

Abstract
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder and the most common cause of dementia among the elderly population. The good correlation between the density and neocortical spread of neurofibrillary tangles (NFTs) and the severity of cognitive impairment offers an opportunity to use a noninvasive imaging technique such as positron emission tomography (PET) for early diagnosis and staging of the disease. PET imaging of NFTs holds promise not only as a diagnostic tool but also because it may enable the development of disease-modifying therapeutics for AD. In this review, we focus on the structural diversity of tau PET tracers, the challenges related to identifying high-affinity and highly selective NFT ligands, and recent progress in the clinical development of tau PET radioligands.
Positron emission tomography (PET), a noninvasive imaging technique, holds promise for monitoring tau aggregates in Alzheimer’s disease. Recent progress has been made in the development of tau PET radioligands for this purpose.
Alzheimer’s disease (AD), a chronic neurodegenerative disorder and the most common cause of dementia among the elderly population, is characterized by memory loss, spatial disorientation, and cognitive impairment (Khachaturian 1985; Citron 2010). Considerable progress has been made by numerous groups and organizations to understand the pathogenic mechanisms underlying AD, resulting in a number of promising disease-modifying therapeutic approaches that are currently under development and clinical evaluation (Anand et al. 2014).
Multiple factors are considered to be involved in the pathogenic mechanism leading to AD, such as age, genetic makeup, environmental factors, head trauma, depression, diabetes mellitus, hyperlipidemia, and vascular factors (Fargo and Bleiler 2014). The development of a disease-modifying therapy for AD has been hampered by the lack of reliable tools for early diagnosis, staging, and accurately monitoring disease progression.
The initial neuropathological changes in the brain are thought to begin 20 years or more before symptoms appear in AD, and during this period individuals are able to function normally. The long duration of this presymptomatic phase offers an opportunity to intervene with disease-modifying therapy, provided that tools for early diagnosis and presymptomatic staging are made available.
Despite the recent advent of amyloid positron emission tomography (PET) imaging agents, definitive diagnosis of AD can only be made by postmortem histological analysis of human brain samples (Delacourte 1998). Postmortem studies of AD brains reveal two pathological hallmarks of the disease: (1) senile plaques, composed of amyloid-β (Aβ) peptides, and (2) neurofibrillary tangles (NFTs), composed of paired helical filaments (PHFs) of hyperphosphorylated aggregated tau protein (Trojanowski et al. 1997). Aβ peptide deposits are commonly found outside neurons in the brain, while tau protein tangles are formed inside neurons but eventually end up in the intercellular space after neuronal death (Karran et al. 2011).
Evidence for AD pathology can be obtained from cerebrospinal fluid (CSF) samples and brain imaging. The latter technique is noninvasive and may be used to identify patients at risk of developing AD and/or to monitor disease progression (DeKosky and Marek 2003; Reiman and Jagust 2012; Zhou et al. 2014). Specifically, PET is a sensitive imaging technique that enables in vivo and in situ noninvasive visualization, characterization, and quantification of physiological processes at the molecular level (Valk et al. 2005). As such, PET has become an extremely powerful diagnostic tool that has been increasingly used in drug discovery and development to show target engagement and to monitor disease progression (Matthews et al. 2011; Piel et al. 2014).
Several PET probes for in vivo imaging of Aβ plaques have been approved by regulatory agencies (Fig. 1) (Kung 2012; Mason et al. 2013; Noёl et al. 2013; Rowe and Villemagne 2013; Holland et al. 2014). However, amyloid imaging has not been approved to diagnose AD or to measure the extent of cognitive impairment but rather as a diagnostic method to exclude AD in subjects who are cognitively impaired and amyloid PET-negative (www.fda.gov). Amyloid deposits have been shown to accumulate in the brains of individuals at risk of developing AD (Sperling et al. 2011), but the density of amyloid plaques does not correlate well with neurodegeneration or cognitive impairment in AD (Villemagne et al. 2012).
Figure 1.

Structures of amyloid-β positron emission tomography (PET) probes for Alzheimer’s disease (AD).
In contrast to Aβ plaques, the density and neocortical spread of NFTs correlate with progressive neuronal degeneration and cognitive impairment (Arriagada et al. 1992; Maccioni et al. 2010; Villemagne et al. 2012), thus making PET imaging of NFTs a desirable biomarker for AD. Recent findings provide additional support for the central role of tau in the pathogenesis of AD (Maccioni et al. 2010; Ke et al. 2012), supporting this protein as both a diagnostic and a therapeutic target (Medeiros et al. 2011). NFT imaging may allow early detection of AD, possibly even before the onset of cognitive symptoms, and presymptomatic staging. PET imaging of NFTs, especially when performed in conjunction with amyloid diagnosis, may also provide a means to distinguish between AD and non-AD dementias. The absence of NFTs and amyloid would provide support for the latter diagnosis, whereas the presence of NFTs and amyloid would provide support for the former. For researchers, NFT imaging provides insights into the relationship between the accumulation of tau aggregates and their spread as a function of time, cognition, and brain structure across the continuum from normal aging to AD. NFT imaging also allows researchers to investigate the effect of disease-modifying therapies on tau aggregation. In addition to its involvement in AD, tau aggregation is a characteristic of other neurodegenerative diseases known as “tauopathies” (Spillantini and Goedert 2013), which include tangle-only dementia (TD), argyrophilic grain disease, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease, and frontotemporal dementia and parkinsonism linked to chromosome 17. The heterogeneity of these disorders is closely related to the wide range of human tau isoforms and posttranslational modifications. Tau aggregates may appear ultrastructurally as PHFs, straight filaments, randomly coiled filaments, or twisted filaments. This variability translates into polymorphism, whereby either the same tau isoform adopts different conformations and tau-aggregate ultrastructure or different isoforms appear with the same ultrastructure. For example, in AD and TD, tau inclusions appear as both 4R and 3R isoforms in the shape of NFTs, whereas 3R isoforms predominate in Pick’s disease as Pick bodies in the shape of randomly coiled and straight filaments. In CBD, tau deposits are found as astrocytic plaques composed of 4R tau isoforms in the form of straight and twisted filaments (Clavaguera et al. 2014).
Consequently, the development of radiotracers for tau aggregates has been pursued by numerous academic and industrial groups (Mach 2014; Okamura et al. 2014a; Shah and Catafau 2014; Villemagne and Okamura 2014; Zimmer et al. 2014; Villemagne et al. 2015) and, recently, we have also reviewed those efforts (Ariza et al. 2015). The aim of this work is to review the progress that has been made in the field of tau-aggregate imaging and the recent evolution in the development of selective tau PET radioligands currently undergoing clinical evaluation.
TAU PET LIGANDS
An ideal tau PET ligand, like any other radiotracer for brain imaging, must fulfill a wide range of general requirements, which have been extensively reviewed on multiple occasions (Ariza et al. 2015). Tau ligand discovery is largely based on compound screening and is guided by the structural hypothesis that fused aromatic systems, which are needed to interact with the β-sheet fibrillar aggregates present in PHF-tau. Because Aβ plaques and NFTs share a similar β-sheet structure, it is not surprising that structural similarities exist between imaging agents for both species. Compound screening is generally based on competitive in vitro assays using either synthetic heparin-induced tau polymers (HITPs) or postmortem AD human-brain homogenates (AD-PHFs) or sections. HITPs are composed only of 3R and/or 4R tau and are unlikely to have similar hyperphosphorylation or glycated or ubiquitinated residues as the native AD-PHFs. Furthermore, HITPs and AD-PHFs differ with respect to the structure and variability in twist in their observed tau filaments (Wegmann et al. 2010). Such differences could be the reason why screening campaigns based on synthetic HITPs have not been successful in identifying ligands for native AD-PHF tau (Janke et al. 1999; Ramachandran and Udgaonkar 2013). Assays based on native protein, as found in brain homogenates or slices from AD patients, have yielded the most promising tau ligands (Mach 2014; Shah and Catafau 2014; Villemagne and Okamura 2014; Zimmer et al. 2014; Villemagne et al. 2015).
Lead Compounds
One of the earliest ligand screening campaigns targeting tau aggregates for PET-tracer discovery was performed by Okamura et al. (2005). In this study, more than 2000 small molecules were tested, which resulted in the discovery of quinoline and benzimidazole lead compounds, such as 6 (BF-126), 7 (BF-158), and 8 (BF-170) (Fig. 2) (Okamura et al. 2005). In vitro fluorescence binding-affinity assay data and neuropathological staining results suggested quinolines 7 and 8 to be better tau ligands than the benzimidazole 6, yet all compounds showed a relatively low selectivity over amyloid plaques (Table 1). Autoradiography studies with 11C-7 confirmed binding of this tracer in tangle-rich brain regions, and in vivo biodistribution studies in mice showed good brain uptake (11.3% ID/g at 2 min postinjection) but rather slow clearance from the brain (3.1% ID/g at 30 min postinjection). Despite their low affinity (EC50 >200 nM) and high nonspecific binding, these compounds first highlighted the potential utility of quinolines and benzimidazoles as tau PET radioligands in AD (Okamura et al. 2005).
Figure 2.

Structures of benzimidazoles 6, 12, and 13; quinolines 7 and 8; benzothiazoles 9–11; and the tau PET ligands 14 and 15.
Table 1.
Log P, affinity for tau, selectivity Aβ/tau, and brain uptake for compounds 6–13
| Compound | Log P | Tau affinity (nM) | Selectivity tau/Aβ | Brain uptake (% ID/g) | |||
|---|---|---|---|---|---|---|---|
| HITP | AD-PHF | 2 min | 30 min | 60 min | |||
| 6 | 1.09 | EC50 = 583 | N/A | 2.20a | 7.2 | 0.16 | N/A |
| 11C-7 | 1.67 | EC50 = 399 | N/A | 1.60a | 11.3 | 3.1 | N/A |
| 8 | 1.85 | EC50 = 221 | N/A | 3.50a | 9.1 | 0.25 | N/A |
| 9 | N/A | EC50 = 83 | N/A | 1.70a | N/A | N/A | N/A |
| 125I-10 | 3.84 | Ki = 0.48 | N/A | 17.20b | 0.94 | N/A | 2.89 |
| 18F-11 | 2.05 | Ki = 13.0 | N/A | 1.54b | 4.28 | N/A | 2.53 |
| 125I-12 | N/A | Kd = 300 | N/A | 2.73c | 3.28 | N/A | 0.33 |
| 13 | N/A | Ki = 3.9 | N/A | 1.60b | 4.8 | 0.12 | N/A |
Selectivity tau versus Aβ. Log P values are the partition coefficient (octanol/water) measured for each compound as reported in the respective references.
HITP, Heparin-induced tau polymer; AD-PHF, Alzheimer’s disease paired helical filament; N/A, not available.
aEC50 (Aβ)/EC50 (tau).
bKi (Aβ)/Ki (tau).
cKd (Aβ)/Kd (tau).
On the basis of phenyldiazenyl benzothiazole (PDB) compounds (9), identified by the Kuret group (Honson et al. 2007), Matsumura et al. (2011) developed two novel PDB agents by substituting the methoxy group with iodine-125 (10, 125I-PDB-3) (Matsumura et al. 2011) and with a fluoro-polyethylene glycol chain, respectively (11, 18F-FPPDB) (Fig. 2) (Matsumura et al. 2012). Further lead optimization eventually led to the butadienyl benzimidazole derivative 13 (Harada et al. 2014) as a modestly tau-selective multispectral fluorescent imaging probe, which gives different fluorescent spectra when bound to tau or Aβ fibrils (Fig. 2, Table 1). Although compound 13 was specifically developed for fluorescence-based imaging, it is an equally promising lead for the development of new tau PET probes.
Besides the planar aromatic bicycle, compounds 6–13 are characterized by the presence of an aniline terminus either directly linked to the aromatic ring or via an sp2 hybridized 2 or 4 atom spacer. Interestingly, the aniline moiety is also present in previously reported Aβ ligands, which may explain the low selectivity achieved with those compounds and illustrates the challenge in differentiating between both fibrillar proteins.
Recently, Hoffmann-La Roche filed several patent applications claiming structurally different potential tau PET tracers. Thus, a first publication in 2014 claimed imidazo[2,1]thiazol-3-one derivatives (14, Fig. 2) (Gobbi et al. 2014) as potent (Ki [HITP tau] ∼0.01 nM) and exceptionally selective tau-aggregate binders (tau/Aβ ∼100- to 1000-fold). These compounds feature a highly polar phenolic terminus on the one side and a lipophilic aromatic terminus on the opposite side. Previous studies with amphiphilic molecules have suggested the possibility of increasing potency and selectivity by incorporating lipophilic and hydrophilic features in the same compound (Taghavi et al. 2011). However, the true potential of this chemical class has yet to be shown because the affinity and selectivity in the human AD brain have not yet been disclosed. Roche has recently published three additional patent applications claiming compounds with different structures (Borroni et al. 2014; Gobbi et al. 2015a,b). Some of these compounds might be the subject of a newly registered clinical phase I study, “Evaluation of [11C]RO6924963, [11C]RO6931643, and [18F]RO6958948 as Tracers for Positron Emission Tomography (PET) Imaging of Tau in Healthy and Alzheimer's Disease Participants” (clinicaltrials.gov/ct2/show/record/NCT02187627), which does not, however, disclose the exact chemical nature of the test compounds. These new potential tau PET tracers by Roche will be discussed further in the next section.
Finally, the naphthalene derivative 2-[1-[6-[N-(2-18F-fluoroethyl)-N-methylamino]naphthalen-2-yl]ethylidene]malononitrile (15, 18F-FDDNP) is an older radioligand, which was first described by Barrio et al. (1999) as a potential PET tracer for Aβ and NFTs with the aim of visualizing amyloid plaques in AD (Barrio et al. 1999; Small et al. 2006, 2013; Nelson et al. 2011; Shin et al. 2011). However, the low affinity for amyloid structures (Thompson et al. 2009) and the low Aβ/tau selectivity of 18F-FDDNP limits its utility as a PET tracer. Various studies with this radioligand have been covered in several recent review articles (Villemagne et al. 2012, 2015; Villemagne and Okamura 2014; Zimmer et al. 2014).
Selective Tau Binders Evaluated in Human Subjects
Recently, considerable progress in the development of high-affinity, tau-selective ligands has been made independently by three groups: the Tohoku University, Sendai, Japan/Austin Hospital, Melbourne, Australia, group, led by N. Okamura, has described a series of 18F-labeled THK compounds; the National Institute of Radiological Sciences (NIRS) group in Chiba, Japan, led by M. Maruyama and H. Shimada, has described a new series of 11C-labeled PBB compounds; and the Siemens group, led by H. Kolb, has reported two 18F-labeled compounds, which are now being developed by Avid/Lilly. In addition, there are tau PET-tracer development efforts at Hoffmann-La Roche, Merck & Co., and Piramal Imaging (Stephens 2015).
THK Compounds
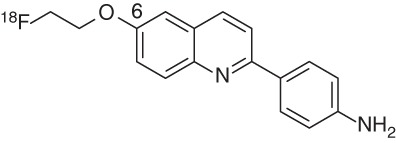
The THK tau imaging probes (Table 2) comprise a family of arylquinolines first reported by Okamura et al. (2005) after structural optimization of lead compounds 7 and 8. The first candidate described with good affinity for synthetic tau fibrils and reasonable selectivity over amyloid plaques was 4-[6-(2-18F-fluoroethoxy)quinolin-2-yl]phenylamine (16, THK-523) (Fodero-Tavoletti et al. 2011). The introduction of an alkyl ether at the 6-position of the arylquinoline not only allowed introduction of 18F but also was accompanied by an improvement in affinity and selectivity compared with the initial hits 7 and 8.
Table 2.
Binding affinities and pharmacokinetics of THK compounds
| Compound | Log P | Tau affinity (nM) | Selectivity tau/Aβ | Brain uptake (% ID/g) | ||
|---|---|---|---|---|---|---|
| HITP | AD-PHF | 2 min | 30 min | |||
 18F-THK-523 (16) |
2.40 |
Kd1 = 1.67 Kd2 = 21.74 Ki = 59.30 |
Kd = 86.50 | 10a | 2.72 | 1.47 |
 18F-THK-5105 (17) |
3.03 |
Kd1 = 1.45 Kd2 = 7.40 Ki = 7.80 |
Kd = 2.63 | 25a | 9.20 | 3.61 |
 18F-THK-5117 (18) |
2.32 | Ki = 10.50 | Kd = 5.19 | 30b,c | 6.06 | 0.59 |
 18F-THK-5351 (19) |
n.r. | n.r. | Kd = 2.9 | n.r. | n.r. | n.r. |
Selectivity tau versus Aβ. Log P values are the partition coefficient (octanol/water) measured for each compound as reported in the respective references.
HITP, Heparin-induced tau polymer; AD-PHF, Alzheimer’s disease paired helical filament; n.r., not reported.
aKd (Aβ)/Kd (tau).
bKi (Aβ)/Ki (tau).
cUnpublished data, AAIC meeting, Copenhagen, 2014.
Initial in vitro saturation binding studies with THK-523 suggested two different binding sites (Fodero-Tavoletti et al. 2011), and further competition studies revealed the affinity for recombinant tau fibrils to be relatively low (Ki = 59.30 nM) (Table 2; Okamura et al. 2013). Unfortunately, the binding affinity for AD-PHF, present in AD brain homogenates, was even lower (Kd = 86.50 nM) than for synthetic HITPs, which again showed the shortcomings of synthetic tau preparations. Even though tau/Aβ selectivity was initially reported to be 10-fold based on the synthetic tau-affinity data (Kd1 = 1.67 nM), THK-523 is actually nonselective based on the other tau-binding data (Okamura et al. 2013).
Interestingly, in vivo retention of this tracer in tau transgenic mice (rTg4510) was significantly higher compared with the analog model for Aβ plaques (APP/PS1 mice) (Fodero-Tavoletti et al. 2011), suggesting that THK-523 may be selective for tau pathology in vivo, with a favorable brain uptake and washout.
In vivo PET imaging with THK-523 showed higher cortical retention in AD patients compared with healthy controls (HCs), and tracer distribution was in agreement with the reported histopathological brain distribution of PHFs in AD (Villemagne et al. 2014). However, the high white matter retention of this radioligand hampers a clear visualization of PET scans and consequently precluded further development.
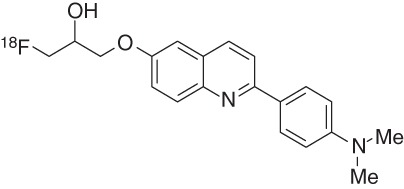
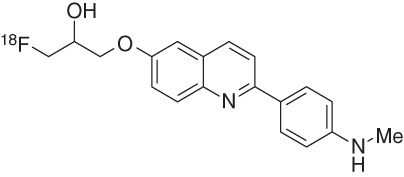
The optimization of the alkyl chain at the quinoline 6-position provided two novel arylquinolines, 1-([2-[4-(dimethylamino)phenyl]quinolin-6-yl]oxy)-3-18F-fluoropropan-2-ol (17, THK-5105) and 1-([2-[4-(methylamino)phenyl]quinolin-6-yl]oxy)-3-18F-fluoropropan-2-ol (18, THK-5117) (Okamura et al. 2013). Both compounds showed higher in vitro tau affinities than THK-523 for synthetic HITP tau fibrils and for human AD-PHF tau aggregates (Table 2), with Kd values of 2.6 nM (17) and 5.2 nM (18) for the latter. Additionally, these two tracers show higher in vitro Aβ/tau selectivity. In vitro autoradiography staining of human AD brain sections revealed that the distribution of THK-5105 and THK-5117 in the mesial temporal sections coincided with Gallyas–Braak staining and tau immunostaining but not with the distribution of Pittsburgh compound B (PiB). Compared with THK-523, both compounds (THK-5105 and THK-5117) showed higher contrast of tau pathology, mostly because of their higher affinity and selectivity. Additionally, autoradiography with the tritiated ligand 3H-THK-5117 on human-brain slices displayed good agreement with AT8 immunostaining, and the Bmax was calculated to be 704 fmol/mg (Lemoine et al. 2015).
First-in-human PET studies of THK-5105 revealed its ability to differentiate between AD patients and HCs (Okamura et al. 2014b). Furthermore, the pattern distribution shown in the mesial and lateral lobes of AD patients agrees with the reported NFT distribution in AD brain (Braak and Braak 1991) and does not correlate with 11C-PiB retention. These results suggest that THK-5105 is selective for NFTs over Aβ plaques, although nonspecific binding to the brainstem, thalamus, and subcortical white matter hampers data interpretation (Bakhti et al. 2014). The relatively slow kinetics and clearance displayed by this ligand compared with other known PET ligands has limited further use.
First-in-human PET studies with THK-5117 are currently underway (Fig. 3) (Okamura et al. 2014a), and preliminary results show faster kinetics and a better signal-to-noise ratio (SNR) than observed for THK-5105, as expected from its higher hydrophilicity and preclinical pharmacokinetic studies. The S-enantiomer was selected as the clinical candidate based on faster kinetics and a better SNR than the R-enantiomer. Tracer retention was associated with the severity of dementia and human-brain atrophy (Fig. 3).
Figure 3.

18F-THK-5117 positron emission tomography (PET) images in mild, moderate, and severe Alzheimer’s disease (AD) patients. In the mild AD case, specific THK-5117 binding is confined to the medial, anterior, and inferior temporal cortex. The moderate AD case shows additional THK-5117 retention in association areas. The severe AD case shows more extensive and higher THK-5117 retention in the neocortex (Okamura et al. 2014a). MMSE, Mini Mental State Examination; CDR, Clinical Dementia Rating; y.o., years old. (From Okamura et al. 2014a; reproduced, with permission, from Springer Science and Business Media 2014.)
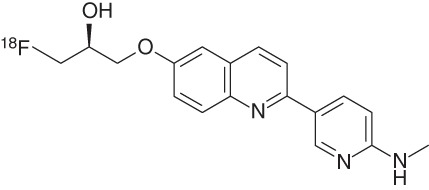
Further optimization provided 18F-THK-5351 (19), the 4-methylaminopyridyl-analog of THK-5117 (Table 2), which displays a high binding affinity for tau in AD brain homogenates (Kd = 2.9 nM) combined with faster kinetics, lower white matter retention, and a higher SNR compared with earlier THK compounds (Okamura et al. 2014c; Villemagne et al. 2015). 18F-THK-5351 autoradiography of AD brain sections showed a good match with tau immunostaining and a high SNR in gray versus white matter. Data from the first human trial of this tracer were disclosed in January 2015 at the 9th Annual Human Amyloid Imaging (HAI) Conference in Miami Beach, Florida (2015). The Tohoku team evaluated THK-5351 in 16 HCs, five subjects with mild cognitive impairment, and 13 AD patients. THK-5351 possesses an excellent pharmacokinetic profile for PET imaging, with the peak target-to-cerebellum standardized uptake value ratio (SUVR) achieved within 5 min after injection and exit from the brain by 90 min. The SUVR values, using the cerebellum as the reference, stabilized as early as 50 min postinjection at ∼1.5 in the control group and 2.5 in the AD group. Thus, 18F-THK-5351 was able to distinguish AD from controls. A careful comparison of 11C-PiB (Aβ) and 18F-THK-5351 binding across the AD brain was in agreement with the areas where tau pathology would be expected according to Braak staging, and this comparison suggested that 18F-THK-5351 does not bind to Aβ (Fig. 4). These cross-sectional data also showed increasing tracer retention with age within the HC group in the hippocampus and, to a lesser degree, the inferior temporal cortex because of increasing tau pathology with age. In addition to AD, the first images of a patient with PSP were shown. The scan showed uptake in the patient’s midbrain, where tau would be expected in this movement disorder (Alzforum 2015).
Figure 4.

18F-THK-5351 positron emission tomography (PET) images in mild, moderate, and severe Alzheimer’s disease (AD) patients. This new tau PET tracer shows low uptake in controls, intermediate uptake in mild cognitive impairment (MCI), and intense tau pathology spreading across the frontal and temporal cortex in AD. SUVR, Standardized uptake value ratio. (Image courtesy of Nobuyuki Okamura, Tohoku University.)
Phenyl-Butadienyl-Benzothiazole Compounds
Phenyl- and pyridyl-butadienyl-benzothiazoles (PBBs) have been identified as potential tau imaging probes by the NIRS group in Chiba, Japan (Fig. 5) (Maruyama et al. 2013). These scaffolds are characterized by a π-electron-conjugated backbone with a specific extent ranging from 13 Å to 19 Å, which the investigators deem essential for gaining selectivity for AD and non-AD tau aggregates. Structurally, the PBB series can be seen as analogs of fluorescent amyloid dye (ThT) in which the two aromatic moieties are spaced with an all-trans butadiene bridge.
Figure 5.

Structure of 11C-PBB3 (20) and a novel fluorinated 18F-PBB3 derivative (21).
Taking advantage of the fluorescent properties of PBBs, NFT-like pathology in the brainstem and spinal cord of transgenic mouse models could be visualized ex vivo in both PS19 and rTg4510 strains. Among five different PBB compounds, 2-[(1E,3E)-4-[6-(11C-methylamino)pyridin-3-yl]buta-1,3-dien-1-yl]-1,3-benzothiazol-6-ol (20, PBB3) (Maruyama et al. 2013) was found to be the best candidate, with a good tau affinity in the nanomolar range (Kd [HITP] = 2.55 nM) and selectivity for tau over Aβ (50-fold). This compound showed sufficient uptake and clearance from mouse brain, yet its biostability was relatively poor (Maruyama et al. 2013). Metabolite analysis in mouse and human plasma of 11C-PBB3 revealed a polar radioactive metabolite (Hashimoto et al. 2014). The percentage of unchanged PBB3 was <2% as early as 1 min after injection in mice, although it was relatively slower in humans (20 < 8% at 3 min postinjection). This may explain the brain's lower uptake of PBB3 (1.92% ID/g at 1 min postinjection) when compared with other radiotracers. One of the key disadvantages of PBB3 stems from the presence of this undesirable radioactive metabolite, which confounds image analysis.
Based on promising preclinical findings in PS19 and especially Tg4510 mice (Maruyama et al. 2013), 11C-PBB3 was tested in humans. It showed greater accumulation in the medial temporal region of AD patients when compared with controls. Furthermore, these early results suggested the possibility of capturing the spreading of neurofibrillary tau pathologies from the limbic system (Braak stage III/IV) to neocortical areas (Braak stage V/VI). The distribution of 11C-PBB3 in AD human brains was different from that of the amyloid tracer 11C-PiB, which showed minimal nonspecific binding to white matter, whereas 11C-PBB3 accumulated in dural venous sinuses in both control and AD brains. Additional PET scans using 11C-PBB3 with a CBD patient showed tracer retention in the basal ganglia, suggesting that this tracer may be useful for imaging non-AD tauopathies as well (Maruyama et al. 2013).
Despite these promising results for in vivo imaging of tau pathology with 11C-PBB3, issues with metabolism and chemical instability need to be addressed. PBB3 undergoes rapid interconversion of E/Z isomers in the presence of light (photoisomerization) (Hashimoto et al. 2014), which is a well-known phenomenon in stilbenes (Waldeck 1991; Lei et al. 2014). This photoisomerization can be largely suppressed by shielding the compound from light during chemical synthesis, radiosynthesis, and purification, yet this adds complexity to the imaging work flow.
As 18F is preferred as an imaging radionuclide over 11C because of its longer half-life, new fluorinated PBB compounds are currently under investigation, such as compound 21 (M Higuchi, H Shimada, N Sahara, et al., unpubl., AAIC meeting, Copenhagen, July 2014).
Siemens-Avid/Lilly Compounds
An in vitro autoradiography screening campaign of more than 800 compounds on human AD brain sections at Siemens MI Biomarker Research revealed two lead series, carbazoles and benzimidazoles. Compound optimization eventually provided two novel tracers: 7-[6-18 F-fluoropyridin-3-yl]-5H-pyrido[4,3-b]indole (22, AV-1451, 18F-T807) (Szardenings et al. 2012) and 2-[4-(2-18F-fluoroethyl)piperidin-1-yl]pyrimido[1,2-a]benzimidazole (23, AV-680, 18F-T808) (Cashion et al. 2011). As PET imaging probes, T807 and T808 satisfy all criteria for quantitative imaging of tau pathology, which include a high affinity for tau in the nanomolar range, more than 25-fold selectivity for AD-PHF over Aβ, lack of white matter binding, and a favorable pharmacokinetic profile (Table 3; Zhang et al. 2012; Xia et al. 2013).
Table 3.
Binding affinity and pharmacokinetics of T807 and T808
| Compound | Tau affinitya (nM) | Brain uptake (%ID/g) | |||
|---|---|---|---|---|---|
| Log P | AD-PHF | Selectivity tau/Aβ | 2 min | 20 min | |
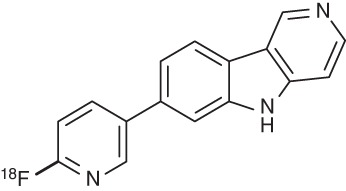 18F-T807 (AV-1451, 22) |
1.67 | Kd = 14.6 | 25 | 4.16 | 1.1 |
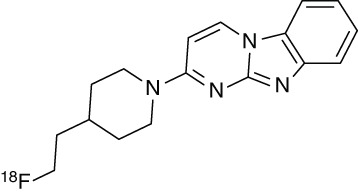 18F-T808 (AV-680, 23) |
N/A | Kd = 22 | 27 | 6.7 | 2.3 |
aBased on Scatchard plot analysis of autoradiography staining of human AD brain slices, containing PHF-Tau. Log P values are the partition coefficient (octanol/water). Selectivity tau versus Aβ: Kd (Aβ)/Kd (tau).
AD-PHF, Alzheimer’s disease paired helical filament.
Comparison of T807/AV-1451 human-brain-section autoradiography to immunostaining shows good agreement with tau pathology but not with the Aβ pattern (Fig. 6), suggesting that this tracer binding colocalizes with tau but not Aβ. A linear correlation was observed between immunostaining-based NFT loads of brain sections from 26 human donors and the T807-based autoradiography staining intensity. Such a correlation did not exist between T807 and Aβ immunostaining (Xia et al. 2013).
Figure 6.

18F-T807/AV-1451 autoradiography (middle) on Alzheimer’s disease (AD) brain sections and a comparison with paired-helical filament (PHF)-tau (left) and Aβ (right) double immunohistochemistry (IHC) (Szardenings et al. 2012). 18F-T807 colocalized with PHF-tau but not with Aβ plaques. (Top) Low magnification. (Bottom) High magnification from the framed areas. Images of PHF-tau (left) and Aβ (right) IHC double immunostaining and autoradiogram image (middle) from two adjacent sections (10 µm) from a PHF-tau-rich AD brain (frontal lobe). Fluorescent and autoradiographic images were obtained using a Fuji Film FLA-7000 imaging instrument. Scale bars, 2 mm. (From Ariza et al. 2015; reprinted, with permission, from the American Chemical Society 2015.)
Interestingly, preclinical in vivo studies with T807 and T808 using Aβ-plaque-bearing APPSWE-PS19 transgenic mice (Radde et al. 2006) showed no noticeable differences in retention between tau transgenic mice and wild-type animals, suggesting that these compounds are specific binders for human aggregated tau, but not the aggregated Aβ PHF-tau present in these mouse models.
The initial clinical PET scans of 18F-T807 with AD and mild cognitive impairment (MCI) patients and HCs showed a radiotracer accumulation in agreement with the known distribution of PHFs in the brain according to Braak (Chien et al. 2013), where increasing neocortical spread was clearly associated with severity of dementia (Fig. 7) (Mintun et al. 2013).
Figure 7.

18F-T807/AV-1451 positron emission tomography (PET) images in a healthy subject (left), mild cognitive impairment (MCI), and mild and severe Alzheimer’s disease (AD) patients. Standardized uptake value ratio (SUVR) means the target-to-cerebellum standardized uptake value. The PET data show increasing tau spread with increasing disease severity. The sensorimotor cortex and the primary visual cortex (not visible in this projection) are largely spared even in the severe case, in agreement with Braak staging (Alzforum 2013; Mintun et al. 2013). MMSE, Mini Mental State Examination; y.o., years old. (From Ariza et al. 2015; reprinted, with permission, from the American Chemical Society 2015.)
The radiotracer displays favorable kinetics with rapid brain uptake and clearance, as well as minimal nonspecific binding to white matter and cortical gray matter of HC subjects.
First-in-human PET studies of 18F-T808 showed faster kinetics than T807, matching earlier preclinical rodent PET studies. Thus, 18F-T808 images stabilized as early as 30 min after tracer injection, whereas the 18F-T807 SUVR values still changed after 80 min. The cortical retentions of T807 (Fig. 8) (Mintun et al. 2013) and T808 (Chien et al. 2014) were correlated with increasing disease severity. Additionally, postmortem human-brain analysis by tau immunostaining of one subject who died from unrelated causes a few weeks after undergoing a PET scan with 18F-T808 was consistent with the observed pattern of regional in vivo uptake, with the exception of the putamen, where increased tracer retention was not matched by the degree of NFT pathology (Kolb et al. 2013). In addition, accumulation of 18F in the skull because of metabolic defluorination of the tracer was observed in some cases, especially at late time points. For this reason, the more stable T807/AV-1451 was selected for further clinical development.
Figure 8.

18F-T807/AV-1451 target-to-cerebellum standardized uptake value ratio (SUVR). The SUVR was measured from 80 to 100 min postinjection. The severe Alzheimer’s disease (AD) subject showed the highest cortical tracer retention. The increasing neocortical spread detected in the positron emission tomography (PET) scans with increasing dementia severity agrees with the pathology-derived Braak staging system (Mintun et al. 2013). NFT, Neurofibrillary tangle; MMSE, Mini Mental State Examination; HC, healthy control. (From Ariza et al. 2015; reprinted, with permission, from the American Chemical Society 2015.)
18F-T807/AV-1451 has generated most of the early tau PET data known thus far, and a number of new results were reported at the 2015 HAI conference (Ninth Annual Human Amyloid Imaging (HAI) Conference 2015). An ongoing multicenter phase II trial has enrolled 230 cognitively normal and clinically affected subjects, some of whom will be subjected to repeat tau scans at 9 and 18 months.
Earlier pathology-based data reveal a linear correlation between the NFT burden and cognitive scores (Arriagada et al. 1992). This relationship has also been shown based on 18F-T807 PET imaging, which showed that AD subjects had more tau pathology than people with MCI, who had more than controls. In general, tau correlates more strongly than amyloid with neurodegeneration and cognitive decline in AD. Together, the data support the hypothesis that although amyloid and tau pathology may begin independently in separate regions in an aging person’s brain, the presence of amyloid intensifies and accelerates an otherwise limited tauopathy.
18F-T807 PET imaging results for 75 older subjects who participated in the Harvard Aging Brain Study, led by Keith Johnson, showed that, in early stages of dementia, tau pathology appears in the hippocampus, parahippocampus, and the entorhinal cortex (Alzforum 2015). Increased tau accumulation in the inferior temporal lobe was associated with worse memory. Elevated amyloid burden was often accompanied by increased tau pathology, but the correlation between cognitive decline and tau burden is stronger in people with amyloid. Chhatwal and Johnson (2015) were able to compare CSF tau results with 18F-T807/AV-1451 PET imaging data in 31 older, cognitively normal participants, identifying statistically significant correlations between CSF tau levels and tau imaging in six brain regions, consistent with the Braak staging criteria for tau pathology: the entorhinal and parahippocampal regions and the inferior temporal, middle temporal, and superior temporal cortices.
Test/retest reproducibility for T807/AV-1451 PET imaging, reported by Avid, was determined to be ∼4%–5% across neocortical and mesial temporal lobe structures with variability in neocortical regions, suggesting that the tracer may be useful for following longitudinal changes in tau burden (Alzforum 2015). Indeed, 18F-T807/AV-1451 scans repeated an average of 14 months apart showed an ∼10% year-over-year increase in mean cortical SUVR in six subjects with high amyloid burden, whereas two subjects with negative amyloid scans showed minor changes in their tau scans. Increased tau pathology by tau PET was accompanied by lower Mini Mental State Examination (MMSE) performance.
18F-T807/AV-1451 has also been evaluated in subjects with nontypical AD tauopathies and non-AD tauopathies (Alzforum 2015). G. Rabinovici of UCSF determined that the regional pattern of tau PET, but not amyloid PET, agrees with the regions thought to underlie the particular clinical symptoms in patients with atypical forms of AD (Ossenkoppele et al. 2016). Interestingly, brain metabolism by fluorodeoxyglucose (FDG) PET was abnormally low where tau pathology was high. A study led by Dickerson (McGinnis et al. 2015) in primary progressive aphasia patients showed that the 18F-T807/AV-1451 tau signal appeared to be spreading through the brain's language networks. Another PET imaging study conducted by G. Rabinovici in PSP patients showed that positive tau signals largely agreed with affected brain regions based on the published neuropathological staging system. Thus, in a group of 5 PSP cases and 16 controls, the suspected PSP regions (the substantia nigra, globus pallidus, and subthalamic nucleus) did have more 18F-T807/AV-1451 binding in patients, yet there was some overlap with the control scans. This might limit the ability of T807/AV-1451 to detect the early stages of PSP (Fig. 9).
Figure 9.

Voxel-wise comparison of T807 retention in five progressive supranuclear palsy (PSP) subjects versus normal controls shows increased signal in the globus pallidus, the substantia nigra, and the subthalamic nucleus in this tau disease. PSPRS, Progressive Supranuclear Palsy Rating Scale; HC, healthy controls. (Image courtesy of Gil Rabinovici and Bill Jagust.)
With the aid of a powerful autoradiography method, which provides subcellular resolution, M. Marquie at the Massachusetts General Hospital was able to show that T807 was perfectly colocalized with tau-containing neurons and with dystrophic neurites (Alzforum 2015). This finding is in agreement with standard autoradiography (Zhang et al. 2012; Xia et al. 2013) and confirms strong T807 binding to tau pathology in AD but not to cerebral amyloid, dementia in Lewy bodies (DLB), multiple system atrophy (MSA), or TDP-43. The overall conclusion from this study was that tangles and dystrophic neurites appear to account for most of the in vivo T807 signal.
There are, however, some issues that will need to be addressed with larger cohort studies and neuropathological confirmation of the imaging findings with T807/AV-1451 to fully validate and characterize this tracer. There is an apparent absence of signal in some tauopathies such as Pick's disease or CBD. In addition, several groups (including Kolb et al. 2013) reported that T807/AV-1451 and T808 generate surprisingly strong signals in parts of the brain's basal ganglia (e.g., the putamen and substantia nigra) regardless of the patient's diagnosis, suggesting that tracer binding in these particular brain regions is unrelated to tau pathology.
Roche Compounds
Hoffmann-La Roche scientists, in collaboration with PET specialists led by Dean Wong at Johns Hopkins University School of Medicine, presented in vitro and in vivo results at the 2015 HAI conference with three potential tau tracers, RO6931643, RO6924963, and RO6958948 (Alzforum 2015). Although the structures have not been disclosed, they are most likely covered by the three recently published patents (Fig. 10) (Borroni et al. 2014; Gobbi et al. 2015a,b). The compounds were shown to display high-affinity binding to tau in the sub-10-nM range, combined with strong selectivity over Aβ in human brain tissue; they displace the binding of the Siemens tau tracer T808 to aggregated tau in human-brain slices at Braak stages V/VI; and their binding is colocalized with the immunohistochemistry of aggregated tau but not Aβ. Baboon PET studies at Johns Hopkins revealed good brain permeability and washout combined with low white matter binding. A phase I trial of all three tracer candidates in HCs and AD patients at Johns Hopkins is ongoing.
Figure 10.

Structures disclosed in recent patents by Roche.
CONCLUSIONS
The development of tau PET radioligands complements earlier work on Aβ imaging because it is now possible to detect both pathological hallmarks of AD in living patients. Together, both types of tracer may enable earlier diagnosis of AD, possibly even before cognitive symptoms arise, and more accurate staging of this neurodegenerative disease and differentiation from non-AD dementia. This may pave the way for early intervention with a disease-modifying therapy, which may have a better chance of halting irreversible neurodegenerative processes. The well-established correlation between the density and neocortical spread of NFTs with memory decline in AD, in combination with the observed excellent test/retest reproducibility and the relatively large longitudinal change in tau PET imaging with 18F-T807/AV-1451, may provide a diagnostic means for monitoring disease progression or success of therapy. This may be applicable not only to therapies that target amyloid and concomitant tau pathology in AD but also, importantly, in tau-centric therapies.
Considerable progress has been made in the development of high-affinity, tau-selective ligands, and four series of tracers, T807/AV-1451, THK-5117, and analogs such as THK-5351, PBB3, and Roche’s radioligands are currently undergoing clinical evaluation. The heterogeneity of tau-protein aggregates and the fact that the binding sites are probably ill-defined (Landau et al. 2011) complicate the interpretation of binding data and classification of ligands. For instance, saturation binding studies with 18F-THK-523 suggested the presence of two different binding sites (Fodero-Tavoletti et al. 2011). Further study of the structure and binding characteristics of tau aggregates isolated from preclinical models and human tauopathies at various disease stages could be instrumental to gain a better understanding of the binding characteristics of tau ligands. To establish tau PET as a diagnostic tool, there is a need to further investigate longitudinal imaging in the context of disease progression and to compare in vivo PET results with ex vivo pathology. Additionally, the dynamic range of tracers needs to be evaluated, as well as their test/retest reproducibility and specificity. These tracers are currently being studied across various tauopathies, and resulting insights will guide the development of disease-modifying therapies, enable early intervention, and, we are hopeful, improve outcomes.
Footnotes
Editor: Stanley B. Prusiner
Additional Perspectives on Prion Biology available at www.cshperspectives.org
REFERENCES
- Alzforum. 2013. HAI—Spotlight on Tau Tracers at Human Amyloid Imaging Meeting. January 25. www.alzforum.org/news/conference-coverage/hai-spotlight-tau-tracers-human-amyloid-imaging-meeting?
- Alzforum. 2015. Human Amyloid Imaging Meeting Was Abuzz with Talk of Tau. February 6. www.alzforum.org/news/conference-coverage/human-amyloid-imaging-meeting-was-abuzz-talk-tau?
- Anand R, Dip Gill K, Ali Mahdi A. 2014. Therapeutics of Alzheimer's disease: Past, present and future. Neuropharmacology 76: 27–50. [DOI] [PubMed] [Google Scholar]
- Ariza M, Kolb HC, Moechars D, Rombouts F, Andrés JI. 2015. Tau positron emission tomography (PET) imaging: Past, present and future. J Med Chem 58: 4365–4382. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. 1992. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42: 631–639. [DOI] [PubMed] [Google Scholar]
- Bakhti M, Aggarwal S, Simons M. 2014. Myelin architecture: Zippering membranes tightly together. Cell Mol Life Sci 71: 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrio JR, Huang SC, Cole G, Satyamurthy N, Petric A, Phelps ME, Small G. 1999. PET imaging of tangles and plaques in Alzheimer disease with a highly hydrophobic probe. J Labelled Compd Radiopharm 42: S194–S195. [Google Scholar]
- Borroni E, Gobbi L, Honer M, Knust H, Koerner M, Muri D. 2014. 2-Phenyl or 2-hetarylimidazo[1,2-a]pyridine derivatives. International Patent 2014. WO2014/177458 A1.
- Braak H, Braak E. 1991. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82: 239–259. [DOI] [PubMed] [Google Scholar]
- Cashion DK, Cheng G, Kasi D, Kolb HC. 2011. Imaging agents for detecting neurological disorders. International Patent 2011. WO 2011/119565 A1.
- Chhatwal J, Johnson K. 2015. Human Amyloid Imaging Conference 2015. www.alzforum.org/print-series/390816.
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, Kolb HC. 2013. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis 34: 457–468. [DOI] [PubMed] [Google Scholar]
- Chien D, Katrin A, Bahri S, Walsh JC, Mu F, Xia C, Shankle WR, Lerner AJ, Su MY, Elizarov A, et al. 2014. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimers Dis 38: 171–184. [DOI] [PubMed] [Google Scholar]
- Citron M. 2010. Alzheimer's disease: Strategies for disease modification. Nat Rev Drug Disc 9: 387–398. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Grueninger F, Tolnay M. 2014. Intercellular transfer of tau aggregates and spreading of tau pathology: Implications for therapeutic strategies. Neuropharmacology 76: 9–15. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Marek K. 2003. Looking backward to move forward: Early detection of neurodegenerative disorders. Science 302: 830–834. [DOI] [PubMed] [Google Scholar]
- Delacourte A. 1998. Diagnosis of Alzheimer's disease. Ann Biol Clin 56: 133–142. [PubMed] [Google Scholar]
- Fargo K, Bleiler L. 2014. Alzheimer's Association report. Alzheimers Dement 10: e47–e92. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O'Keefe G, et al. 2011. 18F-THK-523: A novel in vivo tau imaging ligand for Alzheimer's disease. Brain 134: 1089–1100. [DOI] [PubMed] [Google Scholar]
- Gobbi L, Knust H, Koblet A. 2014. Imidazo[2,1]thiazol-3-one derivatives useful as diagnostic agents for Alzheimer's disease. International Patent 2014. WO2014/026881 A1. [DOI] [PMC free article] [PubMed]
- Gobbi L, Knust H, Koerner M, Muri D. 2015a. Imidazo[1,2-a]pyridin-7-amines as imaging tools. International Patent 2015. WO2015/044095 A1.
- Gobbi L, Knust H, Koerner M, Muri D. 2015b. Diazacarbazole derivatives as Tau-PET-ligands. International Patent 2015. WO2015/052105 A1.
- Harada R, Okamura N, Furumoto S, Yoshikawa T, Arai H, Yanai K, Kudo Y. 2014. Use of a benzimidazole derivative BF-188 in fluorescence multispectral imaging for selective visualization of tau protein fibrils in the Alzheimer's disease brain. Mol Imaging Biol 16: 19–27. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Kawamura K, Igarashi N, Takei M, Fujishiro T, Aihara Y, Shiomi S, Muto M, Ito T, Furutsuka K, et al. 2014. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J Nucl Med 55: 1532–1538. [DOI] [PubMed] [Google Scholar]
- Holland JP, Liang SH, Rotstein BH, Collier TL, Stephenson NA, Greguric I, Vasdev N. 2014. Alternative approaches for PET radiotracer development in Alzheimers disease: Imaging beyond plaque. J Label Compd Radiopharm 57: 323–331. [DOI] [PubMed] [Google Scholar]
- Honson NS, Johnson RL, Huang W, Inglese J, Austin CP, Kuret J. 2007. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol Dis 28: 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Beck M, Stahl T, Holzer M, Brauer K, Bigl V, Arendt T. 1999. Phylogenetic diversity of the expression of the microtubule-associated protein tau: Implications for neurodegenerative disorders. Mol Brain Res 68: 119–128. [DOI] [PubMed] [Google Scholar]
- Karran E, Mercken M, De Strooper B. 2011. The amyloid cascade hypothesis for Alzheimer's disease: An appraisal for the development of therapeutics. Nat Rev Drug Disc 10: 698–712. [DOI] [PubMed] [Google Scholar]
- Ke YD, Suchowerska AK, van der Hoven J, De Silva DM, Wu CW, van Eersel J, Ittner A, Ittner LM. 2012. Lessons from tau-deficient mice. Int J Alzheimers Dis 2012: 873270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian ZS. 1985. Diagnosis of Alzheimer's disease. Arch Neurol 42: 1097–1105. [DOI] [PubMed] [Google Scholar]
- Kolb HC, Attardo G, Mintun M, Chien D, Elizarov A, Conti P, Miller C, Joshi A, Skovronsky D. 2013. First case report: Image to autopsy correlation for tau imaging with 18F-T808 (AV-680). J Alzheimer's Assoc 9: P844–P845. [Google Scholar]
- Kung HF. 2012. The β-amyloid hypothesis in Alzheimer's disease: Seeing is believing. ACS Med Chem Lett 3: 265–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau M, Sawaya MR, Faull KF, Laganowsky A, Jiang L, Sievers SA, Liu J, Barrio JR, Eisenberg D. 2011. Towards a pharmacophore for amyloid. PLoS Biol 9: e1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Yu L, Zhou B, Zhu C, Wen Z, Lin SH. 2014. Landscapes of four-enantiomer conical intersections for photoisomerization of stilbene: CASSCF calculation. J Phys Chem A 118: 9021–9031. [DOI] [PubMed] [Google Scholar]
- Lemoine L, Saint-Aubert L, Marutle A, Antoni G, Eriksson JP, Ghetti B, Okamura N, Nennesmo I, Gillberg PG, Nordberg A. 2015. Visualization of regional tau deposits using (3)H-THK5117 in Alzheimer brain tissue. Acta Neuropathol Commun 3: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni RB, Farías G, Morales I, Navarrete L. 2010. The revitalized tau hypothesis on Alzheimer's disease. Arch Med Res 41: 226–231. [DOI] [PubMed] [Google Scholar]
- Mach RH. 2014. New targets for the development of PET tracers for imaging neurodegeneration in Alzheimer disease. J Nucl Med 55: 1221–1224. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VMY, Ono M, et al. 2013. Imaging of tau pathology in mouse model and in Alzheimer patients compared to normal controls. Neuron 79: 1094–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason NS, Mathis CA, Klunk WE. 2013. Positron emission tomography radioligands for in vivo imaging of Aβ plaques. J Label Compd Radiopharm 56: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K, Ono M, Hayashi S, Kimura H, Okamoto Y, Ihara M, Takahashi R, Moric H, Saji H. 2011. Phenyldiazenyl benzothiazole derivatives as probes for in vivo imaging of neurofibrillary tangles in Alzheimer's disease brains. Med Chem Commun 2: 596–600. [Google Scholar]
- Matsumura K, Ono M, Kimura H, Ueda M, Nakamoto Y, Togashi K, Okamoto Y, Ihara M, Takahashi R, Saji H. 2012. 18F-Labeled phenyldiazenyl benzothiazole for in vivo imaging of neurofibrillary tangles in Alzheimer's disease brains. ACS Med Chem Lett 3: 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews PM, Rabiner EA, Passchier J, Gunn RN. 2011. Positron emission tomography molecular imaging for drug development. Br J Clin Pharmacol 73: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis SM, Makaretz S, Caso C, Stepanovic M, Johnson KA, Dickerson B. 2015. Longitudinal 18F-T807 imaging in a case of nonfluent variant primary progressive aphasia. J Alzheimer's Assoc 11: P791–P792. [Google Scholar]
- Medeiros R, Baglietto-Vargas D, LaFerla FM. 2011. The role of tau in Alzheimer's disease and related disorders. CNS Neurosci Ther 17: 514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun M, Schwarz A, Joshi A, Shcherbinin S, Chien D, Elizarov A, Su MY, Shankle W, Pontecorvo M, Tauscher J, et al. 2013. Exploratory analyses of regional human brain distribution of the PET tau tracer F18-labeled T807 (AV-1541) in subjects with normal cognitive function or cognitive impairment thought to be due to AD. Alzheimers Dement 9: P842. [Google Scholar]
- Nelson LD, Siddarth P, Kepe V, Scheibel KE, Huang SC, Barrio JR, Small GW. 2011. Positron emission tomography of brain β-amyloid and tau levels in adults with Down syndrome. Arch Neurol 68: 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noёl S, Cadet S, Gras E, Hureau C. 2013. The benzazole scaffold: A SWAT to combat Alzheimer's disease. Chem Soc Rev 42: 7747–7762. [DOI] [PubMed] [Google Scholar]
- Okamura N, Suemoto T, Furumoto S, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Fujiwara H, Nemoto M, Maruyama M, et al. 2005. Quinoline and benzimidazole derivatives: Candidate probes for in vivo imaging of tau pathology in Alzheimer's disease. J Neurosci 25: 10857–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N, Furumuto S, Harada R, Tago T, Yoshikawa T, Fodero-Tavoletti M, Mulligan RS, Villemagne VL, Akatsu H, Yamamoto T, et al. 2013. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med 54: 1420–1427. [DOI] [PubMed] [Google Scholar]
- Okamura N, Harada R, Furumoto S, Arai H, Yanai K, Kudo Y. 2014a. Tau PET imaging in Alzheimer's disease. Curr Neurol Neurosci Rep 14: 500. [DOI] [PubMed] [Google Scholar]
- Okamura N, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Harada R, Yates P, Pejoska S, Kudo Y, Masters CL, Yanai K, et al. 2014b. Non-invasive assessment of Alzheimer's disease neurofibrillary pathology using 18F-THK5105 PET. Brain 137: 1762–1771. [DOI] [PubMed] [Google Scholar]
- Okamura N, Furumoto S, Harada R, Tago T, Iwata R, Tashiro M, Furukawa K, Arai H, Yanai K, Kudo Y. 2014c. Characterization of 18F-THK-5351, a novel PET tracer for imaging tau pathology in Alzheimer's disease. Eur J Nucl Med Mol Imaging 41: S260. [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, et al. 2016. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 139: 1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel M, Vernaleken I, Rösch F. 2014. Positron emission tomography in CNS drug discovery and drug monitoring. J Med Chem 57: 9232–9258. [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaeggi F, Wolburg H, Gengler S, et al. 2006. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7: 940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G, Udgaonkar JB. 2013. Mechanistic studies unravel the complexity inherent in tau aggregation leading to Alzheimer's disease and the tauopathies. Biochemistry 52: 4107–4126. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Jagust WJ. 2012. Brain imaging in the study of Alzheimer's disease. Neuroimage 61: 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, Villemagne VL. 2013. Amyloid imaging with PET in early Alzheimer disease diagnosis. Med Clin North Am 97: 377–398. [DOI] [PubMed] [Google Scholar]
- Shah M, Catafau AM. 2014. Molecular imaging insights into neurodegeneration: Focus on tau PET radiotracers. J Nucl Med 55: 871–874. [DOI] [PubMed] [Google Scholar]
- Shin J, Kepe V, Barrio JR, Small GW. 2011. The merits of FDDNP-PET imaging in Alzheimer's disease. J Alzheimers Dis 26: 135–145. [DOI] [PubMed] [Google Scholar]
- Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, et al. 2006. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med 355: 2652–2663. [DOI] [PubMed] [Google Scholar]
- Small GW, Kepe V, Siddarth P, Ercoli LM, Merrill DA, Donoghue N, Bookheimer SY, Martinez J, Omalu B, Bailes J, et al. 2013. PET scanning of brain tau in retired National Football League players: Preliminary findings. Am J Geriatr Psychiatry 21: 138–144. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr, Kaye J, Montine TJ, et al. 2011. Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging and the Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7: 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. 2013. Tau pathology and neurodegeneration. Lancet Neurol 12: 609–622. [DOI] [PubMed] [Google Scholar]
- Stephens A. 2015. Novel tau PET tracers: Characterization and development. ADPD Imaging Satellite Symposium. Nice, France. [Google Scholar]
- Szardenings AK, Kolb HC, Walsh JC, Chen G, Gangadharmath UB, Kasi D, Liu C, Sinha A, Wang E, Yu C, et al. 2012. Imaging agents for detecting neurological dysfunction. International Patent 2012. US 2012/0302755 A1.
- Taghavi A, Nasir S, Pickhardt M, Heyny-von Haussen R, Mall G, Mandelkow E, Mandelkow EM, Schmidt B. 2011. N-Benzylidene-benzohydrazides as novel and selective tau-PHF ligands. J Alzheimers Dis 27: 835–843. [DOI] [PubMed] [Google Scholar]
- Thompson PW, Ye L, Morgenstern JL, Sue L, Beach TG, Judd DJ, Shipley NJ, Libri V, Lockhart A. 2009. Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer's disease pathologies. J Neurochem 109: 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Clark CM, Schmidt ML, Arnold SE, Lee VM. 1997. Strategies for improving the postmortem neuropathological diagnosis of Alzheimer's disease. Neurobiol Aging 18: S75–S79. [DOI] [PubMed] [Google Scholar]
- Valk PE, Bailey DL, Townsend DW, Maisey MN. 2005. Positron emission tomography: Basic science. Springer, New York. [Google Scholar]
- Villemagne VL, Okamura N. 2014. In vivo tau imaging: Obstacles and progress. Alzheimers Dement 10: S254–S264. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Furumoto S, Fodero-Tavoletti M, Harada R, Mulligan RS, Kudo Y, Masters CL, Yanai K, Rowe CC, Okamura N. 2012. The challenges of tau imaging. Future Neurol 7: 409–421. [Google Scholar]
- Villemagne VL, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Hodges J, Harada R, Yates P, Piguet O, Pejoska S, Doré V, et al. 2014. In vivo evaluation of a novel tau imaging tracer for Alzheimer's disease. Eur J Nucl Med Mol Imaging 41: 816–826. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. 2015. Tau imaging: Early progress and future directions. Lancet Neurol 14: 114–124. [DOI] [PubMed] [Google Scholar]
- Waldeck DH. 1991. Photoisomerization dynamics of stilbene. Chem Rev 91: 415–436. [Google Scholar]
- Wegmann S, Jung YJ, Chinnathambi S, Mandelkow EM, Mandelkow E, Muller DJ. 2010. Human tau isoforms assemble into ribbon-like fibrils that display polymorphic structure and stability. J Biol Chem 285: 27302–27313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, et al. 2013. 18F-T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement 9: 666–676. [DOI] [PubMed] [Google Scholar]
- Zhang W, Arteaga J, Cashion DK, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, et al. 2012. A highly selective and specific PET tracer for imaging of tau pathologies. J. Alzheimers Dis 31: 601–612. [DOI] [PubMed] [Google Scholar]
- Zhou M, Wang X, Liu Z, Yu L, Hu S, Chen L, Zeng W. 2014. Advances of molecular imaging probes for the diagnosis of Alzheimer's disease. Curr Alzheimer Res 11: 221–231. [DOI] [PubMed] [Google Scholar]
- Zimmer ER, Leuzy A, Gauthier S, Rosa-Neto P. 2014. Developments in tau PET imaging. Can J Neurol Sci 41: 547–553. [DOI] [PubMed] [Google Scholar]