

Here, Lobanova et al. examine the roles of telomeres at distinct stages of murine brain development by using lineage-specific genetic ablation of TRF2, an essential component of the shelterin complex that protects chromosome ends from the DNA damage response machinery. These results suggest that telomeres are dispensable in terminally differentiated neurons and provide mechanistic insight into cognitive abnormalities associated with aberrant telomere length in humans.
Keywords: dentate gyrus, neurons, telomeres
Abstract
Telomeres have been studied extensively in peripheral tissues, but their relevance in the nervous system remains poorly understood. Here, we examine the roles of telomeres at distinct stages of murine brain development by using lineage-specific genetic ablation of TRF2, an essential component of the shelterin complex that protects chromosome ends from the DNA damage response machinery. We found that functional telomeres are required for embryonic and adult neurogenesis, but their uncapping has surprisingly no detectable consequences on terminally differentiated neurons. Conditional knockout of TRF2 in post-mitotic immature neurons had virtually no detectable effect on circuit assembly, neuronal gene expression, and the behavior of adult animals despite triggering massive end-to-end chromosome fusions across the brain. These results suggest that telomeres are dispensable in terminally differentiated neurons and provide mechanistic insight into cognitive abnormalities associated with aberrant telomere length in humans.
Telomeres are nucleoprotein structures at chromosome ends. Telomeric DNA is maintained by telomerase, which catalyzes the addition of TTAGGG repeats to counteract the erosion of terminal sequences due to the “end replication problem” (Chan and Blackburn 2004). These repeats are the unique binding site for a multiprotein complex, termed shelterin, that protects chromosome ends from the DNA damage response machinery (de Lange 2005). The shelterin complex is comprised of TRF1, TRF2, POT1, TPP1, TIN2, and RAP1, of which TRF2 plays a major role (de Lange 2005). Studies in peripheral tissues have shown that loss of TRF2 alone renders telomeres dysfunctional, resulting in end-to-end chromosome fusions, activation of TRP53, and, depending on cellular context, apoptosis or senescence (Smogorzewska et al. 2002; Jacobs and de Lange 2004; Hockemeyer et al. 2006; Wu et al. 2006; Denchi and de Lange 2007).
Genetic mutations that affect telomere elongation have been implicated in human disorders with a broad spectrum of clinical manifestations (Hoyeraal et al. 1970; Hreidarsson et al. 1988; Dokal 2000; Anderson et al. 2012; Polvi et al. 2012; Savage 2014). While characterized by cellular attrition of highly proliferative tissues such as hematopoietic and epithelial, a poorly understood feature of telomere biology disorders (TBDs) is that they affect the central nervous system (CNS). Children and adults diagnosed with TBDs suffer from intellectual disabilities, progressive decline of memory, anxiety, psychoses, and seizures (Savage 2014). Similarly, late-generation telomerase-deficient mice display reduced levels of adult neurogenesis (Ferron et al. 2004), short-term memory defects (Rolyan et al. 2011), and anxiety-like behaviors (Lee et al. 2010). However, the underlying mechanisms are unclear, since telomere dysfunction arises stochastically in multiple organs and cell types.
While gradual shortening of terminal sequences occurs during DNA replication, several lines of evidence support the notion that telomeres undergo acute erosion in post-mitotic neurons. For example, TTAGGG repeats are prone to oxidative damage (8-oxodG), reactive oxygen species (ROS)-induced DNA breaks, and other exogenous signals that trigger DNA lesions by replication-independent mechanisms (Oikawa 2005; Fumagalli et al. 2012; Cardin et al. 2013; Rossiello et al. 2014). Therefore, it is conceivable that, with age, the long-lived neuronal cells progressively accumulate telomere dysfunction. This could explain some of the progressive onset of cognitive defects associated with age. However, the physiological roles of telomeres in post-mitotic neurons are largely unknown.
Here we show that, whereas functional telomeres are required for embryonic and adult neurogenesis, acute telomere dysfunction in post-mitotic neurons has virtually no detectable consequences on circuit development, neuronal gene expression, or animal behavior. These surprising results indicate that telomeres are dispensable in terminally differentiated neurons and reveal clues to cognitive abnormalities of TBD patients.
Results
Uncapping of telomeres in neural stem cells (NSCs) and Doublecortin (Dcx)-positive progenitors has different consequences on brain development
To investigate the role of telomeres in the brain, we examined the outcomes of conditional knockout (cKO) of TRF2 at different stages of neuronal differentiation (Fig. 1A). First, we crossed the “floxed” mouse Terf2 allele (Terf2F/F) (Fig. 1B; Celli and de Lange 2005) with a strain that expresses Cre recombinase under the control of the Nestin promoter (Nes:Cre) (Tronche et al. 1999) to induce uncapping of chromosome ends in proliferating NSCs. These experiments were prompted by a recent report demonstrating that NSC-specific ablation of another telomere-associated protein, POT1a, had an unexpectedly mild effect on murine brain development. POT1a cKOs had a shorter life span but nonetheless lived into adulthood and only exhibited a loss of interneurons and granule cell precursors in the cerebellum (Lee et al. 2014). We reasoned that the ability of POT1a-deficient NCSs to produce neurons in other brain regions is attributed to limited contribution of this protein to telomere protection instead of stochastic and/or spatially restricted induction of DNA damage response. Indeed, we found that all Terf2F/F/Nes:Cre homozygotes died in utero during gestation (Fig. 1C). Histological analyses of mutant embryos revealed a strong up-regulation of DNA damage signaling and apoptosis in their brains, indicating that functional telomeres are essential for early stages of brain development (Supplemental Fig. S1A).
Figure 1.
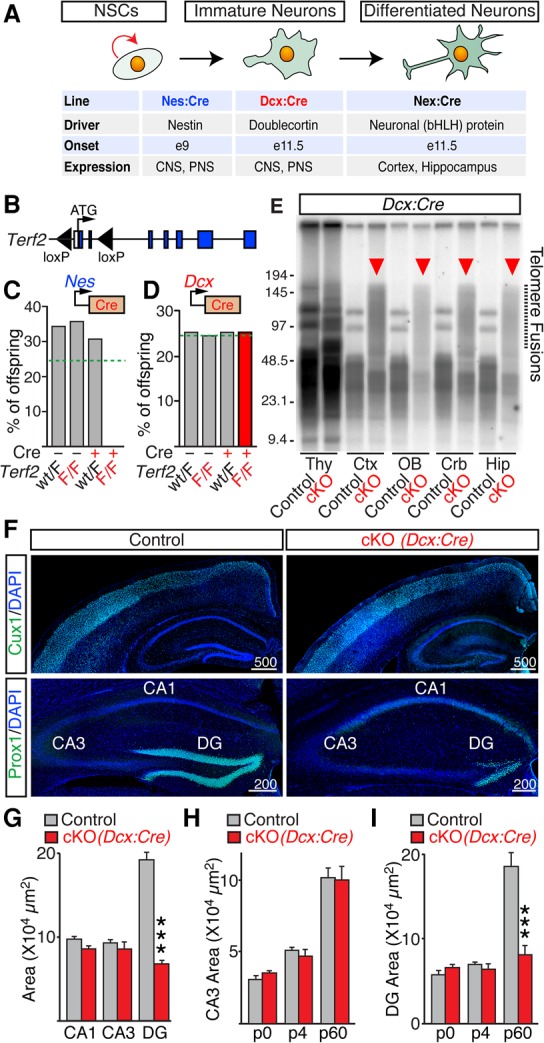
Ablation of TRF2 in NSCs and Dcx-positive neural progenitors has different consequences on animal survival and brain anatomy. (A) Cellular specificity and developmental onsets of Cre-mediated recombination in the Nes:Cre, Dcx:Cre, and Nex:Cre lines. (PNS) Peripheral nervous system. (B) Schematic diagram of the Terf2F/F allele. (C) Percentages of surviving offspring (n = 163 mice) derived from Terf2F/F and Terf2wt/F/Nes:Cre founders. The dashed line marks expected Mendelian ratios. (D) Percentages of surviving offspring (n = 248) derived from Terf2F/F and Terf2wt/F/Dcx:Cre founders. The dashed line marks expected Mendelian ratios. (E–G) Homozygous Terf2F/F/Dcx:Cre mutants (cKO) and their control Cre-negative littermates were analyzed at postnatal day 60 (P60). (E) Genomic DNA was isolated from the thymus (Thy), cortex (Ctx), olfactory bulb (OB), cerebellum (Crb), and hippocampus (Hip); digested with MboI; separated by gel electrophoresis; and hybridized with a radioactive probe for telomeric TTAGGG repeats. Note the abundant fragments of high molecular weight in cKO brains, which reflect end-to-end chromosome fusions. (F) Coronal brain sections were labeled with DAPI and antibodies to principal neurons in superficial cortical layers II/III (Cux1) or granule cells (GCs) in the dentate gyrus (DG; Prox1). Representative confocal images are shown. (G) Averaged areas of hippocampal CA1, CA3, and DG, measured in DAPI-stained sections. n = 5 mice per genotype. (H,I) Averaged areas of CA3 (H) and the DG (I) at different stages of postnatal development. n ≥ 3 mice per genotype. All graphs are plotted as mean ± SD. (***) P < 0.001, Student's t-test.
We then knocked out TRF2 in post-mitotic Dcx-positive neural progenitors. This was accomplished by crossing the Terf2F/F allele with another well-characterized line, Dcx:Cre, which drives widespread recombination in precursors of excitatory neurons in the telencephalon and Purkinje cells in the cerebellum (Zhang et al. 2010; Harris et al. 2014). Terf2F/F/Dcx:Cre cKOs survived, had a normal life span, and appeared indistinguishable from control littermates in the standard laboratory environment (Fig. 1D; data not shown). In agreement with the established role of TRF2 in telomere protection (van Steensel et al. 1998; Celli and de Lange 2005; Denchi and de Lange 2007), ablation of TRF2 (Supplemental Fig. S1B) resulted in high levels of end-to-end chromosome fusions (Fig. 1E). Despite this strong chromosomal abnormality, adult Terf2F/F/Dcx:Cre cKOs had undistorted anatomies of the cerebral cortex, areas CA1/CA3 of the hippocampus, and cerebellar hemispheres (Fig. 1F; Supplemental Fig. S1C). However, their dentate gyrus (DG) was drastically smaller (Fig. 1F,G; Supplemental Fig. S1C). The disproportional shrinkage of the DG was due to reduction in the number of principal granule cell (GC) neurons along the lateral axes, as evidenced by immunofluorescent labeling of GC-specific transcription factor Prox1 at postnatal day 60 (P60) (Fig. 1F). In contrast, staining for ubiquitous and cell type-specific neuronal markers showed no apparent changes in the lamination of other brain regions (Fig. 1F,G; data not shown). To define the onset of GC loss during development, we examined Terf2F/F/Dcx:Cre mice shortly after birth. Unlike adults, P0 and P4 cKO pups had unaltered distribution of Prox1-immunoreactive cells and overall morphology of the DG, suggesting that differentiation of the initial pool of their GCs was preserved (Fig. 1H,I; Supplemental Fig. S1D).
Functional telomeres are essential for adult neurogenesis
Since embryonically born GCs are replaced gradually over the course of postnatal life (Kuhn et al. 1996; Aimone et al. 2014), the observed distortion of the DG in Terf2F/F/Dcx:Cre cKOs may reflect a selective impairment of adult-born neurons that transiently express Dcx and therefore become TRF2-deficient prior to exiting the cell cycle. Alternatively, this phenotype may be associated with degeneration of mature GCs due to their unusually high susceptibility to telomere dysfunction. To distinguish between these scenarios, we monitored cellular proliferation using in vivo labeling with bromodeoxyuridine (BrdU), a synthetic analog of thymidine that incorporates into replicating DNA (Kuhn et al. 1996). Brains were isolated, sectioned, and imaged 48 h following BrdU injection (Fig. 2A; Supplemental Fig. S2A). Whereas cKOs had abundant BrdU-positive cells in the DG at P0 and P4, no detectable nucleoside incorporation occurred in adults (Fig. 2A,C). Consistent with these results, immunostaining with an antibody against Dcx demonstrated that P60 cKOs completely lacked dentate progenitors (Fig. 2B,E; Supplemental Fig. S2B).
Figure 2.
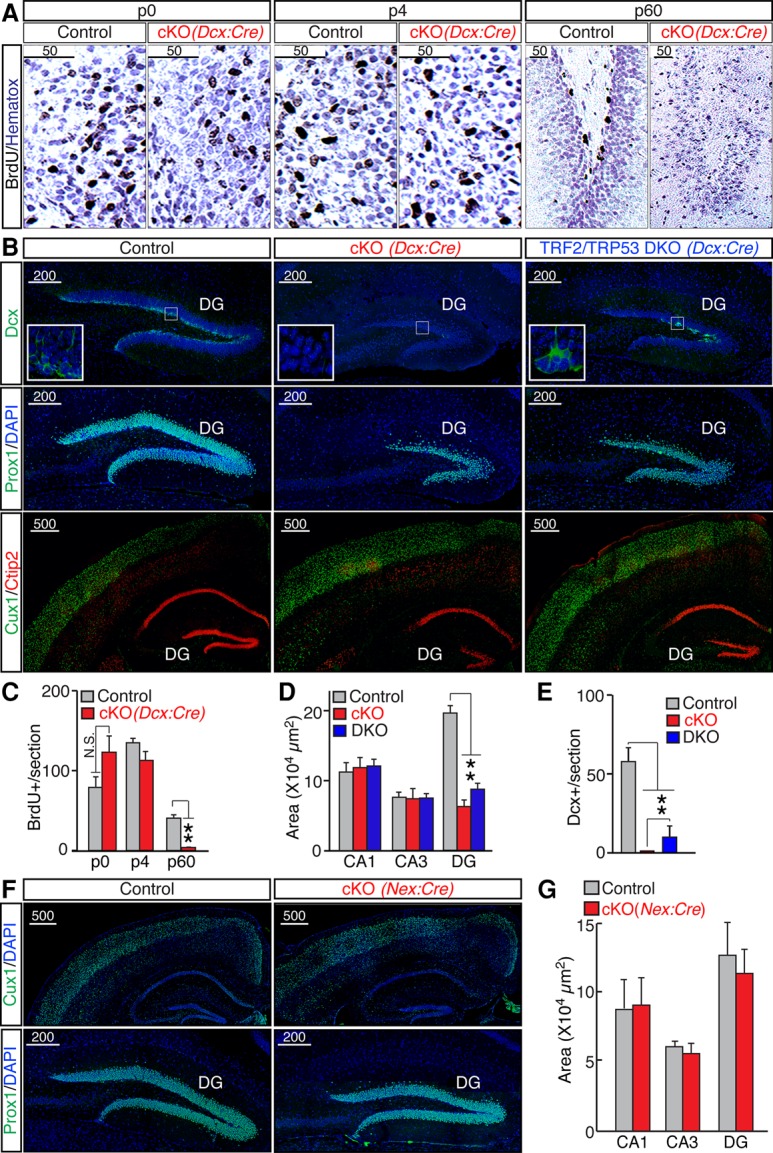
TRF2 depletion inhibits adult neurogenesis. (A) Control and Terf2F/F/Dcx:Cre cKO mice were analyzed at the indicated developmental stages 2 d after in vivo injections of BrdU. BrdU-positive cells were identified by immunohistochemistry using an antibody against BrdU. High-magnification images of the DG are shown. (B) Brain sections from P60 Terf2F/F/Dcx:Cre cKO and Terf2F/F/Trp53F/F/Dcx:Cre double-knockout (DKO) mice were imaged after labeling with the indicated antibodies. (Top row) Dcx-positive progenitors in the DG. (Middle and bottom rows) Distribution of differentiated Prox1-, Cux1-, and Ctip2-expressing neurons. (C) Averaged densities of BrdU-positive cells in the DG of control and Terf2F/F/Dcx:Cre cKO mice of the indicated ages. n ≥ 3 mice per genotype. (D) Sizes of CA1, CA3, and the DG in P60 control, cKO, and double-knockout mice, plotted as averaged areas. n = 5 mice per genotype for wild-type and cKO; n = 3 for double knockout. (E) Averaged densities of Dcx-positive cells in the DG. (F,G) TRF2 was knocked out in post-mitotic immature excitatory forebrain neurons using the Nex:Cre line. Terf2F/F/Nex:Cre mutants (cKO) and control littermates were examined at P60. (F) Representative confocal images of brain sections labeled with DAPI and either Cux1 or Prox1. (G) Averaged sizes of CA1, CA3, and the DG. n = 4 mice per genotype. All graphs are plotted as mean ± SD. (***) P < 0.001; (**) P < 0.01; (*) P < 0.05, Student's t-test.
To test whether TRF2 depletion affects adult neurogenesis through the induction of p53, which plays a key role in mediating cycle arrest and apoptosis in response to telomere dysfunction (Smogorzewska and de Lange 2002), we generated compound Terf2F/F/Trp53F/F/Dcx:Cre double-knockout mice. Double knockouts display a partial rescue of both DG size (Fig. 2B,D) and the Dcx-positive progenitor pool (Fig. 2B,E), suggesting that the loss of GCs upon TRF2 depletion involves both p53-dependent and p53-independent mechanisms (Fig. 2B; Supplemental Fig. S2B).
Next, we explored the possibility that TRF2 is uniquely required for the survival or differentiation of dentate GCs. To this end, we depleted TRF2 in post-mitotic cortical and hippocampal neurons using the well-established Nex:Cre driver line (Goebbels et al. 2006). In contrast to Terf2F/F/Dcx:Cre mice, adult Terf2F/F/Nex:Cre cKOs had intact DGs (Fig. 2F,G) despite efficient recombination of “floxed” alleles in GCs (Supplemental Fig. S2C).
Functional telomeres are dispensable for morphological and functional maturation of post-mitotic neurons
While our findings imply that functional telomeres are not required for migration and survival of post-mitotic neurons, their relevance in mature circuits is unclear. How does telomere dysfunction impact the neuronal morphologies, wiring, gene expression, and capacity of a given circuit to process information? To address these questions, we first evaluated the structures and connectivity of persistent GCs in Terf2F/F/Dcx:Cre cKOs mice. To visualize dendrites and long-range projections, sections from P60 animals were stained with an antibody against calbindin. In parallel, we injected juveniles with adeno-associated virus (AAV) that encodes a Cre-inducible version of membrane-bound GFP (AAV2.2 DIO-mGFP). This previously described technique permitted selective labeling and subsequent tracing of fully differentiated GCs in adult mice due to restricted penetrance of AAV2.2 (Burger et al. 2004; Pieraut et al. 2014). Inspection of calbindin-positive and genetically tagged neurons showed that, similar to intact GCs, TRF2-deficient cells developed extensive dendritic trees and projected axons to downstream area CA3 (Fig. 3A,B; Supplemental Fig. S3A). Moreover, these neurons formed characteristic dendritic spines and large presynaptic mossy fiber terminals (LMTs) (Fig. 3B; Supplemental Fig. S3B–E). Quantitative analyses of single cells in three-dimensional (3D) image stacks revealed no significant differences in dendrite complexity, spine density, and axonal densities of LMTs (Fig. 3C).
Figure 3.
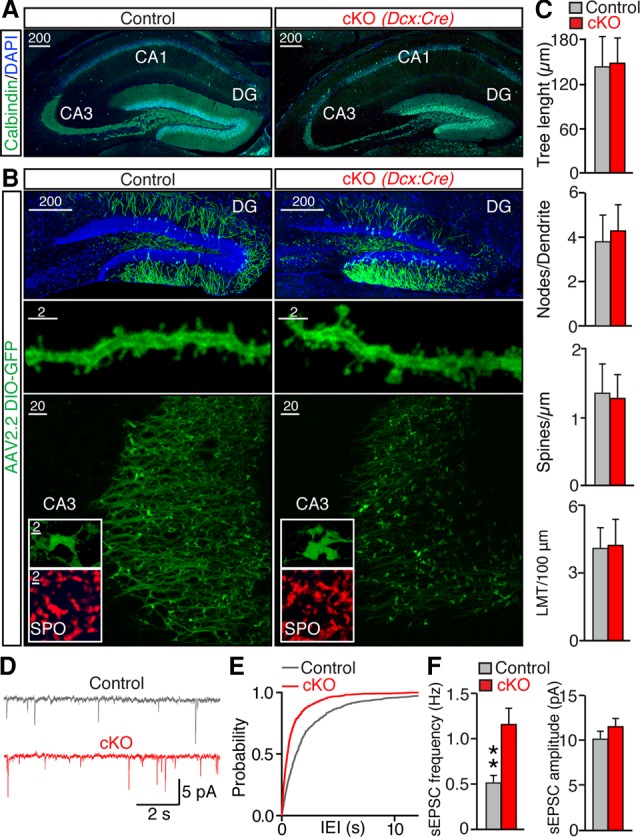
Functional telomeres are not required for development of post-mitotic neurons. (A) Representative images of brain sections labeled with DAPI and the marker of GCs, calbindin. (B,C) Animals were injected in vivo with AAV to achieve selective Cre-inducible expression of membrane-bound GFP in mature GCs (AAV2.2 DIO-mGFP). (B) Representative images of GFP-tagged neurons in the DG (top row), fragments of their dendrites (middle row), and axons with LMTs in the CA3 (bottom row). The inserts show LMTs stained with an antibody to GC-specific presynaptic marker [synaptoporin (SPO)] and GFP-labeled LMTs. (C) Quantitative analyses of axonal and dendritic morphogenesis. Graphs represent averaged dendrite length (tree length) (n = 2 mice/18 neurons for control; n = 3 mice/19 neurons for knockout), numbers of branch nodes (nodes per dendrite) (n = 2 mice/18 neurons for control; n = 3 mice/19 neurons for knockout), linear densities of postsynaptic spines (spines per micrometer) (n = 3 mice/17 neurons/1030 spines for control; n = 4 mice/25 neurons/1063 spines for cKO), and densities of LMTs per axon length (LMTs per 100 μm) (n = 2 mice/42 axons for control; n = 4 mice/60 axons for cKO) in the CA3. (D–F) Spontaneous excitatory postsynaptic currents (sEPSCs) were monitored from GCs in acute brain slices. Holding potential was −70 mV. GABA receptors were blocked with 100 µM picrotoxon. (D) Samples of sEPSCs from GCs of control and cKO mice. (E) Cumulative probability plot of interevent intervals (IEIs). (F) Mean sEPSC frequencies and amplitudes. n = 4 mice/10 neurons for control; n = 3 mice/12 neurons for cKO. All graphs are plotted as mean ± SD. (**) P < 0.01, Student's t-test.
Having established that TRF2-deficient neurons resemble normal neurons at a structural level, we examined their synapses using electrophysiological readouts. Whole-cell recordings from acute brain slices demonstrated that GCs of cKO mice had a twofold higher frequency of spontaneous excitatory postsynaptic currents (sEPSCs) (Fig. 3D–F). This increase in synaptic strength could represent homeostatic plasticity that compensates for cell loss. Nonetheless, we observed no differences in EPSC kinetics and amplitudes (Fig. 3D–F). These results suggest that TRF2 depletion in post-mitotic GCs has no detrimental effect on their morphological and likely functional maturation.
Telomere uncapping and chromosome fusion have a negligible effect on neuronal gene expression
Considering that depletion of TRF2 results in massive levels of end-to-end chromosome fusions (Fig. 1E; Celli and de Lange 2005), one could predict that TRF2-deficient neurons might undergo global changes in gene expression. Moreover, studies in mouse testes and human cell lines have proposed that TRF2 influences transcription by interacting with interstitial chromosomal sites (Biroccio et al. 2006; Simonet et al. 2011; Yang et al. 2011; El Mai et al. 2014; Martianov et al. 2016). We therefore performed genome-wide analysis of mRNA levels in the cerebral cortex and hippocampus of P60 Terf2F/F/Dcx:Cre mice. RNA deep sequencing (RNA-seq) revealed that <0.3% of detectable transcripts (n = 24,374) were significantly different between TRF2-deficient animals and littermate controls (Fig. 4A,B; Supplemental Table S2). As expected, this group included Terf2 and genes that are highly expressed in the DG, such as Prox1 (Fig. 4A,B; Supplemental Table S2). Down-regulation of GC-specific genes most likely reflects neuron loss in the DG of cKO mice rather that transcriptional repression. The remaining hits appeared to be false positives, since they could not be confirmed using quantitative real-time PCR (Supplemental Fig. S4A,B). Taken together, these results indicate that telomere dysfunction and chromosome fusion have a negligible (if any) effect on neuronal gene expression.
Figure 4.
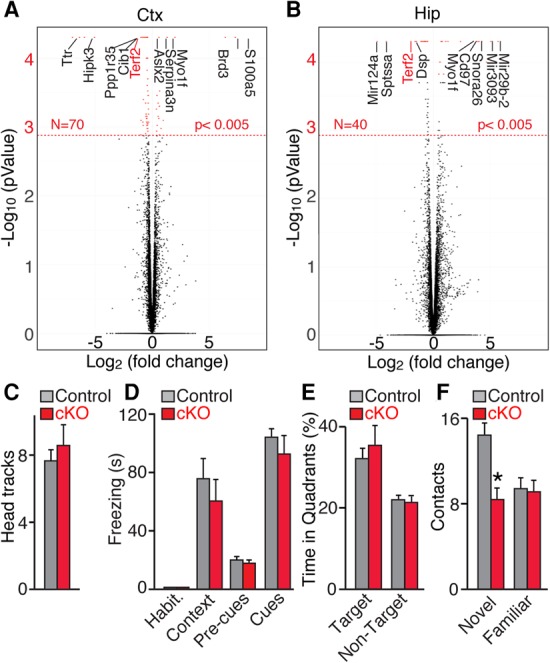
Consequences of telomere erosion on neuronal gene expression and animal behavior. Control and Terf2F/F/Dcx:Cre cKO mice were examined at P60. n = 4 mice per genotype. (A,B) Genome-wide analyses of transcription (RNA-seq) in the cerebral cortex (Ctx; A) and hippocampus (Hip; B). Volcano plots show changes in mRNA levels in cKOs relative to controls. The dashed line marks genes with P < 0.005. (C–F) Quantitative analyses of sensory processing, learning, and memory. n = 16 mice per genotype for all behavioral trials. Graphs are plotted as mean ± SD. (*) P < 0.05, Student's t-test. (C) Vision was assessed as number of head tracks in a stationary platform surrounded by a rotating drum with striped walls. (D) Associative learning and memory expressed as freezing after contextual and cued fear conditioning. (E) Spatial learning and memory were tested in a Barnes maze. The graphs show averaged times spent in correct and other targets of the maze during the probe test. (F) Recognition memory was measured as number of contacts with novel and familiar objects.
Behavior of mice with dysfunctional telomeres
To further evaluate the role of telomeres in neural circuit development and function, we systematically analyzed the behavior of adult TRF2-deficient mice. Series of trials were designed to assess the ability of animals to process sensory information and perform various cortical- and hippocampal-dependent tasks. Both homozygous Terf2F/F/Dcx:Cre and Terf2F/F/Nex:Cre mutants had unaltered vision, hearing, anxiety, and locomotion in open fields (Fig. 4C; Supplemental Fig. S4C,D). Moreover, Terf2F/F/Dcx:Cre mutants showed no detectable changes in associative and spatial memory, as evidenced by quantitative analysis of freezing in a Pavlovian fear-conditioning paradigm and escape times in a Barnes maze probe test, respectively (Fig. 4D,E; data not shown). Notably, Terf2F/F/Dcx:Cre cKOs were unable to distinguish between familiar and novel objects (Fig. 4F), consistent with our analyses of DG anatomy and the previously established role of adult-born GCs in pattern separation (McHugh et al. 2007; Clelland et al. 2009; Nakashiba et al. 2012). Indeed, this phenotype was likely due to loss of the Dcx progenitor pool that gives rise to new dentate GCs rather than the aberrant wiring and/or activity of established circuits in other brain regions, since recognition memory was preserved in Terf2F/F/Nex:Cre mice (Supplemental Fig. S4E).
Discussion
Here, we report genetic analyses of telomere function in the mammalian brain. Through lineage-specific ablation of TRF2, a protein that protects chromosome ends from the DNA damage response machinery (Denchi 2009), we demonstrate that telomeres play contrasting roles in proliferating stem cells and terminally differentiated neurons.
Requirement of telomeres for embryonic and adult neurogenesis
Gradual replication-dependent loss of TTAGGG repeats as well as acute telomere dysfunction have been shown to be detrimental to various dividing nonneuronal cell types (Lee et al. 1998; Chin et al. 1999; Rudolph et al. 1999; Jacobs and de Lange 2004; Celli and de Lange 2005; Zhang et al. 2006; Minty et al. 2008; Martinez et al. 2014). Our observations that Terf2F/F/Nes:Cre cKOs die in utero and exhibit massive cell death across the telencephalon indicate that telomere dysfunction impacts the neurogenesis of a broad spectrum of neuron classes in the developing brain. In addition, we present several lines of evidence that progressive shrinkage of the DG in animals lacking TRF2 in Dcx-positive progenitors is associated with the nearly complete absence of replenishment of the initial GC pool with new neurons. Normal differentiation of GCs and other neuron subtypes in Terf2F/F/Dcx:Cre cKO embryos and neonates can be explained by differences in the timing of cell cycle arrest relative to the onset of induction of Dcx that drives Cre (Harris et al. 2014). However, it is also conceivable that telomere dysfunction selectively induces apoptosis of differentiating adult-born GCs independently of cell cycle progression because these neurons undergo prolonged maturation (Overstreet-Wadiche et al. 2006).
Functional telomeres are dispensable in post-mitotic neurons
While the necessity of intact telomeres for embryonic and adult neurogenesis is reconcilable with prior studies in cells from somatic tissues that proliferate in vitro and in vivo, the outcomes of telomere dysfunction in post-mitotic neurons are truly remarkable. We cannot exclude that a subset of post-mitotic neurons, beyond the analysis carried out in this study, might be affected by TRF2 depletion. Nevertheless, our experiments with Terf2F/F/Dcx:Cre and Terf2F/F/Nex:Cre cKO mice demonstrate that terminally differentiated neurons with fused telomeres survive, appropriately migrate to target zones, form circuitry, and exhibit virtually no significant changes in gene expression. The latter is particularly surprising, considering that depletion of TRF2 induces a cellular response that recognizes chromosome ends as sites of DNA damage. In addition, TRF2 has been implicated in transcriptional regulation through interaction with interstitial loci. For instance, in testes whose transcriptional programs partially overlap with neurons (this phenomenon has been appreciated since early discoveries of synaptic proteins), TRF2 associates with promoters in a complex with RNA polymerase II (Martianov et al. 2016). Moreover, TRF2 is believed to interact with REST, which represses neuronal genes (Ballas and Mandel 2005; Zhang et al. 2008). Nevertheless, our deep sequencing data are at odds with the notion that neuronal TRF2 acts in pathways other than telomere protection. Viewed in a broader scope, our results support the model that spatial distribution of chromosomes within neuronal nuclei is negligible. Indeed, one could anticipate dramatic repercussions on derepression and activity-dependent control of transcriptional programs essential for circuit development and function if chromosome fusions presented topological constraints for intrinsic and experience-driven remodeling of chromatin. However, we found that irreversible uncapping and fusion of neuronal chromosomes during embryogenesis does not interfere with processing of sensory information and memory acquisition/recall in adult mice (with the exception of DG-dependent object discrimination in Terf2F/F/Dcx:Cre cKOs).
Other implications of our work
Apart from elucidating the basic biological roles of telomeres in the nervous system, our work reveals important clues to cognitive abnormalities of human subjects diagnosed with TBDs and explains the variability in the onset of their memory loss. We postulate that inherited TBDs may broadly influence the wiring of the developing brain, whereas negative consequences of acute damage and/or uncapping of terminal TTAGGG repeats in adults should be largely restricted to dentate and olfactory circuits. Given the susceptibility of adult neurogenesis to DNA damage response elicited by telomere dysfunction, our results also provide insight into neurological side effects of platinum-based chemotherapeutic drugs (Mizumatsu et al. 2003; Duffner 2006). Indeed, these treatments induce, similarly to telomere dysfunction, DNA damage activation. Therefore, we speculate that some of the side effects associated with these treatments could be caused by the selective impairment of adult neurogenesis and depletion of GCs in the DG. In agreement with this notion, irradiation of mice leads to a significant reduction in adult neurogenesis (Clelland et al. 2009).
Finally, the phenotypes of TRF2-deficient mice raise questions related to fundamental principles of information processing in the brain. It is noteworthy that Terf2F/F/Dcx:Cre cKOs have preserved spatial and associative memory despite pronounced loss of dentate GCs. Considering the well-established role of the hippocampus in these cognitive tasks (Burgess et al. 2002; Strange et al. 2014; Tonegawa et al. 2015), imaging and functional analyses of active neuronal ensembles in the DG of Terf2F/F/Dcx:Cre mouse model may facilitate further understanding of the cellular mechanisms of learning and memory storage.
Materials and methods
Mice
Nes:Cre, Dcx:Cre, Nex:Cre, Terf2F/F, Trp53F/F, and Ai9 reporter alleles were described previously (Tronche et al. 1999; Marino et al. 2000; Celli and de Lange 2005; Goebbels et al. 2006; Madisen et al. 2010; Harris et al. 2014). Mice carrying these alleles were mated to produce cKO lines, housed, and analyzed according to protocols approved by the Institutional Animal Care and Use Committee.
Immunohistochemistry
Anesthetized mice were perfused with PFA. Brains were incubated overnight in PFA and sliced on a vibratome (Leica Biosystems). Coronal sections (Bregma −1.4 to −2.5) were washed in PBS; blocked in a buffer containing 4% BSA, 2% donkey serum, and 0.2% Triton; and incubated with primary and corresponding fluorescently labeled secondary antibodies. Samples were washed and mounted on glass slides for imaging.
Confocal imaging
Samples were imaged under a Nikon A1 confocal microscope with 10×, 20×, 40×, or 60× objectives. Image data analysis was performed with Nikon Elements, FIJI, and Adobe Photoshop software packages using the Allen Brain Atlas as a reference (http://mouse.brain-map.org/static/atlas). Digital manipulations were applied equally to all pixels. 3D images of single virally labeled neurons were collected at 0.2-µm Z intervals and reconstructed in Neurolucida, as we described previously (Pieraut et al. 2014).
Virus production and injection
AAVs were packaged in-house into serotype 2.2 using published protocols (McClure et al. 2011; Pieraut et al. 2014). P1 pups were injected with 0.5 µL of viral stocks via glass micropipette (10-µm tip diameter) and analyzed at P25.
Electrophysiology
Synaptic currents were monitored in acute hippocampal slices. Currents were sampled at 10 Hz in whole-cell mode using a Multiclamp 700B amplifier (Molecular Devices, Inc.). Data were analyzed offline with pClamp10 (Molecular Devices, Inc.) and Origin8 (OriginLab) software packages.
RNA-seq
Libraries were constructed with Next Ultra RNA library preparation kit (New England Biolabs). Paired-end 100-base reads were generated at The Scripps Research Institute Next-Generation Sequencing Core on an Illumina NextSeq platform. Demultiplexed sequences were mapped to the mouse reference genome with TopHat. Aligned reads were used for final transcriptome assembly in Cufflinks and Cuffmerge. Quantitative analyses of gene expression were performed with Cuffdiff (Trapnell et al. 2013). These can be found under Gene Expression Omnibus accession number GSE95056.
Behavioral trials
Behavioral trials were carried out at The Scripps Research Institute Mouse Behavioral Assessment Core. Vision was tested by measuring the numbers of head tracks in a stationary platform surrounded by a rotating drum with black and white striped walls. Hearing was tested as a startle response to 90- to 120-dB pulses on a 70-dB background level. Spatial memory was examined in a Barnes maze (Barnes 1979). Mice were subjected to four training sessions to allow memorization of the correct path to the escape tunnel. Memory was measured as time spent in each quadrant of the maze when the escape tunnel was removed. For Pavlovian fear conditioning, animals were exposed to white noise paired with foot shocks (0.6 mA). Contextual and cued (tone) fear memory was examined in training and alternative boxes, respectively. Freezing was determined in 0.75-sec bouts and expressed as percentage of time in the context or tone presentation. Recognition memory was assessed by measuring numbers of contacts with familiar and novel objects in the same environment.
Statistics
Means and standard errors were calculated in Excel and Origin8. P-values were determined with Student's t-test. All quantitative analyses of statistics are summarized in Supplemental Table S1.
Supplementary Material
Acknowledgments
We thank Dr. Ulrich Mueller and Dr. Titia de Lange for providing mouse strains and antibodies, members of the Lazzerini Denchi and Maximov laboratories for stimulating discussions, and Dr. Kathy Spencer for technical assistance with confocal microscopy. This study was supported by the Pew Charitable Trust (E.L.D.) and National Institutes of Health grants AG038677 (to E.L.D.) and R01NS087026 (to A.M.). E.L.D., A.L., and A.M. conceived the study. R.S., C.C., and A.L. generated mutant mice and performed imaging and genomic analyses. S.P. assisted A.L. with confocal microscopy and neuron tracing and carried out electrophysiological recordings of synaptic transmission. E.L.D. and A.M. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.295402.116.
References
- Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, Gage FH. 2014. Regulation and function of adult neurogenesis: from genes to cognition. Physiol Rev 94: 991–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O'Sullivan J, et al. 2012. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nature Genetics 44: 338–342. [DOI] [PubMed] [Google Scholar]
- Ballas N, Mandel G. 2005. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol 15: 500–506. [DOI] [PubMed] [Google Scholar]
- Barnes CA. 1979. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93: 74–104. [DOI] [PubMed] [Google Scholar]
- Biroccio A, Rizzo A, Elli R, Koering C-E, Belleville A, Benassi B, Leonetti C, Stevens MFG, D'Incalci M, Zupi G, et al. 2006. TRF2 inhibition triggers apoptosis and reduces tumourigenicity of human melanoma cells. Eur J Cancer 42: 1881–1888. [DOI] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N. 2004. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther 10: 302–317. [DOI] [PubMed] [Google Scholar]
- Burgess N, Maguire EA, O'Keefe J. 2002. The human hippocampus and spatial and episodic memory. Neuron 35: 625–641. [DOI] [PubMed] [Google Scholar]
- Cardin R, Piciocchi M, Tieppo C, Maddalo G, Zaninotto G, Mescoli C, Rugge M, Farinati F. 2013. Oxidative DNA damage in Barrett mucosa: correlation with telomeric dysfunction and p53 mutation. Ann Surg Oncol 20: S583–S589. [DOI] [PubMed] [Google Scholar]
- Celli GB, de Lange T. 2005. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7: 712–718. [DOI] [PubMed] [Google Scholar]
- Chan SR, Blackburn EH. 2004. Telomeres and telomerase. Philos Trans R Soc Lond B Biol Sci 359: 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. 1999. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97: 527–538. [DOI] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD Jr, Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, et al. 2009. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 325: 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. 2005. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19: 2100–2110. [DOI] [PubMed] [Google Scholar]
- Denchi E. 2009. Give me a break: how telomeres suppress the DNA damage response. DNA Repair 8: 1118–1126. [DOI] [PubMed] [Google Scholar]
- Denchi E, de Lange T. 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448: 1068–1071. [DOI] [PubMed] [Google Scholar]
- Dokal I. 2000. Dyskeratosis congenita in all its forms. Br J Haematol 110: 768–779. [DOI] [PubMed] [Google Scholar]
- Duffner PK. 2006. The long term effects of chemotherapy on the central nervous system. J Biol 5: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Mai M, Wagner KD, Michiels JF, Ambrosetti D, Borderie A, Destree S, Renault V, Djerbi N, Giraud-Panis MJ, Gilson E, et al. 2014. The telomeric protein TRF2 regulates angiogenesis by binding and activating the PDGFRβ promoter. Cell Rep 9: 1047–1060. [DOI] [PubMed] [Google Scholar]
- Ferron S, Mira H, Franco S, Cano-Jaimez M, Bellmunt E, Ramirez C, Farinas I, Blasco MA. 2004. Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 131: 4059–4070. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Rosiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, et al. 2012. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14: 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. 2006. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44: 611–621. [DOI] [PubMed] [Google Scholar]
- Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, Bohn P, Mortrud M, Ouellette B, Kidney J, et al. 2014. Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front Neural Circuits 8: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Daniels JP, Takai H, de Lange T. 2006. Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. Cell 126: 63–77. [DOI] [PubMed] [Google Scholar]
- Hoyeraal HM, Lamvik J, Moe PJ. 1970. Congenital hypoplastic thrombocytopenia and cerebral malformations in two brothers. Acta Paediatr Scand 59: 185–191. [DOI] [PubMed] [Google Scholar]
- Hreidarsson S, Kristjansson K, Johannesson G, Johannsson JH. 1988. A syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta Paediatrica 77: 773–775. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, de Lange T. 2004. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol 14: 2302–2308. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. 1996. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 16: 2027–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW II, Greider CW, DePinho RA. 1998. Essential role of mouse telomerase in highly proliferative organs. Nature 392: 569–574. [DOI] [PubMed] [Google Scholar]
- Lee J, Jo YS, Sung YH, Hwang IK, Kim H, Kim SY, Yi SS, Choi JS, Sun W, Seong JK, et al. 2010. Telomerase deficiency affects normal brain functions in mice. Neurochem Res 35: 211–218. [DOI] [PubMed] [Google Scholar]
- Lee Y, Brown EJ, Chang S, McKinnon PJ. 2014. Pot1a prevents telomere dysfunction and ATM-dependent neuronal loss. J Neurosci 34: 7836–7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. 2000. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14: 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Martianov I, Velt A, Davidson G, Choukrallah MA, Davidson I. 2016. TRF2 is recruited to the pre-initiation complex as a testis-specific subunit of TFIIA/ALF to promote haploid cell gene expression. Sci Rep 6: 32069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Ferrara-Romeo I, Flores JM, Blasco MA. 2014. Essential role for the TRF2 telomere protein in adult skin homeostasis. Aging cell 13: 656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure C, Cole KL, Wulff P, Klugmann M, Murray AJ. 2011. Production and titering of recombinant adeno-associated viral vectors. J Vis Exp: e3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA, Tonegawa S. 2007. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science 317: 94–99. [DOI] [PubMed] [Google Scholar]
- Minty F, Thurlow JK, Harrison PR, Parkinson EK. 2008. Telomere dysfunction in human keratinocytes elicits senescence and a novel transcription profile. Exp Cell Res 314: 2434–2447. [DOI] [PubMed] [Google Scholar]
- Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. 2003. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Res 63: 4021–4027. [PubMed] [Google Scholar]
- Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ, Rodriguez Barrera V, Chittajallu R, Iwamoto KS, McBain CJ, et al. 2012. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 149: 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa S. 2005. Sequence-specific DNA damage by reactive oxygen species: implications for carcinogenesis and aging. Environ Health Prev Med 10: 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Bensen AL, Westbrook GL. 2006. Delayed development of adult-generated granule cells in dentate gyrus. J Neurosci 26: 2326–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieraut S, Gounko N, Sando R III, Dang W, Rebboah E, Panda S, Madisen L, Zeng H, Maximov A. 2014. Experience-dependent remodeling of basket cell networks in the dentate gyrus. Neuron 84: 107–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polvi A, Linnankivi T, Kivelä T, Herva R, Keating James P, Mäkitie O, Pareyson D, Vainionpää L, Lahtinen J, Hovatta I, et al. 2012. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet 90: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolyan H, Scheffold A, Heinrich A, Begus-Nahrmann Y, Langkopf BH, Holter SM, Vogt-Weisenhorn DM, Liss B, Wurst W, Lie DC, et al. 2011. Telomere shortening reduces Alzheimer's disease amyloid pathology in mice. Brain 134: 2044–2056. [DOI] [PubMed] [Google Scholar]
- Rossiello F, Herbig U, Longhese MP, Fumagalli M, d'Adda di Fagagna F. 2014. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr Opin Genet Dev 26: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. 1999. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96: 701–712. [DOI] [PubMed] [Google Scholar]
- Savage SA. 2014. Human telomeres and telomere biology disorders. Prog Mol Biol Transl Sci 125: 41–66. [DOI] [PubMed] [Google Scholar]
- Simonet T, Zaragosi LE, Philippe C, Lebrigand K, Schouteden C, Augereau A, Bauwens S, Ye J, Santagostino M, Giulotto E, et al. 2011. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res 21: 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. 2002. Different telomere damage signaling pathways in human and mouse cells. EMBO J 21: 4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. 2002. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol 12: 1635–1644. [DOI] [PubMed] [Google Scholar]
- Strange BA, Witter MP, Lein ES, Moser EI. 2014. Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 15: 655–669. [DOI] [PubMed] [Google Scholar]
- Tonegawa S, Pignatelli M, Roy DS, Ryan TJ. 2015. Memory engram storage and retrieval. Curr Opin Neurobiol 35: 101–109. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. 1999. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99–103. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T. 1998. TRF2 protects human telomeres from end-to-end fusions. Cell 92: 401–413. [DOI] [PubMed] [Google Scholar]
- Wu L, Multani AS, He H, Cosme-Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM, et al. 2006. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126: 49–62. [DOI] [PubMed] [Google Scholar]
- Yang D, Xiong Y, Kim H, He Q, Li Y, Chen R, Songyang Z. 2011. Human telomeric proteins occupy selective interstitial sites. Cell Res 21: 1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang PS, Furukawa K, Opresko PL, Xu XR, Bohr VA, Mattson MP. 2006. TRF2 dysfunction elicits DNA damage responses associated with senescence in proliferating neural cells and differentiation of neurons. J Neurochem 97: 567–581. [DOI] [PubMed] [Google Scholar]
- Zhang P, Pazin MJ, Schwartz CM, Becker KG, Wersto RP, Dilley CM, Mattson MP. 2008. Nontelomeric TRF2–REST interaction modulates neuronal gene silencing and fate of tumor and stem cells. Curr Biol 18: 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Giesert F, Kloos K, Vogt Weisenhorn DM, Aigner L, Wurst W, Couillard-Despres S. 2010. A powerful transgenic tool for fate mapping and functional analysis of newly generated neurons. BMC Neurosci 11: 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.