC CTA CC | Rv-GAG GCC AAG GTC TTC TGC C |
| Exon-7 | Fw-GATTAC AGG CATGAG CCA CTGC | Rv-GAC ACA AAG CAA CCA GGG TC |
Fig. 4.

Pedigree and genotype analysis of a Saudi family with carbonic anhydrase deficiency. Squares indicate male family members, circles female family members; solid symbols indicate affected persons, circles and squares with dots indicate likely carriers, arrow indicates the index case
Fig. 5.

Sequence chromatogram showing both heterozygous mutations identified in the index case; c.232+1G0>A in intron 2 (top) and c.484delG in exon 5 of CA2 gene (down). Arrows indicated altered bases. Small letters in reference sequence indicted intron side
She was treated with NaHCO3, calcium and vitamin D supplements and underwent emergency internal fixation of the bone fracture. She later had another lithotripsy for a large renal stone. Currently, she is being regularly followed up in our clinic and has normal potassium and bicarbonate levels. She did not experience other fractures and is attending college.
Discussion
Inherited RTA disorders are relatively rare in Western populations, and they occur more commonly in areas of the world where rates of parental consanguinity are high. Carbonic anhydrase II is an enzyme encoded by CA2 located on chromosome 8q22, where its deficiency leads to an autosomal recessive disorder [3]. It was described for the first time in 1972 in three separate reports [4–6]. In 1980, Ohlsson and his colleagues described this syndrome in three Saudi families with mental retardation, stunted growth, abnormal teeth, a similar facial appearance, distal renal tubular acidosis, osteopetrosis, and cerebral calcification [7]. In 1983, Sly et al. identified carbonic anhydrase II deficiency as the primary defect of those patients [1]. CAII is known to be expressed not only by osteoclasts but in both proximal and distal nephron segments, explaining the mixed acidosis. The most common of these involves loss of the splice donor site in intron 2 of CA2, nicknamed the Arabic mutation [2].
Interestingly, although the parents of our patient were from the same tribe, they had two different mutations resulting in compound heterozygosity in the patient and her affected siblings. This is consistent with our recently described observation of marked genetic and allelic heterogeneity in the Saudi population despite perceived genetic isolation. It also supports our observation that consanguinity is more important in defining the mutational pattern within a population than the founder effect [8]. Although diagnosis of osteopetrosis is relatively easy, it can get delayed as in our case due to rarity of the disease and lack of clinical suspicion.
The corner stone of the management of the disorder is alkali therapy to correct the condition, allowing normal growth and decreased stone formation and potassium loss. This patient received NaHCO3 supplementation for almost 5 years, and her growth is normal, except for her short stature.
Close monitoring of these patients is important to avoid serious complications such as stone-related obstructive uropathy and cranial nerve entrapment, which includes the optic nerve and the acoustic nerve, causing blindness and deafness, respectively. Respiratory arrest may occur secondary to hypokalemia-related muscle paralysis. Zakzouk et al. reported hearing impairment in association with distal renal tubular acidosis among two Saudi children with osteopetrosis [9]. Al Ibarhim et al. reported a case of carbonic anhydrase II deficiency with recurrent attacks of acute paralysis, which was misdiagnosed initially as Guillain-Barré syndrome and improved with potassium and sodium bicarbonate supplementation [10]. In 2002, Awadet al. described the long-term clinical, biochemical and radiological features of 35 Saudi Arabian children with carbonic anhydrase II deficiency syndrome who have been followed at King Faisal Specialist Hospital and Research Center, Riyadh, since 1979 [11]. Hematological complications, including anemia, leukopenia and thrombocytopenia, are typically present in the recessive malignant lethal form of osteopetrosis and are usually not observed in osteopetrotic patients with carbonic anhydrase II deficiency. Bone marrow transplantation was recently reported to have a role in the radiological and histological resolution of osteopetrosis in these patients. Allogenic bone marrow stem cell replacement was successfully performed in two Irish children with this syndrome, which retarded the development of cerebral calcification; however, it had little effect on the tubular renal acidosis and mental retardation [12].
Carbonic anhydrase II deficiency syndrome is a rare disease that can be missed due to a lack of clinical suspicion. It is important that treatment be introduced at an early stage, and serious complications can be avoided with regular follow up.
Compliance with ethical standards
Conflict of interest
The authors have declared that no conflicts of interest exist.
References
- 1.Sly WS, Hewett-Emmett D, Whyte MP, Yu Y-SL, Tashian RE. Carbonic anhydrase II deficiency identified as the Primary defect in the autosomal recessive syndrome of osteoporosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA. 1983;80:2752–2756. doi: 10.1073/pnas.80.9.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu PY, Roth DE, Skaggs LA, et al. A splice junction mutation in intron 2 of the carbonic anhydrase II gene of osteopetrosis patients from Arabic countries. Hum Mutat. 1992;1(4):288–292. doi: 10.1002/humu.1380010404. [DOI] [PubMed] [Google Scholar]
- 3.Venta PJ, Shows TB, Curtis PJ, Taishan RE. Polymorphic gene for human carbonic anhydrase II: a molecular disease marker located on chromosome 8. Proc Natl Acad Sci USA. 1983;80:4437–4440. doi: 10.1073/pnas.80.14.4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sly WS, Lang R, Avioli L, Haddad J, Lubowitz H, McAlister W. Recessive osteopetrosis: new clinical phenotype. Am J Hum Genet. 1972;24(Suppl):34a. [Google Scholar]
- 5.Guibaud P, Larbre F, Freycon MT, Genoud J. Osteopetrose et acidoserenaletubulaire: deuxcas de cette association dansunefratrie. Archives Francaises de Pediatric. 1972;29:269–286. [PubMed] [Google Scholar]
- 6.Vainsel M, Fondu P, Cadranel S, Rocmans C, Gepts W. Osteopetrosis associated with proximal and distal tubular acidosis. Acta Paediatr Scand. 1972;61:429–434. doi: 10.1111/j.1651-2227.1972.tb15859.x. [DOI] [PubMed] [Google Scholar]
- 7.Ohlsson A, Strak G, Sakati N. Marble brain disease: recessive osteopetrosis, renal tubular acidosis and cerebral calcification in three Saudi Arabian families. Dev Med Child Neurol. 1980;22:72–84. doi: 10.1111/j.1469-8749.1980.tb04307.x. [DOI] [PubMed] [Google Scholar]
- 8.Aldahmesh MA, Abu-Safieh L, Khan AO, Al-Hassnan ZN, Shaheen R, Rajab M, Monies D, Meyer BF, AlKuraya FS. Allelic heterogeneity in consanguineous population: Saudi experience with alstrom syndrome as an illustrative example. Am J Med Genet A. 2009;149(A(4)):662–665. doi: 10.1002/ajmg.a.32753. [DOI] [PubMed] [Google Scholar]
- 9.Zakzouk SM, Sobki SH, Mansour F, Al-Anazi FH. Hearing impairment in association with distal renal tubular acidosis among Saudi children. J Laryngol Otol. 1995;109:930–934. doi: 10.1017/S0022215100131706. [DOI] [PubMed] [Google Scholar]
- 10.Al-Ibrahim A, Al-Harbi M, Al-Musallam S. Paralysis episodes in carbonic anhydrase II deficiency. Saudi J Kidney Dis Transpl. 2003;14:70–74. [PubMed] [Google Scholar]
- 11.Awad M, Al-Ashwal A, Sakati N, Al-Abbad A, Bin-Abbas B. Long-term follow up of carbonic anhydrase II deficiency syndrome. Saudi Med J. 2002;23(1):25–29. [PubMed] [Google Scholar]
- 12.McMahon C, Will A, Hu P, Shah GN, Sly WS, Smith OP. Bone marrow transplantation corrects the osteopetrosis in the carbonic anhydrase II deficiency syndrome. Blood. 2001;97:1947–1950. doi: 10.1182/blood.V97.7.1947. [DOI] [PubMed] [Google Scholar]