

JnBaptiste et al. present analysis of the gene expression and phenotypic changes associated with global miRNA restoration in miRNA-deficient fibroblasts. This analysis uncovers a miRNA-repressed network containing oncofetal genes Imp1–3 that is up-regulated upon Dicer loss and is resistant to resilencing by complete restoration of miRNA activity.
Keywords: miRNA, Dicer, let-7, Imp1–3, oncofetal, cancer
Abstract
MicroRNAs (miRNAs) are post-transcriptional regulators of gene expression critical for organismal viability. Changes in miRNA activity are common in cancer, but how these changes relate to subsequent alterations in transcription and the process of tumorigenesis is not well understood. Here, we report a deep transcriptional, oncogenic network regulated by miRNAs. We present analysis of the gene expression and phenotypic changes associated with global miRNA restoration in miRNA-deficient fibroblasts. This analysis uncovers a miRNA-repressed network containing oncofetal genes Imp1, Imp2, and Imp3 (Imp1–3) that is up-regulated primarily transcriptionally >100-fold upon Dicer loss and is resistant to resilencing by complete restoration of miRNA activity. This Dicer-resistant epigenetic switch confers tumorigenicity to these cells. Let-7 targets Imp1–3 are required for this tumorigenicity and feed back to reinforce and sustain expression of the oncogenic network. Together, these Dicer-resistant genes constitute an mRNA expression signature that is present in numerous human cancers and is associated with poor survival.
MicroRNAs (miRNAs) are an abundant class of noncoding RNAs that repress gene expression typically approximately twofold through target degradation and/or translational inhibition (Baek et al. 2008; Bartel 2009; Ghildiyal and Zamore 2009). miRNAs regulate many processes, including development (Bernstein et al. 2003; Wienholds et al. 2003), differentiation (Kanellopoulou et al. 2005; Xu et al. 2009), apoptosis (Cimmino et al. 2005; Harfe et al. 2005), and proliferation (Wang et al. 2008).
Consistent with such broad influence, miRNA deregulation is commonly observed in cancer, with both oncogenic and tumor-suppressive roles having been reported (Ventura and Jacks 2009; Lin and Gregory 2015). Let-7 miRNAs comprise a tumor suppressor family that is down-regulated in multiple cancers and correlates inversely with patient survival (Calin 2004; Takamizawa et al. 2004; Boyerinas et al. 2010; Nguyen et al. 2014; Kugel et al. 2016; Manier et al. 2016; Powers et al. 2016). Underlying the potent tumor suppressor activity of let-7 is repression of well-established oncogenic mRNAs such as Lin-28, Hmga2, Myc, Ras, and the Igf2 mRNA-binding protein family Imp1, Imp2, and Imp3 (Imp1–3) (Johnson et al. 2005; Lee and Dutta 2007; Sampson et al. 2007; Boyerinas et al. 2008).
Imp1–3 are an RNA-binding protein (RBP) family that control RNA localization, stability, and translation (Bell et al. 2013). Along with Hmga2 and Lin-28, Imp1–3 are categorized as oncofetal genes, defined based on their high expression during embryogenesis and re-expression in diverse cancer types, where they collectively promote stemness, proliferation, invasion, and metastasis (Boyerinas et al. 2008; Janiszewska et al. 2012; Gurtan et al. 2013; Nishino et al. 2013; Degrauwe et al. 2016). Let-7 and its oncofetal targets have been studied intensely in cancer, and several mechanisms for neutralizing let-7 activity have emerged. Besides genetic deletion (Calin 2004), biogenesis of let-7 is suppressed by Lin-28, another developmentally regulated RBP family (Viswanathan et al. 2008; Molenaar et al. 2012; Madison et al. 2013). Furthermore, alternative 3′ untranslated region (UTR) usage by Imp1 and Hmga2 promotes transformation (Mayr et al. 2007; Mayr and Bartel 2009). A more recent study described a novel mechanism in neuroblastoma, where excessive amplification of Myc mRNA might sponge let-7 activity away from other targets (Powers et al. 2016). Additionally, Imp1–3 may sequester Hmga2, Lin-28, and other mRNA targets by directly binding let-7 target sites to impede miRNA activity or modulate the association of transcripts with RISC (Jonson et al. 2014; Busch et al. 2016; Degrauwe et al. 2016; Ennajdaoui et al. 2016). Thus, in cancer, inhibition of let-7 activity and extensive coregulation among its oncofetal targets are thought to synergize to promote oncogenesis.
Beyond perturbation of individual miRNAs in cancer, global miRNA deregulation through impairment of the miRNA processing machinery drives tumor development (Melo et al. 2009, 2010; Walz et al. 2015; Wegert et al. 2015). Mouse models have demonstrated that global miRNA loss through reduced Dicer expression promotes tumorigenesis (Kumar et al. 2007, 2009). In humans, germline or somatic Dicer mutations predispose patients to cancer and are collectively defined as Dicer1 syndrome (Foulkes et al. 2014). Germline loss-of-function mutations in Dicer have been identified in pediatric pleuropulmonary and pituitary blastomas, familial cystic nephroma, and Wilms’ tumor (Hill et al. 2009; Bahubeshi et al. 2010; Foulkes et al. 2011; de Kock et al. 2014). Hypomorphic somatic mutations in Dicer, primarily altering its catalytic residues, are associated with multiple cancer types (Heravi-Moussavi et al. 2012; Rakheja et al. 2014; Chen et al. 2015).
It is unclear why a global reduction in miRNAs promotes cancer (Heravi-Moussavi et al. 2012; Chen et al. 2015). Here, we used a cell-based model of miRNA loss and subsequent reconstitution to identify miRNA-regulated gene networks and characterize their effects on tumorigenesis. In immortalized Dicer-null (knockout) mesenchymal stem cells (MSCs), a fibroblast cell type, we restored global miRNA expression and subsequent post-transcriptional activity by stably re-expressing Dicer. Although post-transcriptional repression by let-7 and other miRNAs is restored, there persists a gene signature of transcriptionally up-regulated oncogenes, including the let-7-regulated Imp1–3 family, whose derepression is largely resistant to Dicer and let-7 restoration. These Dicer-rescued cells are highly tumorigenic, consistent with expression of this oncogenic signature. Expression of this oncogenic network is reinforced in a feedback loop by three of its members; namely, Imp1–3. Demonstrating clinical relevance, this set of irreversibly activated miRNA targets is associated with poor survival in cancer patients.
Results
Human Dicer (hsDicer) restores global miRNA expression in murine Dicer knockout fibroblasts
Previously, we reported the generation of SV40-large T immortalized Dicer wild-type and knockout MSCs as a model system for delineating gene networks regulated by miRNAs (Ravi et al. 2012). miRNA loss through Dicer deletion results in dramatic up-regulation of known oncofetal let-7 targets, such as Hmga2 and Imp1–3, that are highly expressed in both embryos and tumors. Beyond loss of miRNA-mediated post-transcriptional repression, these genes exhibit evidence of transcriptional up-regulation (Gurtan et al. 2013; Gosline et al. 2016).
To investigate the reversibility of previously observed post-transcriptional and transcriptional changes resulting from global miRNA loss, we expressed hsDicer in monoclonal knockout MSCs in which the endogenous murine Dicer had been ablated previously (Dicer−/− in Fig. 1A). Specifically, we HA-Flag-epitope-tagged and cloned wild-type hsDicer into a retroviral construct, infected knockout MSCs, and selected with puromycin for a heterogeneous population of cells. Introduction of Dicer was well tolerated with no evidence of cell death due to re-expression of miRNAs. Protein analysis on these stable lines indicated robust expression of both the tag and hsDicer (Fig. 1B). Northern blots on two monoclonal cell lines (independently isolated from the stable heterogeneous populations) revealed abundant expression of highly expressed miRNAs, including let-7c and miR-22, in the parental wild-type MSCs expressing an empty vector (wild-type Vec). These miRNAs are undetectable in the corresponding knockout MSCs (knockout Vec in Fig. 1B), but stable expression of wild-type hsDicer resulted in levels of mature miRNAs similar to the parental wild-type (Fig. 1B). Thus, hsDicer was stably expressed and functional in processing murine pre-miRNAs to mature miRNAs.
Figure 1.
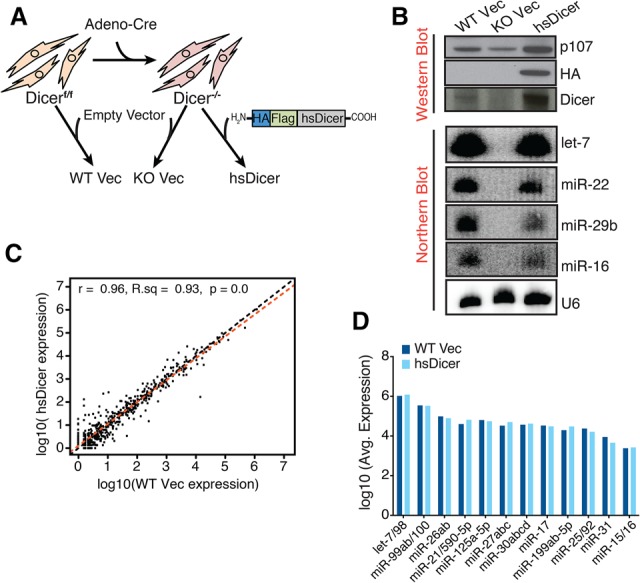
hsDicer rescues murine miRNA expression. (A) Schematized experimental design. Wild-type hsDicer was HA-Flag-epitope-tagged at the N terminus and cloned into pMMP-Puro, a puromycin-resistant vector. SV40-large T immortalized Dicerf/f or Dicer−/− cells were infected with virus encoding the Vec or HA-Flag-hsDicer (hsDicer), drug-selected, and passaged prior to experiments. (B, top panel) Western blot analysis across the heterogeneous population of resistant cells. p107 is shown as a loading control. (Bottom panel) Representative Northern blot analysis across a set of monoclonal lines isolated by low-density seeding from the heterogeneous population of resistant cells. U6 is shown as a loading control. (C) Global miRNA profile comparison between wild-type Vec and hsDicer. miRNAs collapsed by TargetScan family. The indicated values are normalized average expression counts across both replicates within each condition (two wild-type clones and two hsDicer clones). (Black line) y = x; (red line) line of best fit. (D) Normalized expression counts for the 12 TargetScan miRNA families that show significant Ago2 cross-linking at their target sites over background (Bosson et al. 2014). Adjusted P-value = 1 for all comparisons.
To globally profile the miRNAs expressed across these cell lines, we performed sequencing of the small RNA population (small RNA-seq) on each independent clone. We observed strong reproducibility between the clones (Supplemental Fig. 1A,B). On average, mature miRNAs comprised ∼21% of all reads in wild-type cells but only 1% in the Dicer nulls, demonstrating depletion of mature miRNAs globally (Supplemental Fig. 1C). Consistent with the Northern blot results, expression of wild-type hsDicer restored mature miRNAs to levels comparable with wild-type cells (18% of all reads) (Supplemental Fig. 1C; Supplemental Table S1A). More than 250 miRNAs were detectable in both wild-type clones at a threshold of at least five reads per million. The 10 most abundant miRNAs included miR-143, miR-100, miR-22, and multiple members of the let-7 family (Supplemental Table S1B). Upon collapsing by seeds, the let-7 family was most abundant, constituting, on average, 37% of reads mapping to miRNAs (Supplemental Table S1C).
We then directly compared the wild-type and hsDicer reconstituted miRNA libraries using DE-Seq (Anders and Huber 2010). The expression of miRNA families was largely restored to wild-type levels (Fig. 1C). The variation between wild type and hsDicer observed at lowly expressed/nonexpressed miRNAs was not statistically significant, and, globally, only three small RNAs could be assessed as significantly different. These three are all low abundance and together constitute <0.5% of the miRNA population (Supplemental Fig. 1D; Supplemental Table S1D). Notably, let-7, the most abundant seed family, was almost identically expressed between wild type and hsDicer (Fig. 1B,D). To consider other dominant seeds, we evaluated the 12 most active miRNA seeds in MSCs, as defined by statistically significant genome-wide Ago2-dependent cross-linking (iCLIP [individual-nucleotide-resolution UV cross-linking and immunoprecipitation]) at their target sites (Bosson et al. 2014). Importantly, these 12 families were all completely restored at the mature miRNA level with hsDicer reconstitution (P = n.s.) (Fig. 1D; Supplemental Table S1D). Together, these results show that stable wild-type hsDicer expression in a Dicer−/− background returns the mature miRNA expression profile to the parental wild-type (Dicerf/f) state.
hsDicer expression completely reconstitutes endogenous miRNA activity
To determine whether miRNAs generated by hsDicer mediate repression of mRNA targets, we used a previously reported two-color fluorescent reporter (Mukherji et al. 2011; Wilusz et al. 2012) to compare post-transcriptional miRNA activity between parental wild type and hsDicer. A bidirectional promoter drives the expression of mCherry-containing miRNA-binding sites in its 3′ UTR and nontargeted eYFP that serves as an internal control (Fig. 2A). We measured the repression mediated by miRNAs representing a range of expressions. In wild-type Vec cells, compared with a nontargeted mCherry control (“0x”), the addition of miRNA-binding sites resulted in an average of 1.3-fold repression for the lowly expressed miR-24 and 5.3-fold repression for the more abundant let-7 (Fig. 2A). Remarkably, we observed 1.2-fold and 5.5-fold repression for these miRNAs, respectively, in wild-type hsDicer-expressing cells (Fig. 2A). These results are consistent across the second population of clones (data not shown). Hence, the levels of repression across all tested miRNAs are not statistically different between wild-type Vec and hsDicer-expressing cells (P-value = 0.71) (Fig. 2A). Furthermore, there was negligible repression of the reporters in knockout Vec cells, confirming that these cells are indeed miRNA-deficient (Fig. 2A).
Figure 2.
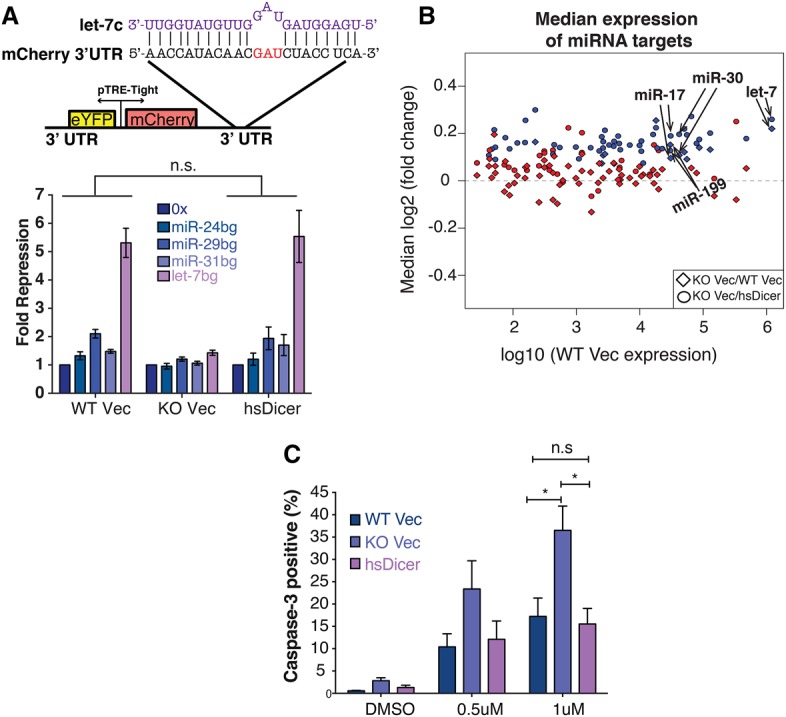
hsDicer expression recovers miRNA activity. (A, top panel) Schematic of the dual-color reporter used to assay miRNA repression. The 3′ UTR of mCherry contains sites imperfectly complementary to the miRNA of interest. The let-7c sequence is shown in purple. (Bottom panel) After transfecting with the reporter, flow cytometry was used to measure mCherry and eYFP levels. For each cell type, fold repression is relative to the nontargeted 0x reporter. Data shown are the mean and standard deviation of three independent experiments. P-values were calculated by paired Student's t-test. (B) Scatter plot of the median change in expression of miRNA targets relative to control genes matched for 3′ UTR length, GC content, and expression. Each point represents conserved targets of a single TargetScan miRNA family. miRNA expression is based on the average wild-type Vec expression reported in this study. Only expressed miRNAs are shown. (Blue) Significant change (Wilcoxon rank sum P ≤ 0.05); (red) not significant. (C) Doxorubicin-induced apoptosis measured by caspase-3 cleavage. The mean ± SEM of three independent experiments is indicated. P-values were calculated by unpaired Students t-test. (*) P < 0.05; (n.s) not significant.
We next examined whether the above results could be extended to endogenous targets. To do so, we carried out mRNA sequencing (mRNA-seq) on poly(A)-selected total RNA isolated from both clones of each cell type. We detected just under 13,000 genes expressed across all conditions (Supplemental Table S2A). A pairwise comparison of the transcriptomes of wild-type Vec and knockout Vec identified a population of 1524 differentially expressed genes that exhibited a median up-regulation of approximately threefold and down-regulation of ∼3.6-fold (Supplemental Fig. 2A; Supplemental Table S2B). The degree of change occurring with Dicer loss showed a strong and significant correlation to previously reported expression changes in MSCs (Supplemental Fig. 2B; Gurtan et al. 2013). Gene set enrichment analysis (GSEA) (Subramanian et al. 2005) confirmed that the most up-regulated genes were enriched for targets of “active” miRNAs (Supplemental Fig. 2C).
To perform a more miRNA-focused comparison of cell-based activity on endogenous targets, we derived empirical cumulative distributive functions for TargetScan miRNA families expressed in wild-type Vec cells. Upon loss of Dicer, conserved targets of most miRNAs were up-regulated, with let-7 showing the most significant change (Fig. 2B, miRNAs whose targets significantly change statistically are shown in blue). The significant up-regulation indicates strong silencing of targets in the wild-type state. hsDicer expression in the Dicer knockout background resulted in significant repression of conserved targets (Fig. 2B), indicating that the restored miRNAs were highly active. Notably, targets of more abundant and “active” miRNA seeds (such as let-7, miR-17, miR-199, and miR-30) that showed significant up-regulation upon Dicer loss exhibited median down-regulation comparable with hsDicer. Hence, hsDicer comprehensively restores miRNA activity on endogenous targets to a degree comparable with the parental wild-type state.
Finally, as miRNAs are known to buffer changes due to environmental stress (Leung and Sharp 2010), we tested for Dicer-dependent changes in stress response by treating the cells with the DNA-damaging agent doxorubicin. Knockout Vec cells were highly sensitive to this drug compared with wild-type Vec (P < 0.05) (Fig. 2C). Expression of hsDicer in knockout cells completely rescued the doxorubicin-induced cell death at all concentrations tested such that the responses between wild-type Vec and hsDicer were indistinguishable (P = 0.77) (Fig. 2C). Together, these results demonstrate that, on a global scale, hsDicer expression completely restores miRNA post-transcriptional activity, which in turn is sufficient to rescue miRNA loss-induced stress defects.
let-7 oncofetal targets remain activated after restoration of Dicer
Next, we examined the expression of oncofetal targets reported previously to contribute to the let-7 signature in Dicer knockout MSCs (Gurtan et al. 2013). Since our data indicate full reconstitution of miRNA expression and activity, we hypothesized that the expression of these targets would revert to levels observed in wild-type Vec. Quantitative PCR (qPCR) analysis showed a dramatic up-regulation of Imp family proteins upon Dicer loss, from 45-fold for Imp2 to upward of 200-fold for Imp1 and Imp3 (Fig. 3A). Hmga2, an additional let-7 oncofetal target that is often correlated with Imp2–3 in expression, was up-regulated to a lesser degree (∼17-fold) (Fig. 3A). These fold changes are consistent with the RNA-seq results (Supplemental Fig. 3A).
Figure 3.
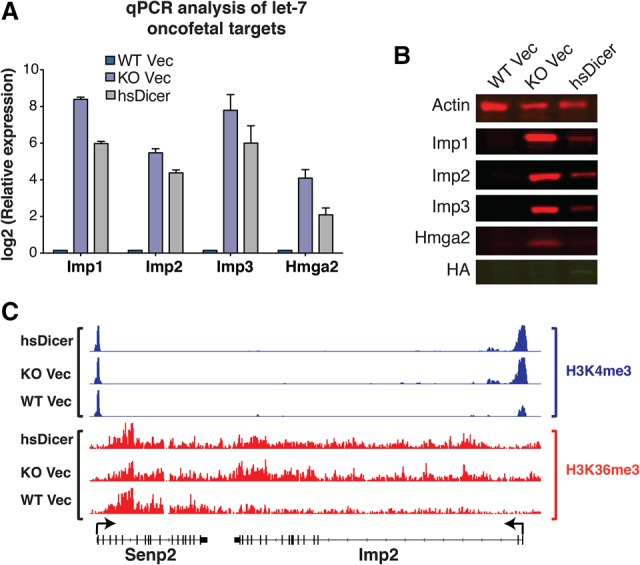
Despite miRNA rescue, let-7 oncofetal targets remain activated. (A) qPCR analysis of four let-7-regulated oncofetal genes. Note the logarithmic scale. For each gene, expression is relative to wild-type Vec levels. Data are plotted as the mean ± SEM of four or more independent experiments. (B) Western blot analysis of the oncofetal gene set. HA tag was used to indicate hsDicer expression. Actin was used as a loading control. (C) Normalized read counts for H3K4me3 and H3K36me3 marks at the Imp2 locus across the three cell lines. Within each chromatin mark, all conditions are set to the same scale. Flanking genes are shown as controls. Arrows indicate transcription start sites (TSSs).
Surprisingly, restoration of miRNA activity with the introduction of hsDicer only marginally down-regulated Imp mRNA expression relative to knockout Vec levels (Fig. 3A). Imp mRNAs that were up-regulated 45-fold to 200-fold were suppressed only twofold to fourfold with restoration of miRNA activity. Less dramatically, Hmga2 mRNA, which increased 17-fold with loss of miRNAs, was suppressed fourfold upon restoration with hsDicer. At the protein level, Western blot analysis mirrored the mRNA results. While Hmga2 was very lowly expressed and the Imp family was undetectable in wild-type Vec, all four genes showed robust protein expression in knockout Vec cells (Fig. 3B). Subsequent to miRNA rescue with hsDicer, Imp1–3 all showed a partial reduction consistent with the typical post-transcriptional repression by miRNAs. Importantly, even after restoration of miRNA activity, all three genes were highly expressed compared with original wild-type cells (Fig. 3B). In contrast, Hmga2 protein expression was more significantly repressed than the Imp family. This is consistent with the mRNA results, where, in hsDicer cells, Hmga2 was only approximately fourfold more highly expressed than in wild-type Vec (Fig. 3A; Supplemental Fig. 3A).
The inability of complete miRNA reconstitution to revert Imp gene expression suggests that somatic Dicer loss activates a self-sustaining “oncofetal-dependent” state. This possibility is consistent with our previous studies showing major transcriptional changes occurring at these loci upon Dicer loss (Gurtan et al. 2013; Gosline et al. 2016). Hence, we evaluated the transcriptional state at these loci through chromatin immunoprecipitation (ChIP) followed by massively parallel DNA sequencing (ChIP-seq) on H3K4me3 and H3K36me3 modifications. Upon inspection of the Imp2 locus, we observed increased H3K4me3 near the transcription start site (TSS) and H3K36me3 toward the 3′ end of the gene body in knockout Vec compared with wild-type Vec cells (Fig. 3C). Intriguingly, add-back of hsDicer effected negligible change from the knockout condition, indicating that there was “miRNA-resistant” transcriptional activity across this locus in hsDicer cells compared with the wild-type Vec. As a control comparison, the flanking gene Senp2 was indistinguishable across all three conditions for both modifications (Fig. 3C). These results indicate a targeted miRNA-resistant transcriptional activation of Imp2 upon Dicer loss.
This trend was observed for the other Imp family members but not for Hmga2, which showed no evidence of Dicer-dependent transcriptional changes (Supplemental Fig. 3B–D). As mentioned above, the degree of up-regulation for Hmga2 at the mRNA level was substantially less than that of the other oncofetal targets. Given that the Hmga2 3′ UTR has seven let-7 seed matches (of which six are conserved), we speculate that the 17-fold up-regulation is due primarily to multiplicative or cooperative post-transcriptional miRNA effects (Doench et al. 2003; Grimson et al. 2007; Saetrom et al. 2007). For the other oncofetal targets, however, we propose that transcriptional changes upon Dicer loss account for most of their up-regulation (beyond direct miRNA effects on mRNA stability), and this change is not reversed upon restoration of miRNA activity.
Transcriptomic profiling reveals a larger, miRNA-resistant gene expression signature
We tested whether the irreversible behavior of the oncofetal let-7 targets Imp1–3 was part of a more global phenomenon. To do this, we used an unsupervised blind source separation approach using independent component analysis (ICA) (Hyvarinen and Oja 2000) to identify distinct gene expression signatures. We used this approach previously to elucidate cancer-associated gene signatures in lung adenocarcinoma (Li et al. 2015; Dimitrova et al. 2016; Papagiannakopoulos et al. 2016). ICA provides the ability to detect statistically independent gene expression signatures within the context of the entire data set (i.e., across all samples), as opposed to performing multiple pair-wise differential expression analyses. When applied to the mRNA-seq data across all cell lines, ICA identified two significant and biologically relevant signatures (P = 0.01) (Fig. 4A): Signature 1 reflects a pattern of gene up-regulation in both knockout Vec and hsDicer conditions relative to parental wild-type, while signature 2 indicates a pattern of higher gene expression in miRNA-deficient conditions relative to miRNA-competent ones (Fig. 4A).
Figure 4.
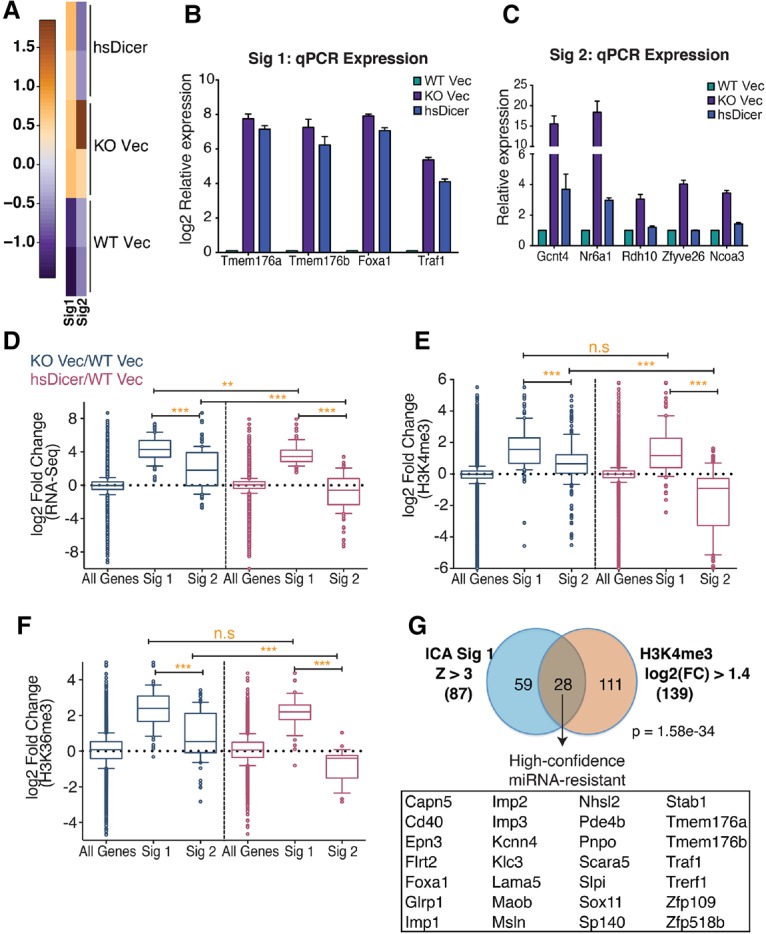
Identification of a high-confidence miRNA-resistant gene signature. (A) Heat map depicting two independent, statistically significant gene signatures detected in the RNA-seq expression data set using ICA. Signature 1 (Sig1) represents an expression pattern of genes up-regulated in knockout Vec and hsDicer conditions relative to wild-type Vec. Signature 2 (Sig2) represents genes that are down-regulated to wild-type Vec levels upon introduction of hsDicer. (B,C) qPCR validation of “irreversible” or “miRNA-resistant” genes identified from signature 1 (B) and “reversible” or “miRNA-sensitive” genes identified from signature 2 (C). Results are plotted relative to wild-type Vec levels. Bars represent mean ± SEM of at least three independent experiments. (D) Box and whisker plots of normalized RNA-seq expression fold changes for all expressed genes (n = 12834), signature 1-correlated genes (n = 87), and signature 2-correlated genes (n = 112). (E,F) Same as in D but for fold changes in normalized counts for gene-associated H3K4me3 peaks (E) and H3K36me3 peaks (F). Whiskers represent the 10th–90th percentile, and all other points are shown as individual dots. The P-values were calculated by Mann-Whitney U-test. (**) P < 0.01; (***) P < 0.0001; (n.s) not significant. (G) Overlap of signature 1-correlated genes and genes with a log2 fold change of at least 1.4 in H3K4me3 peak counts near their TSSs. The H3K4me3 criteria is satisfied by both knockout Vec/wild-type Vec and hsDicer/wild-type Vec comparisons. High-confidence miRNA-resistant genes are listed. The hypergeometric test's P-value is indicated.
qPCR analysis of a sampling of signature 1-correlated genes (z-score >3) (Supplemental Table S3) confirmed that hsDicer reconstitution did not revert their expression to wild-type levels (Fig. 4B). In contrast, signature 2-related genes showed near complete rescue to wild-type expression levels (Fig. 4C). As such, hereafter, signature 1 and signature 2 are referred to as miRNA-resistant and miRNA-sensitive, respectively. Consistent with the observations for the Imp1–3 family, as a group, the miRNA-resistant genes were dramatically up-regulated upon Dicer loss, significantly more so than miRNA-sensitive genes (Fig. 4D). Upon quantification of the ChIP-seq signal at promoters, we observed that the miRNA-resistant genes exhibited significantly higher gains in H3K4me3 than reversible genes (Fig. 4E, cf. second and third box plots) when Dicer was originally lost in these cells. With hsDicer add-back, there was no significant reduction in H3K4me3 at the miRNA-resistant genes (Fig. 4E, cf. second and fifth box plots), while H3K4me3 at the miRNA-sensitive genes was significantly reduced (Fig. 4E, cf. third and sixth box plots). A similar trend was observed for H3K36me3 in the body of genes (Fig. 4F).
Thus, ICA uncovered a broader miRNA-resistant gene set beyond Imp1–3 that exhibits dramatic up-regulation upon Dicer loss followed by incomplete repression to wild-type expression. Our results indicate that increased transcription (measured through ChIP-seq) strongly correlates with this resistance (Fig. 4D–F). Therefore, we defined a “high-confidence” set of miRNA-resistant genes due to changes in transcription using our combined data sets. We overlapped the set of genes defined by ICA (signature 1: z-score >3; n = 87) with genes that show a log2 change of at least 1.4-fold in their associated H3K4me3 peaks in both knockout Vec and hsDicer compared with wild-type Vec (knockout Vec/wild-type Vec ≥ 1.4; hsDicer/wild-type Vec ≥ 1.4; n = 139) (Supplemental Tables S3, S4). This analysis identified a set of 28 genes that we designate as “high-confidence miRNA-resistant” genes (Fig. 4G; Supplemental Table S3). In contrast, an overlap using miRNA-sensitive genes (signature 2: z-score >3; n = 112) with the same H3K4me3 gene set was insignificant and contained only three genes (P = 0.12) (data not shown). These results indicate that transcriptional activation is a feature of miRNA-resistant genes.
miRNA restoration confers a stably inherited transformed phenotype
The high-confidence miRNA-resistant signature includes let-7-regulated oncogenes that are elevated dramatically in various cancers and function to promote tumor growth, drug resistance, and metastasis (Tessier et al. 2004; Boyerinas et al. 2008; Samanta et al. 2013). Thus, we examined whether re-expression of hsDicer and hence miRNAs in Dicer knockout cells resulted in a tumorigenic phenotype. In a soft agar assay, hsDicer cells exhibited significant growth potential not observed for wild-type Vec and knockout Vec cells (Fig. 5A). These results suggest that hsDicer cells acquired properties of anchorage-independent growth and anoikis resistance, which could possibly contribute to tumorigenicity in vivo. To directly test this possibility, we performed subcutaneous injections of wild-type Vec, knockout Vec, and hsDicer cells into the flanks of immune-compromised mice. Strikingly, we observed 100% incidence of tumor formation with hsDicer cells but none for wild-type Vec or knockout Vec (P = 3 × 10−07) (Fig. 5B). Histopathological analysis of the tumors indicated undifferentiated sarcomas, which is consistent with the mesenchymal origin of these cells (Fig. 5C).
Figure 5.
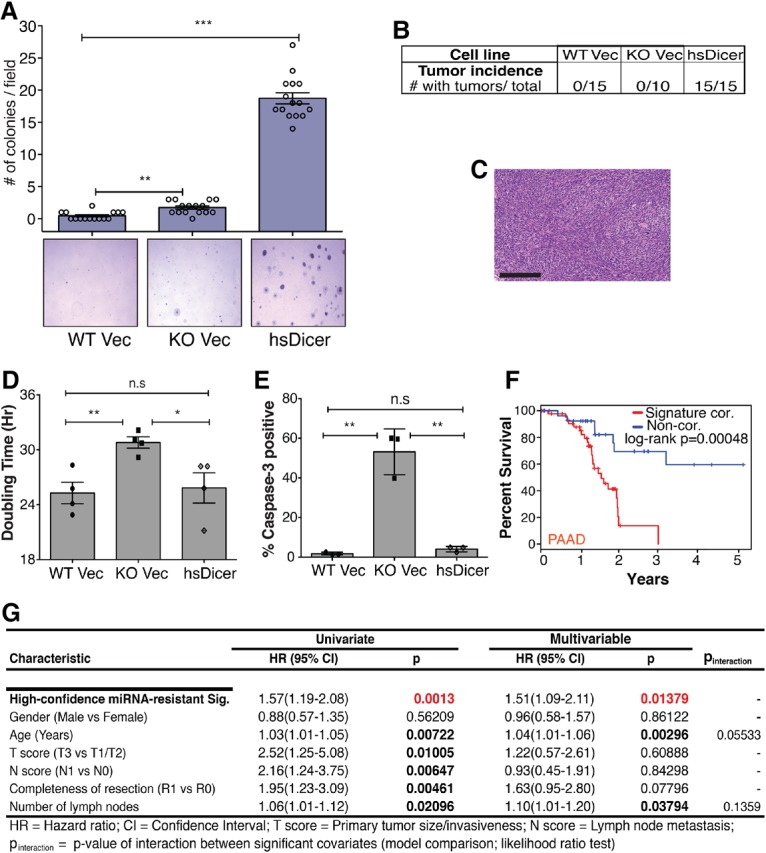
miRNA restoration through hsDicer expression transforms MSCs. (A) Colony formation of MSCs after ∼15 d of growth in agarose-containing medium. Representative 4× bright-field images for each genotype are shown. Colonies were counted by eye in five random fields for three independent experiments. Data are plotted as the mean ± SEM. The Student's t-test P-value is indicated. (B) Frequency of tumor formation by the three parental MSC lines 8 wk after injection of 105 cells into the flanks of immune-compromised mice. (C) Representative hematoxylin and eosin staining of an hsDicer tumor section at 20× magnification. Bar, 200 µm. (D) Proliferation assay indicating the mean ± SEM of two experiments with two replicates each. (E) Apoptosis assayed by capase-3 cleavage. Mean and standard deviation are plotted. P-values were calculated by Student's t-test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.0001; (n.s) not significant. (F) Kaplan-Meier survival analysis of The Cancer Genome Atlas's (TCGA) pancreatic adenocarcinoma (PAAD) patients stratified by their correlation score with the high-confidence miRNA-resistant signature. |z| > 0.5 extremes of score distribution; n = 46 most correlated; n = 28 least correlated. Patients with most correlated gene expression scores (red) exhibit significantly reduced survival times compared with least correlated patients (blue). See the Supplemental Material for details. (G) Results of univariate and multivariable Cox proportional hazard model on overall survival in the TCGA PAAD cohort (all patients). The high-confidence miRNA-resistant signature is found to be independently prognostic within the cohort of TCGA PAAD patients.
To confirm that the tumors were of hsDicer cell origin, we generated cell lines from five tumors and confirmed the absence of endogenous murine Dicer via PCR genotyping (Supplemental Fig. 4A). Furthermore, the tumor cells were all puromycin-resistant due to the vector containing hsDicer (data not shown) and strongly expressed let-7 and other mature miRNAs, thereby confirming the retention of hsDicer in vivo (Supplemental Fig. 4B). Importantly, a sampling of a subset of miRNA-resistant genes indicates that they were highly expressed in all of the tumor cell lines (Supplemental Fig. 4C,D). Thus, we conclude that the observed tumors developed from hsDicer cells.
Our results indicate that only hsDicer cells are tumorigenic despite the fact that knockout Vec cells also have high expression of Imp1–3 and other high-confidence miRNA-resistant genes. To evaluate whether miRNA deficiency adversely impacted growth of knockout Vec cells, we measured doubling times and apoptosis for each genotype. Knockout Vec cells exhibited a significantly slower doubling, while hsDicer completely rescued this phenotype (Fig. 5D). In addition, knockout Vec cells exhibited an elevated basal apoptosis at high culture density compared with wild-type Vec and hsDicer (Fig. 5E). In vivo, these combined defects are probably deleterious to knockout Vec cells and dominate over any oncogenic potential.
The high-confidence miRNA-resistant gene set is a pan-cancer signature
To assess the relevance of the high expression levels of this high-confidence miRNA-resistant network that is enriched in oncofetal genes, we first queried the cBioPortal database (Cerami et al. 2012). A cross-cancer analysis revealed a high degree of alterations in this gene set in breast, lung, bladder, and pancreatic cancers (Supplemental Fig. 4E). Notably, these alterations were predominantly amplifications, suggesting oncogenic activity that is important for human malignancy. To address this more directly, we examined whether the high-confidence miRNA-resistant gene signature could provide prognostic information relevant to human cancer. Using gene expression profiles and clinical information from The Cancer Genome Atlas's (TCGA; http://cancergenome.nih.gov) pancreatic adenocarcinoma (PAAD) cohort, we found the signature to be strongly correlated with clinical outcome: High expression was significantly associated with worse survival in Kaplan-Meier analysis comparing patients at the extremes of the signature correlation score distribution (|z| > 0.5; log-rank P < 0.0005) (Fig. 5F). Furthermore, increasing signature correlation score was also associated with poor prognosis across all patients in the cohort (Hazard ratio = 1.51) in a Cox proportional hazard model while controlling for various other clinical covariates (Fig. 5G), suggesting that this signature is independently prognostic in PAAD. To assess the pan-cancer relevance of this signature, we investigated additional cancer types within the TCGA data set. Strikingly, this signature is also significantly correlated with reduced survival across other solid cancers (lung, kidney, and glioblastoma/low-grade glioma) (Supplemental Fig. 4F), indicating that up-regulation of this high-confidence miRNA-resistant gene set is a more general feature of human cancers.
Cotargeted disruption of Imp1–3 adversely impacts hsDicer tumorigenicity
The miRNA-resistant signature identified in this study is almost certainly oncogenic in human cancer. However, it is unclear whether a core subset of these genes is primarily responsible for the tumorigenicity of the hsDicer cells. To investigate this, we focused on the Imp family because Imp1–3 are not expressed in most adult tissues and are often de novo activated in cancers. To test whether loss of any Imp member would negatively affect tumorigenesis, we used the CRISPR–Cas9 system to generate individual knockouts of Imp1–3 in hsDicer cells and propagated two independent clones of each knockout cell line. Western blot analysis indicated complete loss of the individual Imp proteins in the targeted clones, and the results of genomic DNA sequencing were consistent with a loss of the reading frame (Fig. 6A; data not shown). However, each Imp knockout retained robust expression of the remaining two family members, suggesting a lack of coregulation. We then evaluated the potential of the Imp knockouts to grow in vitro via the soft agar assay. Individual loss of Imp1–3 in hsDicer cells did not confer any meaningful reduction in colony formation (Fig. 6B; Supplemental Fig. 5A). As an alternative measure of tumorigenic potential, we used the growth in low attachment (GILA) assay, which has been shown to capture aspects of transformation that could be missed by soft agar growth (Rotem et al. 2015). We validated that GILA recapitulated the soft agar results; only hsDicer cells were able to grow over a 7-d time course in GILA (Supplemental Fig. 5B). In contrast, wild-type Vec and knockout Vec cells underwent anoikis despite exhibiting strong growth under normal adherent conditions (Supplemental Fig. 5B; data not shown). Consistent with the soft agar results, individual knockouts of Imp1–3 did not dramatically reduce cellular growth via GILA (Fig. 6C). Hence, our data indicate that loss of any one Imp family member on the background of the endogenous expression of the other two Imp proteins is not sufficient to abrogate the tumorigenic properties of hsDicer cells.
Figure 6.

Combined loss of Imp1–3 greatly impairs tumorigenicity in vivo. (A) Western blot of lysates derived from CRISPR–Cas9-generated Imp knockout cells. Two independent clones of each knockout cell type are indicated. (Left panel) Imp1 knockouts (the upper band is the specific band). (Middle panel) Imp2 knockouts. (Right panel) Imp3 knockouts. Vinculin was used as a loading control. (B) Quantification of soft agar colony counts from the cell lines depicted in A. Data are plotted as the mean ± SEM. (C) Growth time course of cells in GILA assay. The ATP yield at each time point is a measure of cell viability. Data are plotted as the mean ± standard deviation. n = 3. (D) Western blot of whole-cell lysates from two independent clones of CRISPR–Cas9-generated Imp1–3 triple-knockout (ImpΔ3) cells. Actin was used as a loading control. (E) Quantification of soft agar colony counts from the cell lines depicted in D. Representative bright-field soft agar images for each genotype are shown at 4× magnification. (F) Same as in C but for ImpΔ3 cells. (G) Cumulative fraction of injected sites where tumors formed as a function of time. Each mouse from this cohort was subcutaneously injected with two cell types: hsDicer cells on the left flank and ImpΔ3 cells on the right flank. n = 10. (H) Kinetics of subcutaneous tumor growth for hsDicer- and ImpΔ3-derived tumors. (*) P < 0.05 from paired Wilcoxon signed rank test. (I) Representative image depicting gross subcutaneous tumor sizes at the experimental end point (64 d after injection). Black and yellow arrows indicate sites of hsDicer or ImpΔ3 cell injection, respectively. (J) Heat map of the 302 genes differentially expressed between hsDicer and ImpΔ3 cells. For each gene (each row), normalized gene expression is z-score standardized. (K) GSEA plots for the high-confidence miRNA-resistant (left panel) and ICA-derived (right panel) signature 1 signatures. Genes are ranked according to descending (ImpΔ3/hsDicer) log2 fold change such that most down-regulated genes are skewed to the right of the plot. Nominal P-values and false discovery rate (FDR) q-values are indicated. (L) Heat map depicting gene-by-gene Spearman correlation coefficients in tumor samples of PAAD patients from TCGA. Depicted genes are the leading edge core genes driving the enrichment of ICA signature 1 in K above. (M) Summary table for GSEA results showing enriched gene sets from the Molecular Signatures Database (MSigDB) curated or hallmark gene set collections. Genes down-regulated with Imp1–3 loss are significantly enriched for gene sets associated with pseudopodia-, cancer-, anoikis-, and inflammation-associated signaling.
Given this result, we next explored the effects of simultaneous loss of all three Imp proteins in the hsDicer condition. We isolated two independent clones of these triple-knockout cells, designated ImpΔ3, and confirmed loss of function by Western blot and genomic DNA sequencing (Fig. 6D; Supplemental Fig. 5C). These clones exhibited a significant reduction in the number of colonies formed in soft agar as compared with the hsDicer cells from which they were derived (Fig. 6E). Similarly, the growth of both ImpΔ3 clones was severely compromised in the low adherent conditions of the GILA assay (Fig. 6F). To extend these results in vivo, we performed subcutaneous tumor formation assays in immune-compromised mice, injecting the parental hsDicer cells and ImpΔ3 cells in the left and right flanks, respectively, of each mouse. hsDicer-derived tumors first appeared after 28 d compared with 35 d for ImpΔ3 (Fig. 6G). ImpΔ3 cells required ∼42 d to form tumors at 50% of the sites of injection, while 90% of sites injected with hsDicer cells had formed tumors at that time point (Fig. 6G). To better assess this difference in tumorigenicity, we measured the growth of tumors with time. The ImpΔ3 tumors that formed grew at a significantly reduced rate, ultimately resulting in dramatically stunted tumors when compared with the parental hsDicer tumors (P < 0.05) (Fig. 6H,I). Histologically, the ImpΔ3 tumors were undifferentiated sarcomas with pleomorphic nuclei, similar to hsDicer-derived tumors (data not shown). Altogether, these data demonstrate that the combined activation of the Imp family is important for the oncogenic switch acquired by Dicer-rescued cells and that additional miRNA-resistant genes may contribute to tumorigenesis.
Imp1–3 reinforce expression of miRNA-resistant genes
To understand the mechanism by which combined loss of Imp1–3 effects this reduced tumorigenicity, we used poly(A)-selected RNA-seq to analyze the steady-state mRNA levels in ImpΔ3 and the parental hsDicer cells. Upon loss of Imp1–3 proteins, 302 genes were significantly differentially expressed (fold change ≥2; false discovery rate [FDR] ≤0.05) (Supplemental Table S5). Two-hundred-seven genes were up-regulated compared with hsDicer with a median fold change of 2.9, while 95 genes were down-regulated approximately sixfold on average (Fig. 6J; Supplemental Fig. 5D). First, we queried whether loss of Imp1–3 predominantly affected other members of the high-confidence miRNA-resistant signature. Only five genes from this signature were significantly differentially expressed (Supplemental Fig. 5E), and, globally, these were not biased to either the highly up-regulated or down-regulated genes (Fig. 6K). Given this result, we extended the analysis to include the larger set of miRNA-resistant genes from which the high-confidence set had been derived (ICA signature 1, z > 3; n = 87) (Fig. 4G). GSEA revealed that ICA signature 1-associated genes were significantly enriched among genes down-regulated by Imp1–3 loss, 12 of which met the significance threshold for differential expression, and, of these, 11 decreased in expression (the exception being Msln) (Fig. 6K; Supplemental Fig. 5F). This indicates that the majority of the genes that were strongly up-regulated in knockout Vec and hsDicer cells, but lacked transcriptional activation, was stabilized by expression of Imp1–3.
We then examined the GSEA leading edge subset, which constitutes the core group of genes accounting for this enrichment and reflects the genes most biologically important for the phenotype of interest (in this case, Imp1–3 loss), and noticed a strong contribution from high-confidence miRNA-resistant genes (Supplemental Table S6). Consistent with expression changes being specific to Imp1–3 activity, in all, 10 of the 44 genes comprising the leading edge have been implicated as direct Imp1–3 targets (statistically significant signal in either CLIP or RNA immunoprecipitation data sets) across studies in pluripotent stem and pancreatic cancer cells (Supplemental Table S6; Taniuchi et al. 2014; Conway et al. 2016; Ennajdaoui et al. 2016). This included, but was not limited to, components of the high-confidence miRNA-resistant signature that we found to be prognostic in human cancers: Sox11, Traf1, and Flrt2. However, the leading edge gene set was also populated by oncogenes that have not been formally linked with Imp1–3, such as Plag1 and Fzd6. To explore potentially novel relationships, we assessed expression patterns of leading edge genes in cancer. Slpi, Plag1, and Fzd6 showed strong correlation with Imp1–3 family members in pancreatic tumors (Fig. 6L), and this trend was consistent in kidney and lung tumors as well (Supplemental Fig. 5G,H). This observation implicates Slpi, Plag1, and Fzd6 as genuine downstream effectors of Imp1–3 oncogenic activity in our cell system and human cancers. In summary, in hsDicer cells, Imp1–3 primarily sustain a post-transcriptional up-regulation of miRNA-resistant genes populated by likely direct Imp targets and other oncogenes.
Finally, we broadened our analysis to investigate externally annotated gene sets from the Molecular Signatures Database (MSigDB) (Liberzon et al. 2011). This unbiased approach implicated multiple biological pathways as being perturbed upon Imp1–3 loss. For genes up-regulated in ImpΔ3, few gene sets were enriched, among which a coherent biological theme was not apparent. However, among down-regulated genes, there was significant enrichment for transcripts localized to pseudopodia, consistent with previously described roles for Imp1 and Imp3 (Stohr et al. 2012; Taniuchi et al. 2014; Ennajdaoui et al. 2016) as well as genes important for avoiding anoikis and for various signaling and cancer pathways (Fig. 6M). In all, this analysis confirmed that the expression changes downstream from Imp1–3 loss are concordant with genes and biological processes with established importance in cancer.
Discussion
We demonstrated that loss of Dicer results in up-regulation of a subset of miRNA targets that are transcriptionally activated and not resilenced upon reconstitution of miRNA expression. The expression of this miRNA-resistant gene set is reinforced by at least three of its members, Imp1–3, and is self-sustaining. The miRNA-resistant signature was observed in independently derived clones of Dicer-restored cells. More important and surprising, these clones were tumorigenic in contrast to their progenitors. Finally, the miRNA-resistant gene set is associated with poor patient prognosis across multiple cancers.
Irreversible up-regulation of oncofetal targets of let-7
Previous work has demonstrated that the most dramatic changes in gene expression upon loss of the post-transcriptional activity of miRNAs in MSCs surprisingly occur at the transcriptional level (Gosline et al. 2016). This dramatic transcription activation was documented as an increase in nascent RNAs, as assessed through intronic reads, and a corresponding increase in H3K4me3 and H3K36me3 chromatin modifications (Gosline et al. 2016). We now extend these findings by showing that rescue of miRNA expression and activity restored the vast majority of changes that occurred with Dicer loss but failed to suppress the transcriptional activation of a core oncogenic gene set, including Imp1–3. The inability of restored miRNAs to completely suppress this oncogenic signature is surprising and reveals important principles regarding the functional organization of miRNAs in gene expression networks. For example, the let-7 targeted genes Imp1–3 are highly expressed during early embryonic development, but their expression dramatically declines in a window that coincides with induction of mature let-7. It is unknown whether their repression is driven solely by let-7-mediated post-transcriptional activity or whether loss/gain of transcriptional regulators acts on them redundantly to facilitate complete silencing in differentiated tissues (Zhu et al. 2011; Nguyen et al. 2014). Our study demonstrates that despite the fact that the sole loss of miRNA activity is sufficient for Imp1–3 transcriptional activation, rescue of this post-transcriptional layer of regulation cannot significantly resilence these genes. This state is distinct from induction of a pluripotent state characterized by expression of Oct4, Sox2, and Lin-28, which are not expressed in any of these MSC-related cells. Thus, loss of miRNA activity in MSCs activates components that are epigenetically resistant to complete restoration of miRNA expression.
Transcriptional and post-transcriptional regulation in an epigenetic switch
The observation that the cells expressing the miRNA-resistant set of genes are highly malignant, whereas their progenitor wild-type cells before loss and restoration of Dicer are not, suggests epigenetic activation of an oncogenic program. A stable inheritance of an acquired transformed phenotype has been described elsewhere (Iliopoulos et al. 2009). That study elucidated a positive feedback loop between transcriptional (NF-κB regulation of IL-6) and post-transcriptional (Lin-28b and let-7) regulators as key components underlying maintenance of transformation. However, neither Lin-28 paralog is expressed in the MSCs. Furthermore, extensive perturbation of candidate transcriptional factors identified by computational approaches did not identify the key mediators of the miRNA-resistant signature's induction (Gosline et al. 2016). While the complete mechanism underlying the switch described in this system is unknown, the positive feedback loop involving Imp1–3 that is activated by temporary loss of Dicer partially underlies this phenomenon.
Reinforcement of sustained oncogene expression by Imp1–3 RBPs
Disruption of let-7 regulation of oncofetal targets (Imp1–3, Hmga2, and Lin-28) is an important aspect of oncogenesis and is achieved through a variety of mechanisms. Let-7 may be genetically deleted or suppressed by Lin-28 (Calin 2004; Viswanathan et al. 2008, 2009), let-7 activity may be sponged (Powers et al. 2016), and alternative 3′ UTR usage may mediate escape of let-7-mediated repression (Mayr et al. 2007; Mayr and Bartel 2009). Based on the above observations on the activation of Imp1–3 in our model system, simultaneous elevated expression of multiple Imp proteins through transcriptional activation may be an additional mechanism overriding let-7 activity. This mechanism is probably independent of observations that Imp1–3 activity converges on post-transcriptionally opposing let-7 and other miRNAs (Jonson et al. 2014; Sheen et al. 2015; Busch et al. 2016). Post-transcriptional silencing by let-7 is fully active in these tumorigenic cells.
There has been a concerted effort toward understanding the mechanisms through which Imp1–3 exert oncogenic effects by use of pull-down experiments to characterize their “RNA interactomes” (Hafner et al. 2013; Taniuchi et al. 2014; Conway et al. 2016; Ennajdaoui et al. 2016). Our work complements such studies by investigating the gene expression changes and functional outcomes downstream from simultaneous inactivation of all Imp paralogs and places Imp1–3 at the core of an oncogenic switch. The protumorigenic phenotype of hsDicer cells was impaired only with combined Imp1–3 loss and not by loss of the individual members, suggesting strong functional redundancy among this family. This observation is consistent with reports of overlapping mRNA binding as well as autoregulation by Imp1–3 (Conway et al. 2016; Ennajdaoui et al. 2016). Additionally, we identified Plag1, Fzd6, and Slpi as novel possible downstream effectors of Imp1–3. These oncogenes belonged to the miRNA-resistant signature, were strongly down-regulated by Imp1–3 loss, and exhibited correlated expression among themselves as well as with Imp1–3 in various human cancers.
In summary, we described an oncogenic switch involving the conversion of nontransformed fibroblasts to stable transformed cells by toggling the activity of miRNAs off and on. Future studies will enhance our understanding of the underlying mechanism and role of Imp1–3 in oncogenic networks.
Materials and methods
All sequencing data are available in the NCBI sequence repository under accession number PRJNA383556.
Cell culture
MSCs were cultured at 37°C with 5% CO2 in α-MEM supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum.
Protein analysis
Cells were harvested in RIPA supplemented with protease inhibitor cocktail (Roche), and samples were diluted in Laemilli buffer with DTT. Samples were electrophoresed using the Nu-PAGE Bis-Tris electrophoresis system (Life Technologies) as per the manufacturer's instructions and transferred to a polyvinylidene fluoride (PVDF) or nitrocellulose membrane in a Mini Trans-Blot wet transfer apparatus (Bio-Rad). Membranes were blocked in 5% milk in TBST (for ECL-based detection) or Odyssey blocking buffer (for Licor-based detection). Primary antibody incubation was performed overnight, and samples were detected on film using ECL (Perkin Elmer) or visualized using Odyssey (Licor).
The Imp1–3 polyclonal antibodies were obtained from MBL International (catalog nos. RN007P, RN008P, and RN009P), anti-Hmga2 was ofrom Cell Signaling (catalog no. 5269), anti-p107 and anti-Actin were from Santa Cruz Biotechnology (catalog nos. sc-318 and sc-130656), anti-HA and anti-vinculin were from Sigma-Aldrich (catalog nos. 11867423001 and V9131), and anti-Dicer was from Bethyl Laboratories (catalog no. A301-936A).
qPCR
Total RNA was isolated using Trizol or the RNeasy minikit (Qiagen) following the manufacturer's instructions, treated with the Turbo DNA-free kit (Life Technologies), and reverse-transcribed with oligo(dT) primers using SuperScript III (Life Technologies). qPCR reactions on resulting cDNAs were performed using Power SYBR Green (Life Technologies) run on an Applied Biosystems 7500 real-time PCR instrument. qPCR primers are listed in Supplemental Table S7.
Northern blot
Total RNA was separated on a 12% polyacrylamide/urea/TBE gel (Sequagel, National Diagnostics) and transferred to a HyBond N+ membrane (GE Healthcare) using the TransBlot SD semidry transfer system (Bio-Rad). RNA was then UV cross-linked to the membrane. DNA oligo probes perfectly complementary to the miRNAs of interest were γ-32P end-labeled using T4 polynucleotide kinase (New England Biolabs) and purified using Illustra G-25 MicroSpin columns (GE Healthcare). Membranes were prehybridized in UltraHyb oligo (Ambion) for at least 30 min at 42°C and then hybridized with a radiolabeled DNA probe overnight at 42°C. Blots were washed twice for 30 min with 2× SSC and 0.5% SDS prewarmed to 42°C. RNA was visualized by exposure to phosphor screens and then imaged on a Storm scanner (Molecular Dynamics).
Animal work
All animal work was performed under the guidelines of the Massachusetts Institute of Technology (MIT) Division of Comparative Medicine with protocols approved by the MIT Committee for Animal Care and were consistent with the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996 (Institutional Animal Welfare Assurance no. A-3125-01).
Soft agar assay
Cells were suspended in 0.4% SeaPlaque agarose (Lonza) in complete DMEM and seeded over a first layer of 0.8% agarose in complete DMEM. Cells were seeded using 2 × 104 to 4 × 104 cells per well of a six-well plate and in triplicate for each cell type. Cells were grown at 37°C and 5% CO2, and colonies were counted 2–3 wk after seeding. For each replicate, five random fields were captured by light microscopy at 4× magnification, and the number of colonies in each field was counted manually by eye.
GILA
Subconfluent cells were trypsinized, counted, and seeded at 1000 cells per well of a 96-well plate in 100 µL of cell culture medium on ultralow-attachment round-bottom plates (Corning, catalog no. 7007). Cells were cultured at 37°C with 5% CO2, and, at the respective time points, plates were transferred to −80°C in order to freeze the cell suspension. At the completion of the time course, cells were assayed for ATP content as a proxy of cell viability using the CellTiter-Glo luminescent cell viability assay (Promega) according to the previously described protocol (Rotem et al. 2015).
Supplementary Material
Acknowledgments
We are grateful to members of the Sharp laboratory for critical discussions and insight. We thank Jacqueline Lees and David Bartel for invaluable discussions. We thank the Robert A. Swanson (1969) Biotechnology Center at the Koch Institute for Integrative Cancer Research at Massachusetts Institute of Technology for technical support, specifically Alla Leshinsky (Biopolymers Core), Glenn Paradis, Michael Jennings, Michele Griffin (Flow Cytometry Core), Bong Kim (Media), and all staff of the Hope Babette Tang (1983) Histology Facility. We thank Roderick Bronson for histopathological analysis of tumor samples. We thank Stuart Levine and the staff of the BioMicro Center at Massachusetts Institute of Technology for sequencing of RNA-seq samples. PX458 was a gift from Feng Zhang. This work was supported by United States Public Health Service grant R01CA133404, National Cancer Institute grant P01CA42063, a generous gift from Fondation MIT to Phillip A. Sharp, and, in part, Koch Institute Support (core) grant P30-CA14051 from the National Cancer Institute.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.296301.117.
References
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. 2008. The impact of microRNAs on protein output. Nature 455: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahubeshi A, Bal N, Rio Frio T, Hamel N, Pouchet C, Yilmaz A, Bouron-Dal Soglio D, Williams GM, Tischkowitz M, Priest JR, et al. 2010. Germline DICER1 mutations and familial cystic nephroma. J Med Genet 47: 863–866. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, Huttelmaier S. 2013. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci 70: 2657–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. 2003. Dicer is essential for mouse development. Nat Genet 35: 215–217. [DOI] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, Sharp PA. 2014. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Shomron N, Hedegaard MM, Vinther J, Andersen JS, Feig C, Xu J, Burge CB, Peter ME. 2008. Identification of let-7-regulated oncofetal genes. Cancer Research 68: 2587–2591. [DOI] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. 2010. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer 17: F19–F36. [DOI] [PubMed] [Google Scholar]
- Busch B, Bley N, Muller S, Glass M, Misiak D, Lederer M, Vetter M, Strauss HG, Thomssen C, Huttelmaier S. 2016. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res 44: 3845–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA. 2004. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci 101: 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wang Y, McMonechy MK, Anglesio MS, Yang W, Senz J, Maines-Bandiera S, Rosner J, Trigo-Gonzalez G, Grace Cheng SW, et al. 2015. Recurrent DICER1 hotspot mutations in endometrial tumours and their impact on microRNA biogenesis. J Pathol 237: 215–225. [DOI] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. 2005. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci 102: 13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway AE, Van Nostrand EL, Pratt GA, Aigner S, Wilbert ML, Sundararaman B, Freese P, Lambert NJ, Sathe S, Liang TY, et al. 2016. Enhanced CLIP uncovers IMP protein-RNA targets in human pluripotent stem cells important for cell adhesion and survival. Cell Rep 15: 666–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degrauwe N, Schlumpf TB, Janiszewska M, Martin P, Cauderay A, Provero P, Riggi N, Suva ML, Paro R, Stamenkovic I. 2016. The RNA binding protein IMP2 preserves glioblastoma stem cells by preventing let-7 target gene silencing. Cell Rep 15: 1634–1647. [DOI] [PubMed] [Google Scholar]
- de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, Hamel N, Choi JH, Park SH, Deal CL, et al. 2014. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol 128: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Gocheva V, Bhutkar A, Resnick R, Jong RM, Miller KM, Bendor J, Jacks T. 2016. Stromal expression of miR-143/145 promotes neoangiogenesis in lung cancer development. Cancer Discov 6: 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Petersen CP, Sharp PA. 2003. siRNAs can function as miRNAs. Genes Dev 17: 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennajdaoui H, Howard JM, Sterne-Weiler T, Jahanbani F, Coyne DJ, Uren PJ, Dargyte M, Katzman S, Draper JM, Wallace A, et al. 2016. IGF2BP3 modulates the interaction of invasion-associated transcripts with RISC. Cell Rep 15: 1876–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes WD, Bahubeshi A, Hamel N, Pasini B, Asioli S, Baynam G, Choong CS, Charles A, Frieder RP, Dishop MK, et al. 2011. Extending the phenotypes associated with DICER1 mutations. Hum Mutat 32: 1381–1384. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Priest JR, Duchaine TE. 2014. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14: 662–672. [DOI] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. 2009. Small silencing RNAs: an expanding universe. Nat Rev Genet 10: 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosline SJ, Gurtan AM, JnBaptiste CK, Bosson A, Milani P, Dalin S, Matthews BJ, Yap YS, Sharp PA, Fraenkel E. 2016. Elucidating microRNA regulatory networks using transcriptional, post-transcriptional, and histone modification measurements. Cell Rep 14: 310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KKH, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtan AM, Ravi A, Rahl PB, Bosson AD, JnBaptiste CK, Bhutkar A, Whittaker CA, Young RA, Sharp PA. 2013. Let-7 represses Nr6a1 and a mid-gestation developmental program in adult fibroblasts. Genes Dev 27: 941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Max KE, Bandaru P, Morozov P, Gerstberger S, Brown M, Molina H, Tuschl T. 2013. Identification of mRNAs bound and regulated by human LIN28 proteins and molecular requirements for RNA recognition. RNA 19: 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. 2005. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci 102: 10898–10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heravi-Moussavi A, Anglesio M, Cheng SW, Senz J, Yang W, Prentice L, Fejes AP, Chow C, Tone A, Kalloger SE, et al. 2012. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 366: 234–242. [DOI] [PubMed] [Google Scholar]
- Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, et al. 2009. DICER1 mutations in familial pleuropulmonary blastoma. Science 325: 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvarinen A, Oja E. 2000. Independent component analysis: algorithms and applications. Neural Netw 13: 411–430. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Struhl K. 2009. An epigenetic switch involving NF-κB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell 139: 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, Stamenkovic I. 2012. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev 26: 1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. 2005. RAS is regulated by the let-7 microRNA family. Cell 120: 635–647. [DOI] [PubMed] [Google Scholar]
- Jonson L, Christiansen J, Hansen TV, Vikesa J, Yamamoto Y, Nielsen FC. 2014. IMP3 RNP safe houses prevent miRNA-directed HMGA2 mRNA decay in cancer and development. Cell Rep 7: 539–551. [DOI] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. 2005. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19: 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugel S, Sebastian C, Fitamant J, Ross KN, Saha SK, Jain E, Gladden A, Arora KS, Kato Y, Rivera MN, et al. 2016. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 165: 1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. 2007. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 39: 673–677. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, Kirsch DG, Golub TR, Jacks T. 2009. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev 23: 2700–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Dutta A. 2007. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 21: 1025–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AKL, Sharp PA. 2010. MicroRNA functions in stress responses. Mol Cell 40: 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CM, Gocheva V, Oudin MJ, Bhutkar A, Wang SY, Date SR, Ng SR, Whittaker CA, Bronson RT, Snyder EL, et al. 2015. Foxa2 and Cdx2 cooperate with Nkx2-1 to inhibit lung adenocarcinoma metastasis. Genes Dev 29: 1850–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. 2011. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 27: 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Gregory RI. 2015. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 15: 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Liu Q, Zhong X, Hahn CM, Lin N, Emmett MJ, Stanger BZ, Lee JS, Rustgi AK. 2013. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes Dev 27: 2233–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manier S, Powers JT, Sacco A, Glavey SV, Huynh D, Reagan MR, Salem KZ, Moschetta M, Shi J, Mishima Y, et al. 2016. The LIN28B/let-7 axis is a novel therapeutic pathway in multiple myeloma. Leukemia 10.1038/leu.2016.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. 2007. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315: 1576–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C, et al. 2009. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet 41: 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Melo SA, Moutinho C, Ropero S, Calin GA, Rossi S, Spizzo R, Fernandez AF, Davalos V, Villanueva A, Montoya G, et al. 2010. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 18: 303–315. [DOI] [PubMed] [Google Scholar]
- Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, Mestdagh P, van Sluis P, Valentijn LJ, van Nes J, et al. 2012. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet 44: 1199–1206. [DOI] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GXY, Tsang JS, Sharp PA, van Oudenaarden A. 2011. MicroRNAs can generate thresholds in target gene expression. Nat Genet 43: 854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LH, Robinton DA, Seligson MT, Wu L, Li L, Rakheja D, Comerford SA, Ramezani S, Sun X, Parikh MS, et al. 2014. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell 26: 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J, Kim S, Zhu Y, Zhu H, Morrison SJ. 2013. A network of heterochronic genes including Imp1 regulates temporal changes in stem cell properties. Elife 2: e00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, Jacks T. 2016. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab 24: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JT, Tsanov KM, Pearson DS, Roels F, Spina CS, Ebright R, Seligson M, de Soysa Y, Cahan P, Theissen J, et al. 2016. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature 535: 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, Khokhar S, Wickiser JE, Karandikar NJ, Malter JS, et al. 2014. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi A, Gurtan AM, Kumar MS, Bhutkar A, Chin C, Lu V, Lees JA, Jacks T, Sharp PA. 2012. Proliferation and tumorigenesis of a murine sarcoma cell line in the absence of DICER1. Cancer Cell 21: 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem A, Janzer A, Izar B, Ji Z, Doench JG, Garraway LA, Struhl K. 2015. Alternative to the soft-agar assay that permits high-throughput drug and genetic screens for cellular transformation. Proc Natl Acad Sci 112: 5708–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetrom P, Heale BSE, Snove O, Aagaard L, Alluin J, Rossi JJ. 2007. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res 35: 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta S, Pursell B, Mercurio AM. 2013. IMP3 protein promotes chemoresistance in breast cancer cells by regulating breast cancer resistance protein (ABCG2) expression. J Biol Chem 288: 12569–12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ. 2007. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res 67: 9762–9770. [DOI] [PubMed] [Google Scholar]
- Sheen YS, Liao YH, Lin MH, Chu CY, Ho BY, Hsieh MC, Chen PC, Cha ST, Jeng YM, Chang CC, et al. 2015. IMP-3 promotes migration and invasion of melanoma cells by modulating the expression of HMGA2 and predicts poor prognosis in melanoma. J Invest Dermatol 135: 1065–1073. [DOI] [PubMed] [Google Scholar]
- Stohr N, Kohn M, Lederer M, Glass M, Reinke C, Singer RH, Huttelmaier S. 2012. IGF2BP1 promotes cell migration by regulating MK5 and PTEN signaling. Genes Dev 26: 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, et al. 2004. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64: 3753–3756. [DOI] [PubMed] [Google Scholar]
- Taniuchi K, Furihata M, Hanazaki K, Saito M, Saibara T. 2014. IGF2BP3-mediated translation in cell protrusions promotes cell invasiveness and metastasis of pancreatic cancer. Oncotarget 5: 6832–6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier CR, Doyle GA, Clark BA, Pitot HC, Ross J. 2004. Mammary tumor induction in transgenic mice expressing an RNA-binding protein. Cancer Res 64: 209–214. [DOI] [PubMed] [Google Scholar]
- Ventura A, Jacks T. 2009. MicroRNAs and cancer: short RNAs go a long way. Cell 136: 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, Gregory RI. 2008. Selective blockade of microRNA processing by Lin28. Science 320: 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O'Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. 2009. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 41: 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, Chen QR, Hsu CH, Yan C, et al. 2015. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. 2008. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet 40: 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegert J, Ishaque N, Vardapour R, Georg C, Gu Z, Bieg M, Ziegler B, Bausenwein S, Nourkami N, Ludwig N, et al. 2015. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27: 298–311. [DOI] [PubMed] [Google Scholar]
- Wienholds E, Koudijs MJ, van Eeden FJ, Cuppen E, Plasterk RH. 2003. The microRNA-producing enzyme Dicer1 is essential for zebrafish development. Nat Genet 35: 217–218. [DOI] [PubMed] [Google Scholar]
- Wilusz JE, Jnbaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, Sharp PA. 2012. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26: 2392–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. 2009. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 137: 647–658. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, et al. 2011. The Lin28/let-7 axis regulates glucose metabolism. Cell 147: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.