

Abstract
Gene-expression profiling and next-generation sequencing have defined diffuse large B-cell lymphoma (DLBCL), the most common lymphoma diagnosis, as a heterogeneous group of subentities. Despite ongoing explosions of data illuminating disparate pathogenic mechanisms, however, the five-drug chemoimmunotherapy combination R-CHOP remains the frontline standard treatment. This has not changed in 15 years, since the anti-CD20 monoclonal antibody rituximab was added to the CHOP backbone, which first entered use in the 1970s. At least a third of patients are not cured by R-CHOP, and relapsed or refractory DLBCL is fatal in ∼90%. Targeted small-molecule inhibitors against distinct molecular pathways activated in different subgroups of DLBCL have so far translated poorly into the clinic, justifying the ongoing reliance on R-CHOP and other long-established chemotherapy-driven combinations. New drugs and improved identification of biomarkers in real time, however, show potential to change the situation eventually, despite some recent setbacks. Here, we review established and putative molecular drivers of DLBCL identified through large-scale genomics, highlighting among other things the care that must be taken when differentiating drivers from passengers, which is influenced by the promiscuity of activation-induced cytidine deaminase. Furthermore, we discuss why, despite having so much genomic data available, it has been difficult to move toward personalized medicine for this umbrella disorder and some steps that may be taken to hasten the process.
INTRODUCTION
Although diffuse large B-cell lymphoma (DLBCL) is the most common lymphoma diagnosis, it is in reality a heterogeneous, overlapping group of subentities defined with varying degrees of precision (Xie et al. 2015). The name is a morphologic description that may cover DLBCL not otherwise specified (DLBCL-NOS), primary mediastinal large B-cell lymphoma (PMBL), intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, ALK-positive DLBCL, Epstein–Barr virus–positive DLBCL of the elderly (EBV+ DLBCL, NOS), T-cell-/histiocyte-rich large B-cell lymphoma (THRLBCL), and others (National Comprehensive Cancer Network 2016). Meanwhile, molecular subtypes of DLBCL-NOS that have been extensively characterized in laboratory studies, particularly those derived from different cells of origin (discussed in detail below), are not yet recognized as separate pathologic diagnoses, despite clear distinctions in their underlying pathogenesis. Moreover, DLBCL may be de novo or may result from transformation of indolent B-lymphomas (Campo et al. 2011; Testoni et al. 2015), a scenario resulting in inevitable relapse of the underlying indolent disease even if the aggressive transformed clone is eliminated by therapy. The CHOP chemotherapy combination (cyclophosphamide, doxorubicin, vincristine, and prednisone), which remains the backbone of frontline therapy, was introduced in the mid-1970s. The early 2000s saw addition of the anti-CD20 monoclonal antibody rituximab, which increased 5-year failure-free survival from 40%–45% to 55%–60% (Coiffier et al. 2002, 2010; Sehn et al. 2005; Habermann et al. 2006; Pfreundschuh et al. 2006; Roschewski et al. 2014; Bachy and Salles 2015). R-CHOP remains the standard of care for newly diagnosed DLBCL, though several clinical, pathologic, and molecular methods reliably identify patients with increased likelihood of failing it.
Such high-risk patients clearly need better options, but in the 15 years since rituximab's United States Food and Drug Administration (FDA) approval as part of frontline therapy for DLBCL (the last drug to win such approval) efforts to improve on R-CHOP have been largely unsuccessful. It has long been thought that intensified chemotherapy regimens might replace the CHOP backbone for many patients once evaluations in randomized clinical trials could be completed. Particular attention has focused on dose-adjusted (da) R-EPOCH, which contains the same drugs as R-CHOP, plus etoposide, and doses the etoposide, doxorubicin, and vincristine infusionally over 4 days each cycle, typically during an in-patient admission (Wilson et al. 2002). A retrospective analysis suggested improved outcomes among patients with the especially high-risk finding of double-hit lymphoma (having dual chromosomal rearrangements involving MYC and either BCL2 or BCL6) if they were treated with da-R-EPOCH rather than R-CHOP (Oki et al. 2014). A high-profile single-arm trial, meanwhile, showed overall survival near 100% for patients with PMBL treated with the regimen (Dunleavy et al. 2013), and another single-arm trial showed 81% overall survival in DLBCL-NOS after more than 5 years follow-up (Wilson et al. 2012). However, data from the ongoing CALGB/Alliance 50303 study presented at the recent 2016 American Society of Hematology (ASH) meeting in San Diego, California did little but reinforce the difficulty of improving on DLBCL's established frontline. This phase III randomized trial compares R-CHOP to da-R-EPOCH in untreated DLBCL. Event-free and overall survival were the same between the treatment arms, whereas toxicities, especially hematologic events, were significantly higher with da-R-EPOCH (Wilson et al. 2016). Additional negative data for frontline DLBCL treatment at ASH 2016 included the GOYA trial, a randomized comparison of R-CHOP versus G-CHOP, the latter regimen containing the second-generation anti-CD20 antibody obinutuzumab, engineered to increase antibody-dependent cellular cytotoxicity of targeted cells. G-CHOP showed no prolonged progression-free survival or any other advantage over R-CHOP in this trial after more than 2 years of median follow-up (Vitolo et al. 2016).
DLBCL patients who are failed by initial therapy—at least one-third of patients overall—constitute another group in urgent need of better therapeutic options. Current salvage therapies lead to long-term disease-free survival in only ∼10% of these relapsed/refractory (rr-DLBCL) patients (Friedberg 2011). Better, more rational therapies are therefore needed both for high-risk patients in the up-front setting and for those with rr-DLBCL. In addition, even those currently cured up-front or, especially, after salvage therapy and stem-cell transplant for rr-DLBCL face long-term toxicities, not the least of which are secondary malignancies that include the almost universally fatal treatment-related acute myeloid leukemia (Sud and Friedberg 2008; Bari et al. 2011; Ng et al. 2011; Raut and Chakrabarti 2014).
Great strides have been made in classifying different subgroups of DLBCL along with identification of their corresponding pathogenic drivers, largely through the discovery of genomic techniques such as next-generation sequencing (NGS) and gene expression profiling (GEP). Although driver identification through genomics can help usher in the era of personalized medicine, potentially improving frontline responses and identifying those likely to fail chemotherapy, few small-molecule targeted inhibitors have gained regulatory approval and exclusively in the rr setting. Here, we review recent advances in understanding of DLBCL through genomics and look to the future of how these data might define targets to enhance therapeutic options for patients.
THE DIVISION OF DLBCL INTO SEPARATE ENTITIES
Standard pathologic analyses have long recognized subentities within DLBCL (e.g., THRLBCL). At the turn of the century, GEP began shedding new light on the underlying biology of subtype distinctions and made further distinctions possible, most notably cell-of-origin (COO) classifications. More recently, NGS has identified many key molecular drivers, defined by recurrently mutated oncogenes and tumor suppressors, some of which are common across subtypes, but many others that further highlight key differences in biology (Orsborne and Byers 2011; Tirado et al. 2012; Intlekofer and Younes 2014; Jardin 2014). The first high-profile GEP study, by Alizadeh et al. (2000), using a microarray of genes preferentially expressed in lymphoid cells, immunology, and cancer called the “Lymphochip” (Alizadeh et al. 1999) established the COO classification of DLBCL. Subsequent studies not only corroborated this classification, but further refined it to consist of three main subgroups—GCB (germinal center B-cell-like; originating from centroblasts in the dark zone), ABC (activated B-cell-like; derived from activated B cells that are in transition to becoming plasmablasts), and PMBL (primary mediastinal B-cell-like lymphoma; from thymic B cells), with prognostic and clear biological differences (Rosenwald et al. 2002; Shipp et al. 2002; Rosenwald and Staudt 2003; Wright et al. 2003; Lossos et al. 2004). Alternate GEP classifications have since been proposed, such as the “consensus cluster” (Monti et al. 2005) and clusters based on stromal gene signatures (Lenz et al. 2008b). Although all these different classifications are reproducible, they do not overlap, highlighting not only the heterogeneous nature of DLBCL and potential contamination from nontumor cells from the microenvironment, but the constraints of interpreting data obtained on different platforms and analyzed with different algorithms.
Responses to treatment may differ among patients because patient factors such as age and comorbidities affect treatments, but the heterogeneous disease entities all included in the same diagnostic grouping is clearly a major reason. Therefore, clearer differentiation of DLBCL into separate entities has been targeted by researchers and clinicians, allowing tailored therapies to be used against specific subtypes by using therapies targeted to their particular drivers (discussed in further detail below). The ABC subtype, for example, is characterized by constitutive activation of the nuclear factor kappa B (NF-κB) pathway (Davis et al. 2001; Compagno et al. 2009; Pasqualucci and Zhang 2016). Inhibitors that interfere with malignant activation of this pathway, such as the immune modulatory agent lenalidomide and the Bruton's tyrosine kinase (BTK) inhibitor ibrutinib, have shown promising preclinical and clinical results and even synergistic ABC cell death (Yang et al. 2012; Chiappella et al. 2013; Tilly et al. 2013; Roschewski et al. 2014; Younes et al. 2014; Zheng et al. 2014; Nowakowski et al. 2015; Wilson et al. 2015), but phase 3 evaluations are still pending (NCT01122472, NCT02285062, NCT02443077, NCT01855750, and NCT01804686).
Opportunities to improve therapeutic options for DLBCL using what we have learned about its pathogenesis in genomic studies therefore depend on substantial additional work to understand how, or even whether, targeting individual molecular alterations will lead to clinically meaningful benefits.
SOMATIC, TARGETABLE ABERRATIONS AFFECTING SUBGROUPS OF DLBCL
The coding genome of DLBCL is more complex (3.3–4.21 mutations per Mb) (Lohr et al. 2012; Morin et al. 2013; Lawrence et al. 2014) than other hematological malignancies (e.g., CLL has approximately one mutation per Mb) (Puente et al. 2011; Quesada et al. 2012), but less so than some solid tumors (e.g., 12.9 mutations per Mb in melanoma) (Hodis et al. 2012; Lawrence et al. 2014). Nevertheless, genomic studies overall have emphasized the complexity of DLBCL with, on average, 30 to more than 100 genetics aberrations (such as point mutations, deletions, amplifications) per case (Pasqualucci 2013, and references therein). These studies have also helped further differentiate the genetic makeup of different DLBCL subtypes as well as the genomes of those that respond to R-CHOP versus rr-DLBCL. Figure 1 represents a snapshot of the frequency of nonsynonymous gene mutations in ∼10% of DLBCL cases pooled across several discovery-genomics studies (Pasqualucci et al. 2011b; Lohr et al. 2012; Morin et al. 2013; Dubois et al. 2016), and the roles of many of these have been extensively characterized and reviewed elsewhere (Table 1; Tirado et al. 2012; Pasqualucci 2013; Watson et al. 2013; Jardin 2014; Bohers et al. 2015; Jiang and Melnick 2015; Pasqualucci and Dalla-Favera 2015; Testoni et al. 2015; Pasqualucci and Zhang 2016). We will only briefly touch upon some examples among these and instead focus primarily on potential DLBCL disease drivers whose roles remain to be clearly defined.
Figure 1.
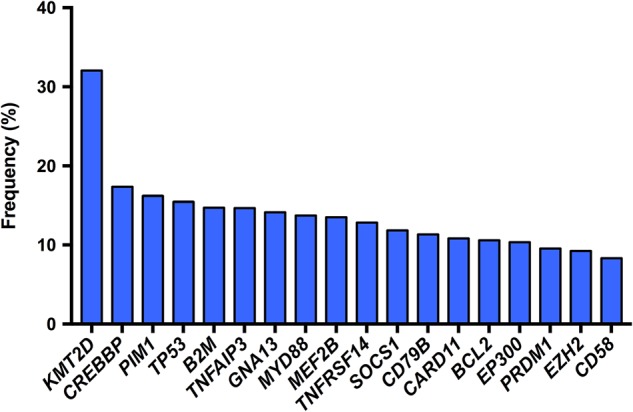
Frequency of nonsynonymous mutations in ∼10% of diffuse large B-cell lymphoma (DLBCL) cases. Data pooled from Dubois et al. (2016), Lohr et al. (2012), Morin et al. (2013), and Pasqualucci et al. (2011b) (see also Table 1).
Table 1.
Subtype distribution of the most mutated genes in diffuse large B-cell lymphoma (DLBCL) (see also Fig. 1)
| Gene | Total (%)a | ABC (%)b | GCB (%) | Functional characterization of gene mutations in DLBCL |
|---|---|---|---|---|
| KMT2D | 32.1 | 40.5 | 46.0 | Ortega-Molina et al. 2015; Zhang et al. 2015 |
| CREBBP | 17.3 | 6.0 | 31.0 | Pasqualucci et al. 2011a |
| PIM1 | 16.4 | 33.0 | 8.5 | Kumar et al. 2005; Peters et al. 2016 |
| TP53 | 15.7 | 18.0 | 15.5 | – |
| B2M | 14.7 | 8.5 | 18.0 | Challa-Malladi et al. 2011 |
| TNFAIP3 | 14.7 | 15.0 | 11.0 | Compagno et al. 2009; Honma et al. 2009 |
| GNA13 | 14.1 | 8.5 | 12.0 | Muppidi et al. 2014 |
| MYD88 | 13.7 | 28.0 | 10.0 | Ngo et al. 2011 |
| MEF2B | 13.5 | 12.0 | 23.0 | Pon et al. 2015; Ying et al. 2013 |
| TNFRSF14 | 12.8 | 2.0 | 17.0 | Boice et al. 2016 |
| SOCS1 | 11.8 | 6.0 | 15.5 | – |
| CD79B | 11.3 | 25.0 | 2.5 | Davis et al. 2010 |
| CARD11 | 10.8 | 13.5 | 7.0 | Lenz et al. 2008a |
| BCL2 | 10.6 | 1.0 | 24.0 | Schuetz et al. 2012 |
| EP300 | 10.4 | 15.0 | 14.5 | Pasqualucci et al. 2011a |
| PRDM1 | 9.6 | 16.0 | 6.0 | Calado et al. 2010; Mandelbaum et al. 2010 |
| EZH2 | 9.2 | 0.0 | 18.0 | Béguelin et al. 2013; Velichutina et al. 2010 |
| CD58 | 8.3 | 6.0 | 10.0 | Challa-Malladi et al. 2011 |
ABC, activated B-cell-like; GCB, germinal center B-cell-like.
aMutation frequencies were pooled from Pasqualucci et al. 2011b; Lohr et al. 2012; Morin et al. 2013; Dubois et al. 2016. All subentities were included.
bMutation frequencies in ABC only or GCB only from Dubois et al. 2016.
Several of these aberrations are putatively targetable, and specific inhibitors are already under evaluation. For example, somatic mutations of histone modifying proteins are a hallmark of DLBCL with increased epigenetic heterogeneity linked to poor outcome (Morin et al. 2011; Pasqualucci et al. 2011a; Jiang and Melnick 2015). The GCB subtype, in particular, is enriched in mutations affecting histone marks (Jiang and Melnick 2015). EZH2, for example, is a histone methyltransferase important for bivalency at key promoters that regulate the GC B-cell phenotype (Raaphorst et al. 2000; Velichutina et al. 2010; Chase and Cross 2011; Béguelin et al. 2013; Caganova et al. 2013). Although present in 21.7% of GCB DLBCLs, EZH2 mutations are absent in the ABC subtype (Morin et al. 2010; Béguelin et al. 2013). The mutations, which increase trimethylation at H3K27 compared with wild-type, occur in the protein's SET domain, maintaining centroblast proliferation while blocking terminal differentiation (Sneeringer et al. 2010; Yap et al. 2011; McCabe et al. 2012a; Béguelin et al. 2013). Several EZH2 inhibitors arrest proliferation and induce apoptosis of EZH2-mutant cells by blocking H3K27me3, resulting in the reactivation of silenced gene targets (Knutson et al. 2012, 2014; McCabe et al. 2012b; Qi et al. 2012; Béguelin et al. 2013). The histone acetyltransferases CREBBP and EP300 are mutated in ∼25% of DLBCLs, with preference but not exclusivity for the GCB subtype (Goodman and Smolik 2000; Morin et al. 2011; Pasqualucci et al. 2011a). Perturbed acetylation of histones and other targets, such as BCL6 and p53, through inactivating mutations of CREBBP and EP300 may drive lymphomagenesis, making histone deacetylase (HDAC) inhibition seem like a promising therapeutic strategy (Bereshchenko et al. 2002; Pasqualucci et al. 2011a; Intlekofer and Younes 2014; Havas et al. 2016). However, in one of many cautionary tales of translating seemingly strong preclinical potential to benefits for patients, HDAC inhibitors show weak single-age clinical activity in DLBCL (Marks and Xu 2009; Prince et al. 2009; Copeland et al. 2010).
The ABC subtype, as mentioned, is characterized by multiple aberrations causing constitutive activation of the NF-κB pathway. This, accordingly, provides potential opportunities for targeted therapies depending on the specific upstream mechanism leading to activation, such as BCR or toll-like receptor (TLR) signaling. Although tonic BCR signaling is required for the maintenance of B cells (Lam et al. 1997; Srinivasan et al. 2009), ABC DLBCL cells exploit chronically active BCR signaling that is sustained, in part, by mutations in CD79A/B (Davis et al. 2010). Downstream activation of BTK suggests potential activity of ibrutinib in the treatment of such cases (Davis et al. 2010; Bohers et al. 2015). The most frequent single-nucleotide variants in ABC DLBCL, however, seen in approximately one-third of cases, are gain-of-function changes in MYD88, stimulating TLR signaling that activates both NF-κB and JAK/STAT (Ngo et al. 2011; Choi et al. 2013; Fernández-Rodríguez et al. 2014; Bohers et al. 2015). The most frequent mutation, MYD88-L265P, activates signaling through the spontaneous assembly and activation of a complex with the IRAK1 and IRAK4 kinases, suggesting that IRAK-inhibitors or inhibitors against the toll-like receptors may be beneficial (Dufner and Schamel 2011; Loiarro et al. 2013; Ansell et al. 2014; Yang et al. 2014; Nowakowski et al. 2015). Furthermore, 24% of ABC DLBCLs promote NF-κB signaling by selecting for truncating mutations and/or deletions of A20 (TNFAIP3), an NF-κB repressor that is involved in curbing activity caused by BCR and/or TLR stimulation (Compagno et al. 2009; Kato et al. 2009; Wang et al. 2014). Such mutations that suppress negative NF-κB pathway regulation pose particular therapeutic challenges, however, as clinical compounds to directly inhibit NF-κB mediators do not exist (Davis et al. 2001, 2010; Lam et al. 2008; Compagno et al. 2009; Ngo et al. 2011; Young et al. 2015; Pasqualucci and Zhang 2016). In addition to the above-mentioned use of lenalidomide and ibrutinib, the activities of enzastaurin and sotrastaurin (AEB071) (protein kinase C-β [PKC-β] inhibitors) (Robertson et al. 2007; Naylor et al. 2011), fostamatinib (SYK inhibitor) (Friedberg et al. 2010; Cheng et al. 2011; Chen et al. 2013b), and IMO-8400 (TLR-8 and -9 inhibitors) (Brenner et al. 2014) are all currently under investigation.
Abnormalities in several targetable pathways have been linked to PMBL, a subtype of DLBCL with many similarities to classical Hodgkin's lymphoma (Dunleavy and Steidl 2015). Aberrant JAK/STAT signaling, for example, may result from mutations in SOCS1, PTPN1, and STAT6 as well as JAK2 amplification (Guiter et al. 2004; Melzner et al. 2005; Weniger et al. 2006; Mottok et al. 2007; Lu et al. 2008; Ritz et al. 2009, 2013; Steidl and Gascoyne 2011; Schif et al. 2013; Gunawardana et al. 2014), suggesting potential exploitation by inhibitors such as pacritinib and ruxolitinib, both of which may also have activity in ABC-DLBCL (Younes et al. 2012). Mutations in IL4R, B2M, and members of the interferon regulator factor gene family affect interferon α, β, and γ signaling (Andersson et al. 2010; Challa-Malladi et al. 2011; Raia et al. 2011; Mareschal et al. 2016). Additionally, acquired immune privilege leading to a loss of major histocompatibility (MHC) class I and II expression along with increased PDL1, and PDL2 expression may be exploited using various immune checkpoint inhibitors, including nivolumab, pembrolizumab, and pidilizumab (Rigaud et al. 2001; Rosenwald et al. 2003; Savage et al. 2003; Wessendorf et al. 2007; Armand et al. 2013; Chen et al. 2013a; Twa et al. 2014; Ansell et al. 2015; Kline and Bishop 2015).
The strategies discussed above represent only the tip of the iceberg of targeted therapies currently under evaluation as single agents, in combination with other such inhibitors,or in combination with chemotherapy (Bachy and Salles 2015; Mehta-Shah and Younes 2015). Importantly, there are also several recurrently altered genes that have yet to be well characterized, and their roles in DLBCL pathogenesis, if any, remain unclear. Disease-driving mutations that arise early in pathogenesis, so-called founder mutations, appear in most or all subsequent clones and are therefore, in theory, the best therapeutic targets (Green et al. 2013). Many of DLBCL's well-characterized driver mutations, however, including EZH2, MYD88, CARD11, and CD79B often are found only in subclonal populations (Morin et al. 2013). This complicates assessment of additional uncharacterized recurrently mutated genes also, because the progression of tumors is much more complex than mere linear acquisition of different mutations (Watson et al. 2013). Further complications arise from the actions of the enzyme activation-induced cytidine deaminase (AID, encoded by AICDA), which can make highly mutated alleles appear to be drivers when they are in fact passengers mutated by aberrant somatic hypermutation (SHM, see next section) (Liu and Schatz 2009; Gu et al. 2012; Jiang et al. 2012; Khodabakhshi et al. 2012; Gu et al. 2016).
DRIVERS VERSUS PASSENGERS: THE AID EFFECT
Somatic hypermutation and class switch recombination are essential processes in the germinal center (GC) that allow for the generation of highly specific antibodies and B cells with distinct effector functions (Di Noia and Neuberger 2007). However, because they induce breaks in the genome of GC B cells, where DNA damage checkpoints are already down-regulated, errors are believed to contribute to lymphomagenesis. AID targets immunoglobulin (Ig) locus genes for SHM, where it deaminates cytosines, resulting in a C:G to U:G mismatch that is subsequently repaired through either the base excision or DNA mismatch repair pathways, with each favoring different substitutions (Di Noia and Neuberger 2007). The targeting of cytosines by AID leads to a high ratio of transition to transversion, and the majority of C mutations fall within WRCY motifs (in which W is an A or T; R is an A or G; and Y is a C or T), whereas the A mutation is more likely when A is in the coding strand, and T mutations are found less frequently (Milstein et al. 1998; Rogozin and Diaz 2004). AID-induced SHM is dependent on transcription and targeted to the 5′ region of the gene, with the majority of mutations lying within the first 1–2 kb downstream from the transcription start site (Both et al. 1990; Meng et al. 2014). Initially the BCL6 and CD95 genes were identified as non-Ig targets of SHM, but with numerous genome- and exome-sequencing studies in the past decade it is clear that more than 40 genes exhibit these hallmarks of AID mutator activity (enrichment in mutations targeted to WRCY motifs, high transition-to-transversion and C:G to A:T ratios) (Pasqualucci et al. 1998, 2011b; Müschen et al. 2000; Khodabakhshi et al. 2012; Lohr et al. 2012; Zhang et al. 2013; Peters et al. 2016). The most well-defined SHM off-target genes based on AID hallmarks include BCL2, BCL6, MYC, RHOH/TTF, PIM1 (see below), PAX5, IRF4, ST6GAL1, BCL7A, CIITA, LRMP, and SOCS1. Recent analyses have identified that some of these frequent AID off-target genes, such as PIM1, MYC, CD79B, and PAX5, are associated with sites of convergent transcription and the presence of super enhancer elements, which supports the finding that transcription is required for targeting by AID (Maul and Gearhart 2010; Meng et al. 2014; Qian et al. 2014). Despite considerable data published in recent years on the targets of aberrant SHM in DLBCL, the significance of most such mutations remains unclear. Specifically, questions linger about whether these mutations are selected for pro-oncogenic functions or simply a secondary result of GC biology. Overexpression of the oncogene MYC by the Ig λ promoter results in pre-GC lymphomas that lack mutations in the Ig variable region, whereas in IμHABCL6 mice, BCL6 mRNA expression is deregulated, driving GC-derived lymphomas. A lack of AID, however, blocks lymphoma development only in the BCL6-dependent GC IμHABCL6 model and has no effect on MYC-driven, pre-GC lymphomas (Pasqualucci et al. 2008). It is possible AID has a role other than the specific mutations it generates, but this remains speculative for now.
PIM1
The PIM1 proto-oncogene serine/threonine kinase was originally identified as a site of frequent proviral insertion by the Moloney leukemia virus that synergizes potently with MYC in lymphomagenesis (Cuypers et al. 1984). Subsequently, two highly homologous additional family members, PIM2 and PIM3, were identified through their abilities to replace PIM1 in oncogenesis models (Breuer et al. 1989; Feldman et al. 1998). All three family members are constitutively active kinases expressed downstream from growth factor and cytokine signaling pathways and control pro-growth and pro-survival signaling outputs. Elevated PIM expression is seen in multiple malignancies, particularly those derived from hematopoietic cells. Surprisingly, the only phenotype of PIM1/2/3 triple-knockout mice is diminished body size and an impaired response of hematopoietic cells to cytokine (Mikkers et al. 2004). PIM1 is among the list of most frequently targeted genes by aberrant SHM found in numerous studies in DLBCL, but the mutational pattern is curious for a constitutively active kinase, given the much higher probability that random point mutations would lead to a nonfunctional protein (Pasqualucci et al. 2011b; Zhang et al. 2013; Meng et al. 2014; Mareschal et al. 2016). We recently asked the question whether these mutations impacted the function of the kinase. We performed a meta-analysis of whole-genome and -exome sequencing and identified 92 substitutions resulting in missense mutations, of which 53 were recurrent (Fig. 2A; Peters et al. 2016). We tested these mutations for their ability to enrich in cytokine-deprived FL 5.12 cells, a murine pro-B cell line for which PIM expression is the major survival target of cytokine stimulation. The vast majority of mutations behaved identically to wild-type PIM1 (and none conferred resistance to the pan-PIM kinase inhibitor PIM447) (Peters et al. 2016). These results suggest that the high frequency of SHM at the PIM1 locus is not a disease-driving process, at least not through the mutations themselves. If anything, it could even be a bottleneck because for the most part only those mutations that preserve kinase function are found in surviving clones.
Figure 2.

Mapping the known mutations for PIM1 and BTG1. (A) PIM1 mutational pattern adapted from Peters et al. 2016. (B) BTG1 mutations mapped using the Catalogue of Somatic Mutations in Cancer (COSMIC; http://cancer.sanger.ac.uk/cosmic) database and Lohr et al. 2012.
BTG1
BTG1 is a member of the BTG/Tob family that regulates cell cycle progression and apoptosis, gene transcription in the nucleus, cytoplasmic mRNA deadenylation and turnover, and protein–protein interactions (Winkler 2010). Several studies have reported mutations in BTG1, with several patients even exhibiting more than one mutation (Morin et al. 2011; Lohr et al. 2012; Zhang et al. 2013). Mutations are predicted to generally favor loss of function of the protein (Morin et al. 2011) and occur in both coding and noncoding regions, suggesting altered codon use or defects in BTG1 mRNA stability (Lohr et al. 2012). A predictive “classification tree model,” however, identified BTG1 as one of 101 high-interest genes that are likely to be targets of AID (Duke et al. 2013). Indeed, modeling BTG1’s mutational pattern using the Catalogue of Somatic Mutations in Cancer (COSMIC; http://cancer.sanger.ac.uk/cosmic) database strongly suggests that this recurrently mutated gene is not a driver of DLBCL, but is instead a consequence of AID-mediated mutations because of the clustering of mutations at the amino terminus (Fig. 2B; Pasqualucci et al. 2001; Qian et al. 2014).
The above are just two examples, but they highlight the care that must be taken when determining the driver status of the large number of frequently mutated DLBCL genes unearthed through large-scale genomics, with considerable efforts required to confirm whether the contribution of AID renders them merely passengers.
BCL10
An important bottleneck for NF-κB activation required for ABC-DLBCL is the CBM complex, consisting of the proteins CARMA1 (CARD11), BCL10, and MALT1 (Thome 2004; Staudt 2010; Yang et al. 2014). BCL10 and MALT1 amplifications, mutations, and translocations are well-established in MALT lymphoma (Willis et al. 1999; Zhang et al. 1999; Isaacson and Du 2004; Sagaert et al. 2006; Hailfinger et al. 2014). Although CARMA1 mutations are well-described in ABC-DLBCL oncogenesis (Lenz et al. 2008a) and MALT1’s protease activity also plays a role (Ferch et al. 2009; Hailfinger et al. 2009; Baens et al. 2014), genomic data suggest BCL10 mutations may also drive DLBCL pathogenesis. Morin et al. (2011, 2016) reported evidence for DLBCL's selection of inactivating mutations in BCL10. Why would DLBCL cells select for mutations that inactivate a protein that is an integral member of a complex required for oncogenic NF-κB signaling? Perhaps the answer to this conundrum lies in the supposed dual functions of BCL10 in activating NF-κB signaling while also inducing apoptosis in some cells, each function carried out by separate unique portions of the protein (Willis et al. 1999; Zhang et al. 1999). Indeed, although the IκB kinase-β is required for CBM complex formation through phosphorylation of the amino terminus of BCL10, it subsequently phosphorylates downstream residues of BCL10 to attenuate this signaling (Wegener et al. 2006; Zeng et al. 2007). Our laboratory is currently investigating whether putative separation-of-function mutations in BCL10 are true drivers of the disease.
FAS
As recently reviewed by Testoni et al. (2015), mutations in FAS may be important contributors to DLBCL pathogenesis. FAS induces apoptosis as part of the death-inducing signaling complex (DISC). DLBCL cells, accordingly, modulate FAS's pro-apoptotic activity via loss-of-function mutations, deletions, and copy loss, protecting the cancer cells from immune surveillance and apoptosis following chemotherapy (Gronbaek et al. 1998; Kojima et al. 2006; Morin et al. 2011, 2016; Mian et al. 2012; Monti et al. 2012; Testoni et al. 2015). The driver status of FAS, however, remains to be established with respect to DLBCL.
SGK1
Like BCL10 and FAS, the PI3K-regulated serine/threonine protein kinase SGK1 (serum and glucocorticoid-regulated kinase 1) shows a propensity for inactivating mutations particularly in GCB DLBCL (Morin et al. 2011) and is in a region of Chromosome 6q that is recurrently deleted in DLBCL, FL, and other B-cell lymphomas (Lenz et al. 2008c; Oricchio et al. 2011; Monti et al. 2012; Morin et al. 2013). SGK1 has several cellular functions, including ion channel activity regulation in renal cells (Lang and Shumilina 2013), FOXO3 phosphorylation to mediate cell survival signals (Brunet et al. 2001), IκB-α and p300 phosphorylation up-regulating NF-κB signaling (Tai et al. 2009), and FBW7 phosphorylation to induce degradation and ubiquitylation of the NOTCH1-intracellular domain, thereby inhibiting the NOTCH1 signaling pathway (Mo et al. 2011). Recently, a t(6;14)(q22;q32) translocation juxtaposing the IGHG3 switch region to an intron of SGK1 was predicted to inactivate SGK1 (Ryan et al. 2015). Furthermore, mutation in DLBCL results in a loss of promoter occupancy of several genes, including SGK1 (Ortega-Molina et al. 2015), and SGK1 confers resistance to PI3K inhibition in breast cancer (Castel et al. 2016). As with BCL10, DLBCL cells seem to paradoxically lose SGK1, despite its role in pro-proliferation pathway activation, with an algorithm suggesting mutations are indeed drivers of DLBCL (Lohr et al. 2012). Our laboratory is currently working to understand how DLBCL's growth might be facilitated through SGK1 loss of function.
TBL1XR1
The E3 ubiquitin ligase activity of transducin β-like 1 X-linked receptor 1 (TBL1XR1) polyubiquitinates the nuclear receptor corepressor 2/silencing mediator for retinoid or thyroid-hormone receptors (NCoR/SMRT) complex, resulting in transcriptional repression through NF-κB and WNT-mediated signaling pathways (Yoon et al. 2003; Zhang et al. 2006; Perissi et al. 2008; Rossi et al. 2012). This putative tumor-suppressor gene has recurrent mutations activating NF-κB in acute lymphoblastic leukemia (ALL), splenic marginal zone lymphoma (SMZL), and primary central nervous system lymphoma (PCNSL), with the latter also exhibiting a 3q26.32 deletion that encompasses the TBL1XR1 locus (Parker et al. 2008; Braggio et al. 2011; Zhang et al. 2011; Gonzalez-Aguilar et al. 2012). Further evidence was presented at ASH 2016 to support the tumor-suppressive role of TBL1XR1 in which in silico modeling and functional characterization of TBL1XR1 mutations indicate their inactivating nature through heightened proliferation and putative perturbed interactions with CD79B/MYD88 in DLBCL as well as PCNSL (Chapuy et al. 2016). It is perhaps unsurprising that TBL1XR1 mutations have been observed in two out of six ABC DLBCL samples (Mareschal et al. 2016), because studies have shown a high degree of correlation between ABC and PCNSL mutational patterns (Bruno et al. 2014). Furthermore, focal deletions and single-nucleotide variants (SNVs) of TBL1XR1 have been previously reported in DLBCL (Pasqualucci et al. 2011b; Lohr et al. 2012; Scott et al. 2012; Morin et al. 2013). Gene fusions are also evident in some GCB DLBCL (6/115 GCB; 0/138 non-GCB) and FL cases (1/81) (Scott et al. 2012). A report that links deletions in TBL1XR1 to glucocorticoid resistance may suggest a role for TBL1XR1 inactivating mutations in facilitating R-CHOP resistance (Jones et al. 2014; Mareschal et al. 2016). Further support for this comes from one study that saw TBL1XR1 mutations only at relapse but not at diagnosis (Morin et al. 2016). This warrants further investigation as combining R-CHOP therapy with agents modifying the E3 ubiquitin ligase activity of TBL1XR1 might prevent the onset of resistance in some cases of DLBCL. The amplification of TBL1XR1 is seen in breast cancer, in which its inactivation is associated with decreased tumor cell invasion (Kadota et al. 2009), suggesting that the oncogenic properties of TBL1XR1 differ between solid tumors and hematological malignancies.
New genomic data presented at ASH 2016 emphasize the power of having large numbers of cases to better contextualize putative drivers. Zhang et al. (2016) carried out what could possibly be the largest whole-exome sequencing study of any individual cancer type, by analyzing the genomes of 1001 de novo DLBCL patients. Forty-two novel drivers were identified, including the aforementioned BTK. This elaborate study further separated genes into functionally related subnetworks and found that the majority of genes within each subnetwork were mutated in mutually exclusive patterns. Furthermore, several new genes emerged as positive or negative prognostic biomarkers. EZH2 and CD70 mutations, for example, associated with favorable prognosis in GCB-DLBCL, whereas mutations in KLHL14 associated with poor prognosis in ABC cases. Interestingly, TP53 mutations were found only to be prognostic when associated with both MLL2 mutation and high BCL2 expression (Zhang et al. 2016). A number of potentially clinically useful findings emerge from this study, aiding therapeutic selections in precision medicine contexts and generating hypotheses for testing in both clinical trials and laboratory studies.
THE IMPORTANCE OF FUNCTIONAL STUDIES TO UNDERSTAND THE ROLES OF RECURRENTLY ALTERED GENES
Laboratory functional studies provide the best initial route to elucidate the roles of recurrently altered genes in lymphomagenesis, exemplified by the recent examples of the histone methyltransferase KMT2D and the TNFRSF14 receptor gene HVEM (herpes virus entry mediator) (Ortega-Molina et al. 2015; Zhang et al. 2015; Boice et al. 2016). KMT2D exerts a widespread effect by controlling the expression of a set of genes that includes the above-mentioned SGK1 and FAS, with KMT2D deficiency resulting in altered promoter occupancy. Mutations in KMT2D, however, were not associated with outcome to R-CHOP therapy (Ortega-Molina et al. 2015). The results obtained for HVEM's role in FL are particularly enticing. The interactions between HVEM and BTLA (B- and T-lymphocyte attenuator) normally oppose lymphoma development (Costello et al. 2003; Cai and Freeman 2009; Steinberg et al. 2011; Pasero et al. 2012; Bjordahl et al. 2013). In a large number of FL cases, however, this interaction is lost, as HVEM or BTLA are mutated or lost in a mutually exclusive manner. By genetically engineering CD19-targeted chimeric antigen receptor (CAR) T cells to locally and continuously produce the ectodomain portion of the HVEM protein, termed “micro-pharmacies,” the HVEM-BTLA inhibitory axis is restored leading to a significant therapeutic response (Boice et al. 2016). Although this therapy is limited to BTLA-expressing FL cells that have defective HVEM, the use of genetically engineered CAR T cells as micro-pharmacies to secrete tumor-suppressive proteins that are mutated in patients, even restoring protein–protein interactions that have been lost by these mutations, is especially alluring, as CAR T cells, in principle, can attack any tumor antigen (Brentjens et al. 2003; Batlevi et al. 2016). Similar functional analyses can, in principle, uncover the roles for other putative drivers of DLBCL.
THE FEASIBILITY OF TUMOR GENOMICS IN REAL TIME
Fast and accurate identification of DLBCL subtypes and biomarkers can aid in risk stratification and could eventually drive choices of alternate therapies for patients predicted to fail standard regimens like R-CHOP. The gene-expression studies that identified and validated ABC versus GCB versus PMBL COO subtypes required fresh-frozen tumor samples from which high-quality RNA could be extracted, techniques that are not practical in routine clinical practice. Efforts to simplify COO identification have been the subjects of intensive efforts ever since. For example, although several thousand cDNA clones were used in the Lymphochip microarray that initially established the COO classification of DLBCL (Alizadeh et al. 1999, 2000), subsequently, 27-gene (Wright et al. 2003), 17-gene (Rosenwald et al. 2002), six-gene (Lossos et al. 2004), and even two-gene (Rimsza et al. 2008) models have been used for predicting overall survival (OS) after chemotherapy. It is necessary that COO be reliably identifiable from formalin-fixed paraffin-embedded (FFPE) tumor samples, and encouraging results have been obtained with the Lymphoma/Leukemia Molecular Profiling Project's digital gene expression (NanoString)-based Lymph2Cx assay (Scott et al. 2014). Here, a 20-gene assay accurately categorized COO subtypes from FFPE tissue samples with a low misassignment score that is on par with currently used immunohistochemistry (IHC)-based algorithms (Hans et al. 2004; Choi et al. 2009; Meyer et al. 2011) and a turnaround time of <36 h. The use of synthetic oligonucleotides that are run alongside patient samples for normalization was designed to increase assay stringency by accounting for potential lot-to-lot probe variation and suggests that routine clinical implementation of such techniques as predictors of OS is within grasp (Scott et al. 2014). Furthermore, OS prediction following CHOP and R-CHOP from paraffin samples has been achieved using real-time PCR on a handful of genes (Malumbres et al. 2008; Alizadeh et al. 2011) providing further options for fast and reliable patient management.
Although identifying patients who are likely to respond to chemotherapy via GEP is helpful, pinpointing targetable pathways and aberrations to treat those who are unresponsive (or resistant) to therapy is a higher priority as the field stands currently. Furthermore, the high degree of stromal contamination found in clinical lymphoma specimens, for example, would require techniques such as deep sequencing to identify relevant alterations in tumors, some of which may be subclonal (Lohr et al. 2012). The use of NGS routinely in the clinic is appealing because of the sheer amount of information one can garner for each patient to direct the most targeted and relevant therapies, with companies, such as Foundation Medicine, making real-time targeted exome sequencing a reality (Wagle et al. 2012; Frampton et al. 2013; Intlekofer and Younes 2014). Despite this, however, the modes of patient treatment have not substantially changed. Researchers and clinicians have capabilities now to generate a plethora of data about any given patient's tumor, but we do not yet understand enough about various alterations, their mechanisms of action, and whether they are indeed relevant enough to change current treatment practices. Once the impact of these genetic changes is better understood, NGS may become useful to tailor therapies that will exploit them.
CONCLUSION
The international prognostic index (IPI), established in 1993 (The International Non-Hodgkin's Lymphoma Prognostic Factors Project 1993; Shipp 1994; Sehn et al. 2007; Zhou et al. 2014), predicts patients likely to fail standard therapy. It gives no insight, however, into tumor biology, nor does it suggest more effective treatments. Furthermore, although current guidelines recognize the importance of the COO classification and the presence of MYC rearrangements (Ghielmini et al. 2013; Zelenetz 2014), R-CHOP or R-EPOCH remain the recommended course of therapy (Testoni et al. 2015). Concerted efforts are underway to identify relevant disease drivers that are therapeutically targetable using the wealth of genomic data that has been amassed through technological advancements and evolving bioinformatic power. Additionally, uncovering potential causes of resistance to chemotherapy is of the utmost importance. Immunohistochemistry is one method that is currently used, with ALDH1A1, for example, identified as causing resistance to CHOP via the JAK/STAT pathway (Jiang et al. 2016). Genomic data can also greatly aid in this process, and, as discussed above, mutations in TBL1XR1 have been identified as a potential cause of R-CHOP resistance—a hypothesis that requires further confirmation. To simplify efforts, we must unify these fragmented global data sets, obtained by different sequencing platforms, using different algorithms to call mutations, stored in different data formats, while preserving ethical practices when using any such data (Grossman et al. 2016; Siu et al. 2016). Various global efforts are ongoing to achieve this, such as President Obama's “Moonshot” program to cure cancer (McCarthy 2016), The Cancer Genome Atlas (TCGA) (The Cancer Genome Atlas Research Network et al. 2013), the Actionable Cancer Genome Initiative (ACGI), which aims to identify “actionable” genes in several cancers (Lawler et al. 2015), the NCI Genomic Data Commons (GDC) established through the collective efforts of the National Cancer Institute, the University of Chicago, the Ontario Institute for Cancer Research, and Leidos Biomedical Research (Grossman et al. 2016), and others (for review, see Siu et al. 2016). Wading through all the genomic data in AML and FL have helped illuminate the complexities of these diseases. The same goal will be attainable in DLBCL through data “harmonization,” providing hope that personalized medicine can soon be implemented to treat each individual.
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Alizadeh A, Eisen M, Davis RE, Ma C, Sabet H, Tran T, Powell JI, Yang L, Marti GE, Moore DT, et al. 1999. The lymphochip: a specialized cDNA microarray for the genomic-scale analysis of gene expression in normal and malignant lymphocytes. Cold Spring Harb Symp Quant Biol 64: 71–78. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. 2000. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403: 503–511. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Gentles AJ, Alencar AJ, Liu CL, Kohrt HE, Houot R, Goldstein MJ, Zhao S, Natkunam Y, Advani RH, et al. 2011. Prediction of survival in diffuse large B-cell lymphoma based on the expression of 2 genes reflecting tumor and microenvironment. Blood 118: 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson E, Schain F, Sjöberg J, Björkholm M, Claesson HE. 2010. Interleukin-13 stimulation of the mediastinal B-cell lymphoma cell line Karpas-1106P induces a phenotype resembling the Hodgkin lymphoma cell line L1236. Exp Hematol 38: 116–123. [DOI] [PubMed] [Google Scholar]
- Ansell SM, Hodge LS, Secreto FJ, Manske M, Braggio E, Price-Troska T, Ziesmer S, Li Y, Johnson SH, Hart SN, et al. 2014. Activation of TAK1 by MYD88 L265P drives malignant B-cell growth in non-Hodgkin lymphoma. Blood Cancer J 4: e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, et al. 2015. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 372: 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Holland HK, Winter JN, Mason JR, et al. 2013. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol 31: 4199–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachy E, Salles G. 2015. Treatment approach to newly diagnosed diffuse large B-cell lymphoma. Semin Hematol 52: 107–118. [DOI] [PubMed] [Google Scholar]
- Baens M, Bonsignore L, Somers R, Vanderheydt C, Weeks SD, Gunnarsson J, Nilsson E, Roth RG, Thome M, Marynen P. 2014. MALT1 auto-proteolysis is essential for NF-κB-dependent gene transcription in activated lymphocytes. PLoS One 9: e103774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bari A, Marcheselli L, Marcheselli R, Liardo EV, Pozzi S, Ferri P, Sacchi S. 2011. Therapy-related myeloid neoplasm in non-Hodgkin lymphoma survivors. Mediterr J Hematol Infect Dis 3: e2011065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlevi CL, Matsuki E, Brentjens RJ, Younes A. 2016. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol 13: 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. 2013. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23: 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereshchenko OR, Gu W, Dalla-Favera R. 2002. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet 32: 606–613. [DOI] [PubMed] [Google Scholar]
- Bjordahl RL, Steidl C, Gascoyne RD, Ware CF. 2013. Lymphotoxin network pathways shape the tumor microenvironment. Curr Opin Immunol 25: 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohers E, Mareschal S, Bertrand P, Viailly PJ, Dubois S, Maingonnat C, Ruminy P, Tilly H, Jardin F. 2015. Activating somatic mutations in diffuse large B-cell lymphomas: lessons from next generation sequencing and key elements in the precision medicine era. Leuk Lymphoma 56: 1213–1222. [DOI] [PubMed] [Google Scholar]
- Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, Jiang M, Mottok A, Denis-Lagache N, Ciriello G, et al. 2016. Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell 167: 405–418 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Both GW, Taylor L, Pollard JW, Steele EJ. 1990. Distribution of mutations around rearranged heavy-chain antibody variable-region genes. Mol Cell Biol 10: 5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braggio E, McPhail ER, Macon W, Lopes MB, Schiff D, Law M, Fink S, Sprau D, Giannini C, Dogan A, et al. 2011. Primary central nervous system lymphomas: a validation study of array-based comparative genomic hybridization in formalin-fixed paraffin-embedded tumor specimens. Clin Cancer Res 17: 4245–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner L, Arbeit RD, Sullivan T. 2014. IMO-8400, an antagonist of Toll-like receptors 7, 8, and 9, in development for genetically defined B-cell lymphomas: safety and activity in Phase 1 and Phase 2 clinical trials. Blood 124: 3101.25170122 [Google Scholar]
- Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Riviere I, et al. 2003. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med 9: 279–286. [DOI] [PubMed] [Google Scholar]
- Breuer ML, Cuypers HT, Berns A. 1989. Evidence for the involvement of pim-2, a new common proviral insertion site, in progression of lymphomas. EMBO J 8: 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. 2001. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21: 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno A, Boisselier B, Labreche K, Marie Y, Polivka M, Jouvet A, Adam C, Figarella-Branger D, Miquel C, Eimer S, et al. 2014. Mutational analysis of primary central nervous system lymphoma. Oncotarget 5: 5065–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, George L, Alberghini F, Ferrarini L, Talukder AK, et al. 2013. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest 123: 5009–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G, Freeman GJ. 2009. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev 229: 244–258. [DOI] [PubMed] [Google Scholar]
- Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, Rodig S, Kutok J, Tarakhovsky A, Schmidt-Supprian M, et al. 2010. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 18: 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. 2011. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 117: 5019–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et al. 2016. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell 30: 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, Dalla-Favera R. 2011. Combined genetic inactivation of β2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 20: 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Kamburov A, Coughlin CA, Stewart C, Dunford A, Aono M, Sauer C, Su A, Roemer MGM, Rodig SJ, et al. 2016. In silico and functional characterization of TBL1XR1 as a tumor suppressor in large B-cell lymphomas. In American Society of Hematology: 58th Annual Meeting & Exposition, San Diego, CA. [Google Scholar]
- Chase A, Cross NC. 2011. Aberrations of EZH2 in cancer. Clin Cancer Res 17: 2613–2618. [DOI] [PubMed] [Google Scholar]
- Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, Yu H, Fletcher CD, Freeman GJ, Shipp MA, et al. 2013a. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res 19: 3462–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Monti S, Juszczynski P, Ouyang J, Chapuy B, Neuberg D, Doench JG, Bogusz AM, Habermann TM, Dogan A, et al. 2013b. SYK inhibition modulates distinct PI3K/AKT- dependent survival pathways and cholesterol biosynthesis in diffuse large B cell lymphomas. Cancer Cell 23: 826–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Coffey G, Zhang XH, Shaknovich R, Song Z, Lu P, Pandey A, Melnick AM, Sinha U, Wang YL. 2011. SYK inhibition and response prediction in diffuse large B-cell lymphoma. Blood 118: 6342–6352. [DOI] [PubMed] [Google Scholar]
- Chiappella A, Tucci A, Castellino A, Pavone V, Baldi I, Carella AM, Orsucci L, Zanni M, Salvi F, Liberati AM, et al. 2013. Lenalidomide plus cyclophosphamide, doxorubicin, vincristine, prednisone and rituximab is safe and effective in untreated, elderly patients with diffuse large B-cell lymphoma: a phase I study by the Fondazione Italiana Linfomi. Haematologica 98: 1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WW, Weisenburger DD, Greiner TC, Piris MA, Banham AH, Delabie J, Braziel RM, Geng H, Iqbal J, Lenz G, et al. 2009. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res 15: 5494–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Kim Y, Lee JH, Kim YS. 2013. MYD88 expression and L265P mutation in diffuse large B-cell lymphoma. Hum Pathol 44: 1375–1381. [DOI] [PubMed] [Google Scholar]
- Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, Morel P, Van Den Neste E, Salles G, Gaulard P, et al. 2002. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346: 235–242. [DOI] [PubMed] [Google Scholar]
- Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M, Sebban C, et al. 2010. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood 116: 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, et al. 2009. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 459: 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland A, Buglio D, Younes A. 2010. Histone deacetylase inhibitors in lymphoma. Curr Opin Oncol 22: 431–436. [DOI] [PubMed] [Google Scholar]
- Costello RT, Mallet F, Barbarat B, Schiano De Colella JM, Sainty D, Sweet RW, Truneh A, Olive D. 2003. Stimulation of non-Hodgkin's lymphoma via HVEM: an alternate and safe way to increase Fas-induced apoptosis and improve tumor immunogenicity. Leukemia 17: 2500–2507. [DOI] [PubMed] [Google Scholar]
- Cuypers HT, Selten G, Quint W, Zijlstra M, Maandag ER, Boelens W, van Wezenbeek P, Melief C, Berns A. 1984. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell 37: 141–150. [DOI] [PubMed] [Google Scholar]
- Davis RE, Brown KD, Siebenlist U, Staudt LM. 2001. Constitutive nuclear factor κB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 194: 1861–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. 2010. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463: 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, Neuberger MS. 2007. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 76: 1–22. [DOI] [PubMed] [Google Scholar]
- Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, Maingonnat C, Jais JP, Peyrouze P, Figeac M, et al. 2016. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res 22: 2919–2928. [DOI] [PubMed] [Google Scholar]
- Dufner A, Schamel WW. 2011. B cell antigen receptor-induced activation of an IRAK4-dependent signaling pathway revealed by a MALT1-IRAK4 double knockout mouse model. Cell Commun Signal 9: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke JL, Liu M, Yaari G, Khalil AM, Tomayko MM, Shlomchik MJ, Schatz DG, Kleinstein SH. 2013. Multiple transcription factor binding sites predict AID targeting in non-Ig genes. J Immunol 190: 3878–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy K, Steidl C. 2015. Emerging biological insights and novel treatment strategies in primary mediastinal large B-cell lymphoma. Semin Hematol 52: 119–125. [DOI] [PubMed] [Google Scholar]
- Dunleavy K, Pittaluga S, Maeda LS, Advani R, Chen CC, Hessler J, Steinberg SM, Grant C, Wright G, Varma G, et al. 2013. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 368: 1408–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JD, Vician L, Crispino M, Tocco G, Marcheselli VL, Bazan NG, Baudry M, Herschman HR. 1998. KID-1, a protein kinase induced by depolarization in brain. J Biol Chem 273: 16535–16543. [DOI] [PubMed] [Google Scholar]
- Ferch U, Kloo B, Gewies A, Pfander V, Duwel M, Peschel C, Krappmann D, Ruland J. 2009. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 206: 2313–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Rodríguez C, Bellosillo B, Garcia-Garcia M, Sanchez-Gonzalez B, Gimeno E, Vela MC, Serrano S, Besses C, Salar A. 2014. MYD88 (L265P) mutation is an independent prognostic factor for outcome in patients with diffuse large B-cell lymphoma. Leukemia 28: 2104–2106. [DOI] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. 2013. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31: 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg JW. 2011. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program 2011: 498–505. [DOI] [PubMed] [Google Scholar]
- Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, Schaefer-Cutillo J, De Vos S, Sinha R, Leonard JP, et al. 2010. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 115: 2578–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghielmini M, Vitolo U, Kimby E, Montoto S, Walewski J, Pfreundschuh M, Federico M, Hoskin P, McNamara C, Caligaris-Cappio F, et al. 2013. ESMO Guidelines consensus conference on malignant lymphoma 2011 part 1: diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL) and chronic lymphocytic leukemia (CLL). Ann Oncol 24: 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Aguilar A, Idbaih A, Boisselier B, Habbita N, Rossetto M, Laurenge A, Bruno A, Jouvet A, Polivka M, Adam C, et al. 2012. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res 18: 5203–5211. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev 14: 1553–1577. [PubMed] [Google Scholar]
- Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, Kela I, Hopmans ES, Myklebust JH, Ji H, et al. 2013. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 121: 1604–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronbaek K, Straten PT, Ralfkiaer E, Ahrenkiel V, Andersen MK, Hansen NE, Zeuthen J, Hou-Jensen K, Guldberg P. 1998. Somatic Fas mutations in non-Hodgkin's lymphoma: association with extranodal disease and autoimmunity. Blood 92: 3018–3024. [PubMed] [Google Scholar]
- Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM. 2016. Toward a shared vision for cancer genomic data. N Engl J Med 375: 1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Shivarov V, Strout MP. 2012. The role of activation-induced cytidine deaminase in lymphomagenesis. Curr Opin Hematol 19: 292–298. [DOI] [PubMed] [Google Scholar]
- Gu X, Booth CJ, Liu Z, Strout MP. 2016. AID-associated DNA repair pathways regulate malignant transformation in a murine model of BCL6-driven diffuse large B-cell lymphoma. Blood 127: 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiter C, Dusanter-Fourt I, Copie-Bergman C, Boulland ML, Le Gouvello S, Gaulard P, Leroy K, Castellano F. 2004. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood 104: 543–549. [DOI] [PubMed] [Google Scholar]
- Gunawardana J, Chan FC, Telenius A, Woolcock B, Kridel R, Tan KL, Ben-Neriah S, Mottok A, Lim RS, Boyle M, et al. 2014. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet 46: 329–335. [DOI] [PubMed] [Google Scholar]
- Habermann TM, Weller EA, Morrison VA, Gascoyne RD, Cassileth PA, Cohn JB, Dakhil SR, Woda B, Fisher RI, Peterson BA, et al. 2006. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol 24: 3121–3127. [DOI] [PubMed] [Google Scholar]
- Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, Penas EM, Dierlamm J, Chan WC, Staudt LM, et al. 2009. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci 106: 19946–19951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailfinger S, Lenz G, Thome M. 2014. Targeting B-cell lymphomas with inhibitors of the MALT1 paracaspase. Curr Opin Chem Biol 23: 47–55. [DOI] [PubMed] [Google Scholar]
- Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Muller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, et al. 2004. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103: 275–282. [DOI] [PubMed] [Google Scholar]
- Havas AP, Rodrigues KB, Bhakta A, Demirjian JA, Hahn S, Tran J, Scavello M, Tula-Sanchez AA, Zeng Y, Schmelz M, et al. 2016. Belinostat and vincristine demonstrate mutually synergistic cytotoxicity associated with mitotic arrest and inhibition of polyploidy in a preclinical model of aggressive diffuse large B cell lymphoma. Cancer Biol Ther 17: 1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. 2012. A landscape of driver mutations in melanoma. Cell 150: 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, Seto M. 2009. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood 114: 2467–2475. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Younes A. 2014. Precision therapy for lymphoma—current state and future directions. Nat Rev Clin Oncol 11: 585–596. [DOI] [PubMed] [Google Scholar]
- Isaacson PG, Du MQ. 2004. MALT lymphoma: from morphology to molecules. Nat Rev Cancer 4: 644–653. [DOI] [PubMed] [Google Scholar]
- Jardin F. 2014. Next generation sequencing and the management of diffuse large B-cell lymphoma: from whole exome analysis to targeted therapy. Discov Med 18: 51–65. [PubMed] [Google Scholar]
- Jiang Y, Melnick A. 2015. The epigenetic basis of diffuse large B-cell lymphoma. Semin Hematol 52: 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Soong TD, Wang L, Melnick AM, Elemento O. 2012. Genome-wide detection of genes targeted by non-Ig somatic hypermutation in lymphoma. PLoS One 7: e40332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Liu Y, Tang Y, Li L, Zeng R, Zeng S, Zhong M. 2016. ALDH1A1 induces resistance to CHOP in diffuse large B-cell lymphoma through activation of the JAK2/STAT3 pathway. Onco Targets Ther 9: 5349–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CL, Bhatla T, Blum R, Wang J, Paugh SW, Wen X, Bourgeois W, Bitterman DS, Raetz EA, Morrison DJ, et al. 2014. Loss of TBL1XR1 disrupts glucocorticoid receptor recruitment to chromatin and results in glucocorticoid resistance in a B-lymphoblastic leukemia model. J Biol Chem 289: 20502–20515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota M, Sato M, Duncan B, Ooshima A, Yang HH, Diaz-Meyer N, Gere S, Kageyama S, Fukuoka J, Nagata T, et al. 2009. Identification of novel gene amplifications in breast cancer and coexistence of gene amplification with an activating mutation of PIK3CA. Cancer Res 69: 7357–7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, Niwa A, Chen Y, Nakazaki K, Nomoto J, et al. 2009. Frequent inactivation of A20 in B-cell lymphomas. Nature 459: 712–716. [DOI] [PubMed] [Google Scholar]
- Khodabakhshi AH, Morin RD, Fejes AP, Mungall AJ, Mungall KL, Bolger-Munro M, Johnson NA, Connors JM, Gascoyne RD, Marra MA, et al. 2012. Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget 3: 1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline J, Bishop MR. 2015. Update on checkpoint blockade therapy for lymphoma. J Immunother Cancer 3: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. 2012. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8: 890–896. [DOI] [PubMed] [Google Scholar]
- Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, et al. 2014. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 13: 842–854. [DOI] [PubMed] [Google Scholar]
- Kojima Y, Tsurumi H, Goto N, Shimizu M, Kasahara S, Yamada T, Kanemura N, Hara T, Sawada M, Saio M, et al. 2006. Fas and Fas ligand expression on germinal center type-diffuse large B-cell lymphoma is associated with the clinical outcome. Eur J Haematol 76: 465–472. [DOI] [PubMed] [Google Scholar]
- Kumar A, Mandiyan V, Suzuki Y, Zhang C, Rice J, Tsai J, Artis DR, Ibrahim P, Bremer R. 2005. Crystal structures of proto-oncogene kinase Pim1: a target of aberrant somatic hypermutations in diffuse large cell lymphoma. J Mol Biol 348: 183–193. [DOI] [PubMed] [Google Scholar]
- Lam KP, Kuhn R, Rajewsky K. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 90: 1073–1083. [DOI] [PubMed] [Google Scholar]
- Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, Chan JW, Rosenwald A, Gascoyne RD, Staudt LM. 2008. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{κ}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 111: 3701–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Shumilina E. 2013. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J 27: 3–12. [DOI] [PubMed] [Google Scholar]
- Lawler M, Siu LL, Rehm HL, Chanock SJ, Alterovitz G, Burn J, Calvo F, Lacombe D, Teh BT, North KN, et al. 2015. All the world's a stage: facilitating discovery science and improved cancer care through the Global Alliance for Genomics and Health. Cancer Discov 5: 1133–1136. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. 2014. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, et al. 2008a. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319: 1676–1679. [DOI] [PubMed] [Google Scholar]
- Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, et al. 2008b. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 359: 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. 2008c. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci 105: 13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Schatz DG. 2009. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol 30: 173–181. [DOI] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, et al. 2012. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci 109: 3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiarro M, Ruggiero V, Sette C. 2013. Targeting the Toll-like receptor/interleukin 1 receptor pathway in human diseases: rational design of MyD88 inhibitors. Clin Lymphoma Myeloma Leuk 13: 222–226. [DOI] [PubMed] [Google Scholar]
- Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, Levy R. 2004. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med 350: 1828–1837. [DOI] [PubMed] [Google Scholar]
- Lu X, Malumbres R, Shields B, Jiang X, Sarosiek KA, Natkunam Y, Tiganis T, Lossos IS. 2008. PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood 112: 4098–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres R, Chen J, Tibshirani R, Johnson NA, Sehn LH, Natkunam Y, Briones J, Advani R, Connors JM, Byrne GE, et al. 2008. Paraffin-based 6-gene model predicts outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Blood 111: 5509–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, Chadburn A, Rajewsky K, Tarakhovsky A, Pasqualucci L, et al. 2010. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 18: 568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareschal S, Dubois S, Viailly PJ, Bertrand P, Bohers E, Maingonnat C, Jais JP, Tesson B, Ruminy P, Peyrouze P, et al. 2016. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer 55: 251–267. [DOI] [PubMed] [Google Scholar]
- Marks PA, Xu WS. 2009. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem 107: 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul RW, Gearhart PJ. 2010. AID and somatic hypermutation. Adv Immunol 105: 159–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, et al. 2012a. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci 109: 2989–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A III, Diaz E, et al. 2012b. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492: 108–112. [DOI] [PubMed] [Google Scholar]
- McCarthy M. 2016. US president endorses “moonshot” effort to cure cancer. BMJ 352: i213. [DOI] [PubMed] [Google Scholar]
- Mehta-Shah N, Younes A. 2015. Novel targeted therapies in diffuse large B-cell lymphoma. Semin Hematol 52: 126–137. [DOI] [PubMed] [Google Scholar]
- Melzner I, Bucur AJ, Bruderlein S, Dorsch K, Hasel C, Barth TF, Leithauser F, Moller P. 2005. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood 105: 2535–2542. [DOI] [PubMed] [Google Scholar]
- Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, Meyers RM, Amor C, Wasserman CR, Neuberg D, et al. 2014. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 159: 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer PN, Fu K, Greiner TC, Smith LM, Delabie J, Gascoyne RD, Ott G, Rosenwald A, Braziel RM, Campo E, et al. 2011. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol 29: 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian M, Scandurra M, Chigrinova E, Shen Y, Inghirami G, Greiner TC, Chan WC, Vose JM, Testoni M, Chiappella A, et al. 2012. Clinical and molecular characterization of diffuse large B-cell lymphomas with 13q14.3 deletion. Ann Oncol 23: 729–735. [DOI] [PubMed] [Google Scholar]
- Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, Berns A. 2004. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol 24: 6104–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein C, Neuberger MS, Staden R. 1998. Both DNA strands of antibody genes are hypermutation targets. Proc Natl Acad Sci 95: 8791–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo JS, Ann EJ, Yoon JH, Jung J, Choi YH, Kim HY, Ahn JS, Kim SM, Kim MY, Hong JA, et al. 2011. Serum- and glucocorticoid-inducible kinase 1 (SGK1) controls Notch1 signaling by downregulation of protein stability through Fbw7 ubiquitin ligase. J Cell Sci 124: 100–112. [DOI] [PubMed] [Google Scholar]
- Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, et al. 2005. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 105: 1851–1861. [DOI] [PubMed] [Google Scholar]
- Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, Inguilizian H, Mermel C, Currie T, Dogan A, et al. 2012. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 22: 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. 2010. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42: 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. 2011. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476: 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, Scott DW, Ding J, Roth A, Chiu R, et al. 2013. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 122: 1256–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, Grewal J, Yu S, Fornika D, Bushell K, et al. 2016. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res 22: 2290–2300. [DOI] [PubMed] [Google Scholar]
- Mottok A, Renne C, Willenbrock K, Hansmann ML, Brauninger A. 2007. Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood 110: 3387–3390. [DOI] [PubMed] [Google Scholar]
- Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, An J, Xu Y, Rosenwald A, Ott G, et al. 2014. Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature 516: 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müschen M, Re D, Jungnickel B, Diehl V, Rajewsky K, Kuppers R. 2000. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. J Exp Med 192: 1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Comprehensive Cancer Network. 2016. NCCN clinical practice guidelines in oncology, non-Hodgkin's lymphomas (Version 3.2106).
- Naylor TL, Tang H, Ratsch BA, Enns A, Loo A, Chen L, Lenz P, Waters NJ, Schuler W, Dorken B, et al. 2011. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res 71: 2643–2653. [DOI] [PubMed] [Google Scholar]
- Ng AK, LaCasce A, Travis LB. 2011. Long-term complications of lymphoma and its treatment. J Clin Oncol 29: 1885–1892. [DOI] [PubMed] [Google Scholar]
- Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. 2011. Oncogenically active MYD88 mutations in human lymphoma. Nature 470: 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski GS, LaPlant B, Macon WR, Reeder CB, Foran JM, Nelson GD, Thompson CA, Rivera CE, Inwards DJ, Micallef IN, et al. 2015. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 33: 251–257. [DOI] [PubMed] [Google Scholar]
- Oki Y, Noorani M, Lin P, Davis RE, Neelapu SS, Ma L, Ahmed M, Rodriguez MA, Hagemeister FB, Fowler N, et al. 2014. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 166: 891–901. [DOI] [PubMed] [Google Scholar]
- Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M, Liu X, Bruno J, Heguy A, Olshen AB, et al. 2011. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell 147: 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsborne C, Byers R. 2011. Impact of gene expression profiling in lymphoma diagnosis and prognosis. Histopathology 58: 106–127. [DOI] [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. 2015. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 21: 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker H, An Q, Barber K, Case M, Davies T, Konn Z, Stewart A, Wright S, Griffiths M, Ross FM, et al. 2008. The complex genomic profile of ETV6-RUNX1 positive acute lymphoblastic leukemia highlights a recurrent deletion of TBL1XR1. Genes Chromosomes Cancer 47: 1118–1125. [DOI] [PubMed] [Google Scholar]
- Pasero C, Speiser DE, Derre L, Olive D. 2012. The HVEM network: new directions in targeting novel costimulatory/co-inhibitory molecules for cancer therapy. Curr Opin Pharmacol 12: 478–485. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L. 2013. The genetic basis of diffuse large B-cell lymphoma. Curr Opin Hematol 20: 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Dalla-Favera R. 2015. The genetic landscape of diffuse large B-cell lymphoma. Semin Hematol 52: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Zhang B. 2016. Genetic drivers of NF-κB deregulation in diffuse large B-cell lymphoma. Semin Cancer Biol 39: 26–31. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, Chaganti RS, Klein U, Kuppers R, Rajewsky K, et al. 1998. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci 95: 11816–11821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, Dalla-Favera R. 2001. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 412: 341–346. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC III, Nussenzweig MC, Dalla-Favera R. 2008. AID is required for germinal center-derived lymphomagenesis. Nat Genet 40: 108–112. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. 2011a. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471: 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et al. 2011b. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 43: 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG. 2008. TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol Cell 29: 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters TL, Li L, Tula-Sanchez AA, Pongtornpipat P, Schatz JH. 2016. Control of translational activation by PIM kinase in activated B-cell diffuse large B-cell lymphoma confers sensitivity to inhibition by PIM447. Oncotarget 7: 63362–63373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfreundschuh M, Trumper L, Osterborg A, Pettengell R, Trneny M, Imrie K, Ma D, Gill D, Walewski J, Zinzani PL, et al. 2006. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 7: 379–391. [DOI] [PubMed] [Google Scholar]
- Pon JR, Wong J, Saberi S, Alder O, Moksa M, Grace Cheng SW, Morin GB, Hoodless PA, Hirst M, Marra MA. 2015. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat Commun 6: 7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince HM, Bishton MJ, Harrison SJ. 2009. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 15: 3958–3969. [DOI] [PubMed] [Google Scholar]
- Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M, et al. 2011. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. 2012. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci 109: 21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G, Tang Z, Mathe E, Benner C, et al. 2014. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 159: 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, et al. 2012. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44: 47–52. [DOI] [PubMed] [Google Scholar]
- Raaphorst FM, van Kemenade FJ, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP, Meijer CJ. 2000. Coexpression of BMI-1 and EZH2 polycomb group genes in Reed–Sternberg cells of Hodgkin's disease. Am J Pathol 157: 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raia V, Schilling M, Bohm M, Hahn B, Kowarsch A, Raue A, Sticht C, Bohl S, Saile M, Moller P, et al. 2011. Dynamic mathematical modeling of IL13-induced signaling in Hodgkin and primary mediastinal B-cell lymphoma allows prediction of therapeutic targets. Cancer Res 71: 693–704. [DOI] [PubMed] [Google Scholar]
- Raut LS, Chakrabarti PP. 2014. Management of relapsed-refractory diffuse large B cell lymphoma. South Asian J Cancer 3: 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud G, Moore PS, Taruscio D, Scardoni M, Montresor M, Menestrina F, Scarpa A. 2001. Alteration of chromosome arm 6p is characteristic of primary mediastinal B-cell lymphoma, as identified by genome-wide allelotyping. Genes Chromosomes Cancer 31: 191–195. [DOI] [PubMed] [Google Scholar]
- Rimsza LM, Leblanc ML, Unger JM, Miller TP, Grogan TM, Persky DO, Martel RR, Sabalos CM, Seligmann B, Braziel RM, et al. 2008. Gene expression predicts overall survival in paraffin-embedded tissues of diffuse large B-cell lymphoma treated with R-CHOP. Blood 112: 3425–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz O, Guiter C, Castellano F, Dorsch K, Melzner J, Jais JP, Dubois G, Gaulard P, Moller P, Leroy K. 2009. Recurrent mutations of the STAT6 DNA binding domain in primary mediastinal B-cell lymphoma. Blood 114: 1236–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz O, Rommel K, Dorsch K, Kelsch E, Melzner J, Buck M, Leroy K, Papadopoulou V, Wagner S, Marienfeld R, et al. 2013. STAT6-mediated BCL6 repression in primary mediastinal B-cell lymphoma (PMBL). Oncotarget 4: 1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson MJ, Kahl BS, Vose JM, de Vos S, Laughlin M, Flynn PJ, Rowland K, Cruz JC, Goldberg SL, Musib L, et al. 2007. Phase II study of enzastaurin, a protein kinase C β inhibitor, in patients with relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol 25: 1741–1746. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Diaz M. 2004. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol 172: 3382–3384. [DOI] [PubMed] [Google Scholar]
- Roschewski M, Staudt LM, Wilson WH. 2014. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol 11: 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Staudt LM. 2003. Gene expression profiling of diffuse large B-cell lymphoma. Leuk Lymphoma 44: S41–S47. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. 2002. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346: 1937–1947. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, et al. 2003. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 198: 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, Monti S, Vaisitti T, Arruga F, Fama R, et al. 2012. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med 209: 1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RJ, Drier Y, Whitton H, Cotton MJ, Kaur J, Issner R, Gillespie S, Epstein CB, Nardi V, Sohani AR, et al. 2015. Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma. Cancer Discov 5: 1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagaert X, Laurent M, Baens M, Wlodarska I, De Wolf-Peeters C. 2006. MALT1 and BCL10 aberrations in MALT lymphomas and their effect on the expression of BCL10 in the tumour cells. Mod Pathol 19: 225–232. [DOI] [PubMed] [Google Scholar]
- Savage KJ, Monti S, Kutok JL, Cattoretti G, Neuberg D, De Leval L, Kurtin P, Dal Cin P, Ladd C, Feuerhake F, et al. 2003. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 102: 3871–3879. [DOI] [PubMed] [Google Scholar]
- Schif B, Lennerz JK, Kohler CW, Bentink S, Kreuz M, Melzner I, Ritz O, Trumper L, Loeffler M, Spang R, et al. 2013. SOCS1 mutation subtypes predict divergent outcomes in diffuse large B-Cell lymphoma (DLBCL) patients. Oncotarget 4: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, Boyle M, Slack GW, Marra MA, Connors JM, et al. 2012. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 26: 1383–1390. [DOI] [PubMed] [Google Scholar]
- Scott DW, Mungall KL, Ben-Neriah S, Rogic S, Morin RD, Slack GW, Tan KL, Chan FC, Lim RS, Connors JM, et al. 2012. TBL1XR1/TP63: a novel recurrent gene fusion in B-cell non-Hodgkin lymphoma. Blood 119: 4949–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DW, Wright GW, Williams PM, Lih CJ, Walsh W, Jaffe ES, Rosenwald A, Campo E, Chan WC, Connors JM, et al. 2014. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 123: 1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehn LH, Donaldson J, Chhanabhai M, Fitzgerald C, Gill K, Klasa R, MacPherson N, O'Reilly S, Spinelli JJ, Sutherland J, et al. 2005. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol 23: 5027–5033. [DOI] [PubMed] [Google Scholar]
- Sehn LH, Berry B, Chhanabhai M, Fitzgerald C, Gill K, Hoskins P, Klasa R, Savage KJ, Shenkier T, Sutherland J, et al. 2007. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood 109: 1857–1861. [DOI] [PubMed] [Google Scholar]
- Shipp MA. 1994. Prognostic factors in aggressive non-Hodgkin's lymphoma: who has “high-risk” disease? Blood 83: 1165–1173. [PubMed] [Google Scholar]
- Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, et al. 2002. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med 8: 68–74. [DOI] [PubMed] [Google Scholar]
- Siu LL, Lawler M, Haussler D, Knoppers BM, Lewin J, Vis DJ, Liao RG, Andre F, Banks I, Barrett JC, et al. 2016. Facilitating a culture of responsible and effective sharing of cancer genome data. Nat Med 22: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. 2010. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci 107: 20980–20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K. 2009. PI3 kinase signals BCR-dependent mature B cell survival. Cell 139: 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt LM. 2010. Oncogenic activation of NF-κB. Cold Spring Harb Perspect Biol 2: a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidl C, Gascoyne RD. 2011. The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood 118: 2659–2669. [DOI] [PubMed] [Google Scholar]
- Steinberg MW, Cheung TC, Ware CF. 2011. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev 244: 169–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud R, Friedberg JW. 2008. Salvage therapy for relapsed or refractory diffuse large B-cell lymphoma: impact of prior rituximab. Haematologica 93: 1776–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai DJ, Su CC, Ma YL, Lee EH. 2009. SGK1 phosphorylation of IκB Kinase α and p300 Up-regulates NF-κB activity and increases N-Methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem 284: 4073–4089. [DOI] [PubMed] [Google Scholar]
- Testoni M, Zucca E, Young KH, Bertoni F. 2015. Genetic lesions in diffuse large B-cell lymphomas. Ann Oncol 26: 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Non-Hodgkin's Lymphoma Prognostic Factors Project. 1993. A predictive model for aggressive non-Hodgkin's lymphoma. The International Non-Hodgkin's Lymphoma Prognostic Factors Project. N Engl J Med 329: 987–994. [DOI] [PubMed] [Google Scholar]
- Thome M. 2004. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol 4: 348–359. [DOI] [PubMed] [Google Scholar]
- Tilly H, Morschhauser F, Salles G, Casasnovas RO, Feugier P, Molina TJ, Jardin F, Terriou L, Haioun C, Coiffier B. 2013. Phase 1b study of lenalidomide in combination with rituximab-CHOP (R2-CHOP) in patients with B-cell lymphoma. Leukemia 27: 252–255. [DOI] [PubMed] [Google Scholar]
- Tirado CA, Chen W, Garcia R, Kohlman KA, Rao N. 2012. Genomic profiling using array comparative genomic hybridization define distinct subtypes of diffuse large B-cell lymphoma: a review of the literature. J Hematol Oncol 5: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twa DD, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, Slack GW, Gunawardana J, Lim RS, McPherson AW, et al. 2014. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood 123: 2062–2065. [DOI] [PubMed] [Google Scholar]
- Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, Elemento O. 2010. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116: 5247–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitolo U, Trneny M, Belada D, Carella AM, Chua N, Abrisqueta P, Demeter J, Flinn IW, Hong X, Kim WS, et al. 2016. Obinutuzumab or rituximab plus CHOP in patients with previously untreated diffuse large B-cell lymphoma: final results from an open-label, randomized phase 3 study (GOYA) [Abstract]. In American Society of Hematology: 58th Annual Meeting & Exposition, San Diego, CA. [Google Scholar]
- Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, Ducar M, Van Hummelen P, Macconaill LE, Hahn WC, et al. 2012. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov 2: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, Jeelall YS, Beutler B, Horikawa K, Goodnow CC. 2014. Consequences of the recurrent MYD88(L265P) somatic mutation for B cell tolerance. J Exp Med 211: 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, Takahashi K, Futreal PA, Chin L. 2013. Emerging patterns of somatic mutations in cancer. Nat Rev Genet 14: 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener E, Oeckinghaus A, Papadopoulou N, Lavitas L, Schmidt-Supprian M, Ferch U, Mak TW, Ruland J, Heissmeyer V, Krappmann D. 2006. Essential role for IκB kinase β in remodeling Carma1-Bcl10-Malt1 complexes upon T cell activation. Mol Cell 23: 13–23. [DOI] [PubMed] [Google Scholar]
- Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K, Mattfeldt T, Barth TF, Moller P. 2006. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene 25: 2679–2684. [DOI] [PubMed] [Google Scholar]
- Wessendorf S, Barth TF, Viardot A, Mueller A, Kestler HA, Kohlhammer H, Lichter P, Bentz M, Dohner H, Moller P, et al. 2007. Further delineation of chromosomal consensus regions in primary mediastinal B-cell lymphomas: an analysis of 37 tumor samples using high-resolution genomic profiling (array-CGH). Leukemia 21: 2463–2469. [DOI] [PubMed] [Google Scholar]
- Willis TG, Jadayel DM, Du MQ, Peng H, Perry AR, Abdul-Rauf M, Price H, Karran L, Majekodunmi O, Wlodarska I, et al. 1999. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 96: 35–45. [DOI] [PubMed] [Google Scholar]
- Wilson WH, Grossbard ML, Pittaluga S, Cole D, Pearson D, Drbohlav N, Steinberg SM, Little RF, Janik J, Gutierrez M, et al. 2002. Dose-adjusted EPOCH chemotherapy for untreated large B-cell lymphomas: a pharmacodynamic approach with high efficacy. Blood 99: 2685–2693. [DOI] [PubMed] [Google Scholar]
- Wilson WH, Jung SH, Porcu P, Hurd D, Johnson J, Martin SE, Czuczman M, Lai R, Said J, Chadburn A, et al. 2012. A Cancer and Leukemia Group B multi-center study of DA-EPOCH-rituximab in untreated diffuse large B-cell lymphoma with analysis of outcome by molecular subtype. Haematologica 97: 758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, Lih CJ, Williams PM, Shaffer AL, Gerecitano J, et al. 2015. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 21: 922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WH, sin-Ho J, Pitcher BN, Hsi ED, Friedberg J, Cheson B, Bartlett NL, Smith S, Johnston NW, Kahl BS, et al. 2016. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303 [Abstract]. In American Society of Hematology: 58th Annual Meeting & Exposition, San Diego, CA. [Google Scholar]
- Winkler GS. 2010. The mammalian anti-proliferative BTG/Tob protein family. J Cell Physiol 222: 66–72. [DOI] [PubMed] [Google Scholar]
- Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. 2003. A gene expression–based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci 100: 9991–9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Pittaluga S, Jaffe ES. 2015. The histological classification of diffuse large B-cell lymphomas. Semin Hematol 52: 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shaffer AL III, Emre NC, Ceribelli M, Zhang M, Wright G, Xiao W, Powell J, Platig J, Kohlhammer H, et al. 2012. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 21: 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, David L, Qiao Q, Damko E, Wu H. 2014. The CBM signalosome: potential therapeutic target for aggressive lymphoma? Cytokine Growth Factor Rev 25: 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. 2011. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117: 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, Califano A, Pasqualucci L, Basso K, Dalla-Favera R. 2013. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol 14: 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. 2003. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J 22: 1336–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A, Romaguera J, Fanale M, McLaughlin P, Hagemeister F, Copeland A, Neelapu S, Kwak L, Shah J, de Castro Faria S, et al. 2012. Phase I study of a novel oral Janus kinase 2 inhibitor, SB1518, in patients with relapsed lymphoma: evidence of clinical and biologic activity in multiple lymphoma subtypes. J Clin Oncol 30: 4161–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A, Thieblemont C, Morschhauser F, Flinn I, Friedberg JW, Amorim S, Hivert B, Westin J, Vermeulen J, Bandyopadhyay N, et al. 2014. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 15: 1019–1026. [DOI] [PubMed] [Google Scholar]
- Young RM, Shaffer AL III, Phelan JD, Staudt LM. 2015. B-cell receptor signaling in diffuse large B-cell lymphoma. Semin Hematol 52: 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenetz AD. 2014. Guidelines for NHL: updates to the management of diffuse large B-cell lymphoma and new guidelines for primary cutaneous CD30+ T-cell lymphoproliferative disorders and T-cell large granular lymphocytic leukemia. J Natl Compr Canc Netw 12: 797–800. [DOI] [PubMed] [Google Scholar]
- Zeng H, Di L, Fu G, Chen Y, Gao X, Xu L, Lin X, Wen R. 2007. Phosphorylation of Bcl10 negatively regulates T-cell receptor-mediated NF-κB activation. Mol Cell Biol 27: 5235–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Siebert R, Yan M, Hinzmann B, Cui X, Xue L, Rakestraw KM, Naeve CW, Beckmann G, Weisenburger DD, et al. 1999. Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32). Nat Genet 22: 63–68. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Chang Q, Zeng L, Gu J, Brown S, Basch RS. 2006. TBLR1 regulates the expression of nuclear hormone receptor co-repressors. BMC Cell Biol 7: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, Buetow KH, Carroll WL, Chen IM, Devidas M, et al. 2011. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118: 3080–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, et al. 2013. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci 110: 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. 2015. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21: 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Reddy A, Love C, Moffitt AB, Rajagopalan D, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, Norgaard P, et al. 2016. Integrative genetic and clinical analysis through whole exome sequencing in 1001 diffuse large B cell lymphoma (DLBCL) patients reveals novel disease drivers and risk groups. In American Society of Hematology: 58th Annual Meeting & Exposition, San Diego, CA. [Google Scholar]
- Zheng X, Ding N, Song Y, Feng L, Zhu J. 2014. Different sensitivity of germinal center B cell-like diffuse large B cell lymphoma cells towards ibrutinib treatment. Cancer Cell Int 14: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Sehn LH, Rademaker AW, Gordon LI, Lacasce AS, Crosby-Thompson A, Vanderplas A, Zelenetz AD, Abel GA, Rodriguez MA, et al. 2014. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123: 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]