

Abstract
In an effort to more fully understand the full spectrum of human genetic variation, we generated deep single-molecule, real-time (SMRT) sequencing data from two haploid human genomes. By using an assembly-based approach (SMRT-SV), we systematically assessed each genome independently for structural variants (SVs) and indels resolving the sequence structure of 461,553 genetic variants from 2 bp to 28 kbp in length. We find that >89% of these variants have been missed as part of analysis of the 1000 Genomes Project even after adjusting for more common variants (MAF > 1%). We estimate that this theoretical human diploid differs by as much as ∼16 Mbp with respect to the human reference, with long-read sequencing data providing a fivefold increase in sensitivity for genetic variants ranging in size from 7 bp to 1 kbp compared with short-read sequence data. Although a large fraction of genetic variants were not detected by short-read approaches, once the alternate allele is sequence-resolved, we show that 61% of SVs can be genotyped in short-read sequence data sets with high accuracy. Uncoupling discovery from genotyping thus allows for the majority of this missed common variation to be genotyped in the human population. Interestingly, when we repeat SV detection on a pseudodiploid genome constructed in silico by merging the two haploids, we find that ∼59% of the heterozygous SVs are no longer detected by SMRT-SV. These results indicate that haploid resolution of long-read sequencing data will significantly increase sensitivity of SV detection.
The comprehensive discovery of genetic variation is central to the field of human genetics and more broadly to the characterization of personalized genomes and the vision of precision medicine. Variant discovery is tightly linked to advances in sequencing technology and computational algorithmic developments (Kruglyak and Nickerson 2001; The 1000 Genomes Project Consortium 2015). Despite several attempts to establish “gold standard” reference genomes over the years, a comprehensive assessment of all the genetic variants in any single human has remained elusive (Venter et al. 2001; Levy et al. 2007; Kidd et al. 2010b; Zook et al. 2014). While our understanding of single-nucleotide variants (SNVs) is beginning to approach nearly complete sensitivity for the euchromatic portion of the genome, structural variants (SVs; insertions, deletions, and inversions ≥50 bp in length) and indels (1–49 bp in length) have fared far worse because of their stronger association with repetitive DNA (Tuzun et al. 2005; Korbel et al. 2007; Mills et al. 2011; Gymrek et al. 2012; Willems et al. 2014; Sudmant et al. 2015a,b). Our inability to understand the complete spectrum of genetic variation stems from the complexity of human genetic variation, biases in the sequencing technology, and difficulties in discovery of variant regions in a diploid genome (Huddleston and Eichler 2016). The relative contribution of each of these effects to limit sensitivity has not been robustly assessed because of an inability to uncouple these aspects during whole-genome sequencing.
We sought to build a verifiable gold standard for human genetic variation by first eliminating the complexity of diploidy and then applying an alternate sequencing technology that improves sensitivity over repetitive regions of the human genome (Chaisson et al. 2015b; English et al. 2015; Shi et al. 2016). To this end, we generated data from two effectively haploid human genomes obtained from complete hydatidiform moles and systematically assessed both indels and SVs by comparing both long- and short-read data sets. Complete hydatidiform moles retain only a single set of homologous chromosomes due to either fertilization of an enucleated egg by a sperm or the subsequent loss of the maternal complement post-fertilization (Jacobs et al. 1980; Destouni et al. 2016). Each therefore represents a functionally haploid equivalent of the human genome lacking allelic variation. As part of this effort, we enhanced and extended the functionality of our previous strategy (Chaisson et al. 2015a) to include inversions as well as indels ≥2 bp. We applied this new pipeline, SMRT-SV, to single-molecule, real-time (SMRT) whole-genome sequence (WGS) data derived from a hydatidiform mole, CHM1, and a second haploid human from a different hydatidiform mole, CHM13. The combination of these two haploid human genomes allowed us to identify and rapidly validate haplotype-specific SVs and indels. We used this to establish the baseline of variation we might expect in a theoretical diploid human compared with the merging of two haploid sequence complements in silico to create a “pseudodiploid” genome. This allowed us to assess the effect of heterozygosity or diploidy on SV detection sensitivity. Finally, we used the sequence-resolved SVs to uncouple discovery from genotyping. Specifically, we investigate the potential to genotype sequence-resolved SVs more generally in deeply sequenced Illumina short-read data sets.
Results
We sequenced two complete hydatidiform mole genomes using SMRT sequencing technology, generating 62.4-fold (9.4-kbp median subread length) and 66.3-fold sequence coverage (7.4-kbp median subread length) for CHM1 and CHM13, respectively (Supplemental Table S1). We developed and applied SMRT-SV (https://github.com/EichlerLab/pacbio_variant_caller) to discover variants in these samples. The tool builds on our previous strategy where we align raw SMRT sequence reads to the human reference (GRCh38), identify signatures of putative structural variation from the alignments, and then generate local assemblies from regions with signatures of variation. We also incorporate a second approach where we tile across the entire euchromatic region of the genome in 60-kbp windows (sliding every 20 kbp) and then construct and align local assemblies back to the human reference to resolve at the single-base-pair level both SVs (insertions, deletions, and inversions ≥50 bp) and indels. These two approaches produced a median of four tiling assemblies per locus and a range of one to 14 assemblies across the genome where the maximum number of assemblies corresponded to hotspots of adjacent signature windows. We focused on SVs where more than one local assembly supported the same breakpoints and indels ≥2 bp due to the known single-base-pair error biases in SMRT sequencing technology (Chin et al. 2013). Each haploid data set was also subjected to WGS assembly using FALCON (Chin et al. 2016) and error self-correction (Chin et al. 2013) in an effort to increase sensitivity of all variants detected.
Haploid variant detection and a theoretical diploid SV call set
By using SMRT-SV, we identified 20,602 SVs in CHM1 (12,998 insertions, 7557 deletions, and 47 inversions) and 20,470 SVs in CHM13 (13,118 insertions, 7306 deletions, and 46 inversions) (Table 1; Supplemental Figs. S1–S5). We could account for 13,788 of the 15,755 euchromatic SVs (88%) previously reported for CHM1 events (Chaisson et al. 2015a). Of the remaining 12% of missing calls (1967), the majority mapped to segmental duplications—a known source of false positives. With the inclusion of the tiled-assembly approach, we recovered an additional 9000 calls not previously called with the signature-only approach. We note, however, that this data set of CHM1 SVs was generated from a new (62-fold) WGS data set using an improved sequencing chemistry (P6C4) where the average read lengths are significantly longer (Supplemental Table S1). As expected (Chaisson et al. 2015a), the majority of SVs for CHM1 (83% or 17,019 of the 20,602 SVs) were not previously reported by other recent SV studies, including the 1000 Genomes Project (Conrad et al. 2010; Kidd et al. 2010a; Mills et al. 2011; Sudmant et al. 2015a,b). We observed a similar pattern for the CHM13 human genome, where 83% (16,939/20,470) of SVs were previously unreported (Supplemental Fig. S12). Repeating the analysis with the SV callers LUMPY and WHAM, based on Illumina WGS data generated from CHM1 and CHM13, produced consistent results with 90% of SMRT-SV variants missed by short-read callers, suggesting that this increased sensitivity is driven primarily by long-read sequencing technology (Supplemental Figs. S15, S16). If we merge CHM1 and CHM13 data sets into a theoretical diploid, we identify a total of 30,062 SVs corresponding to 13.4 Mbp of sequence difference between the two haplotypes (Fig. 1A; Table 1). Half of these inserted or deleted SV sequences (6.5 Mbp) consisted of tandem repeats or complex arrays of different repeat classes (Supplemental Table S2). It is interesting to note that this single diploid identifies 44% as many SVs as reported for 2504 diploid genomes in Phase 3 of the 1000 Genomes Project (Sudmant et al. 2015b) and that 89% of all SVs we detected were missed in the short-read genomes (Fig. 1B; Supplemental Figs. S8–S10). As expected, an assessment of the length distributions shows that the majority of novel SVs (23,444 of 24,890 or 94%) were <1 kbp.
Table 1.
Summary of SVs (≥50 bp) and indels (2–49 bp) called by SMRT-SV
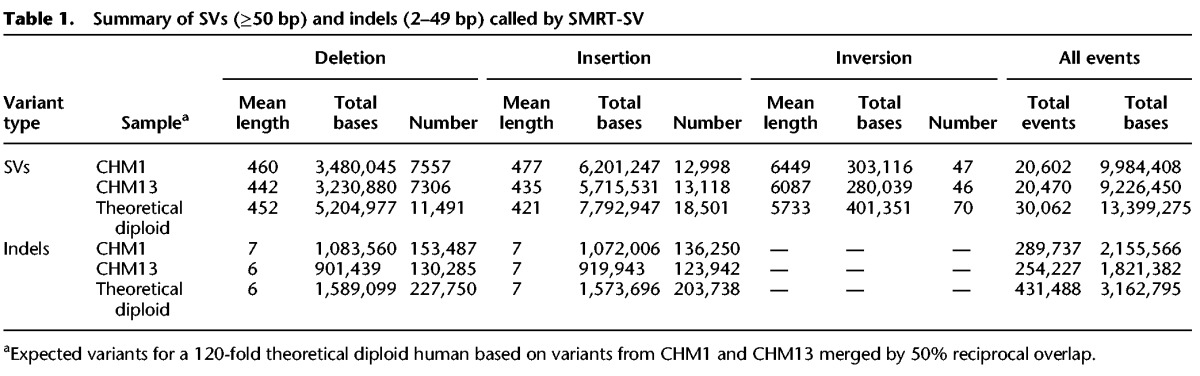
Figure 1.

Structural variant (SV) discovery. (A) SV deletions (red) and insertions (black) identified by SMRT-SV in a theoretical diploid human (CHM1 and CHM13) are classified as either novel (83%) or previously reported (17%) based on their presence in previously published SV call sets (Conrad et al. 2010; Kidd et al. 2010a; Mills et al. 2011; Sudmant et al. 2015a,b). (B) Compared specifically against insertions and deletions from Phase 3 of the 1000 Genomes Project (Sudmant et al. 2015b). Counts per call set are shown with mean and median SV size (base pair) shown in parentheses. The Venn diagram compares one theoretical diploid genome sequenced and analyzed using SMRT sequence data versus 2504 diploid genomes lightly sequenced (approximately sixfold coverage) with Illumina sequence.
We validated the SVs using four different approaches. First, we targeted 38 deletions and 58 insertions by sequence and assembly of large-insert clones and de novo assembly of SMRT WGS derived from CHM1 and CHM13 BAC libraries (CHORI-17 and VMRC59). The combination of BAC and de novo WGS assemblies confirmed the sequence and breakpoints of all 96 SV events (Supplemental Table S3). Next, we targeted 214 random, high-quality SVs <500 bp for PCR amplification and Sanger sequencing in CHM1 and CHM13 and confirmed 200 (93%) variants (Supplemental Table S4). We also compared CHM1 and CHM13 and found that 32% (9682 of 30,062) of the theoretical diploid SVs were shared between the two samples (50% reciprocal overlap), consistent with expectations based on human single-nucleotide polymorphism diversity. Finally, we assessed 30 additional human genomes and confirmed the presence of the alternate allele (see below) for 78% of all variants, suggesting that the majority of missed variants we discovered are common variants in the human population.
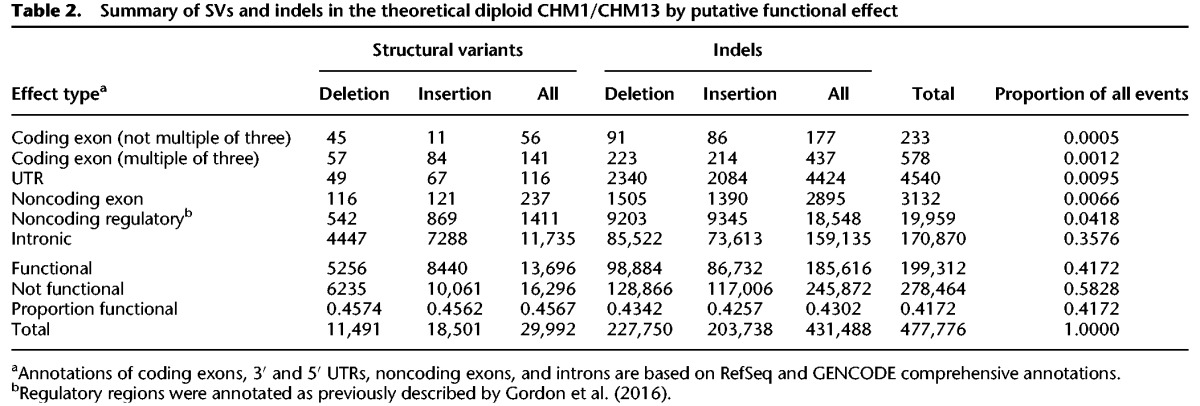
We also assessed the distribution and frequency of indels defined here as variants in the size range of 2–49 bp in both samples compared with the human reference, excluding 1-bp indels because of an enrichment for false positives. We identified 289,737 indels in CHM1, 254,227 indels in CHM13, and 431,488 in the CHM1/CHM13 diploid (Table 1). As with SVs, we selected 51 high-quality indels for PCR amplification and Sanger sequencing in CHM1 and CHM13 and confirmed 50 (98%). We find that, compared to dbSNP Build 146 (Sherry et al. 2001), which includes the 1000 Genomes Project Phase 3 variants, 72% of the indels (309,268/431,488) are novel (Fig. 2A; Supplemental Figs. S13, S14). Overall, we observed an enrichment of indels of 2-bp increments across the genome and multiples of 3 bp within genes, consistent with expected patterns of dinucleotide short tandem repeats (STRs) and selection operating within protein-coding regions, respectively (Weber et al. 2002; Bhangale et al. 2005; Gymrek et al. 2012; Montgomery et al. 2013). BAC-based sequencing and de novo assemblies of SMRT WGS established a validation rate of 95% (1349/1426) (Supplemental Table S3; Supplemental Figs. S6, S7). To more directly compare the sensitivity of short- and long-read data sets, we applied two of the most popular callers, FreeBayes (Garrison and Marth 2012) and GATK HaplotypeCaller (McKenna et al. 2010), to Illumina data generated from CHM1 and CHM13. After excluding regions of low-complexity DNA where Illumina is known to have a higher error rate due to coverage and mapping biases (Li 2014), we observed that 43% (81,640/189,096) of pseudodiploid indels from SMRT-SV were not detected by FreeBayes or GATK (Fig. 2B). Novelty was positively correlated with indel size, with a fivefold increase in sensitivity observed when indel length exceeded 7 bp (Fig. 2A). Interestingly, novel insertions surpassed deletions for the largest of category of indels (20–48 bp).
Figure 2.

Indel discovery. Small indels (2–49 bp) identified by SMRT-SV in a theoretical diploid human (CHM1 and CHM13) from SMRT WGS data are compared with merged FreeBayes and GATK HaplotypeCaller indel calls from CHM1 and CHM13 Illumina WGS. All call sets were filtered to exclude previously defined low-complexity regions (Li 2014) and 1-bp indels that cannot be reliably detected by SMRT sequence data (Gordon et al. 2016). (A) The proportion of SMRT-SV calls that are not observed in Illumina call sets increases linearly with indel size. (B) The total number of calls shared between or distinct to SMRT and Illumina WGS call sets (with mean and median call size in parentheses) highlights that 43% of SMRT-SV indels were not detected by FreeBayes or GATK, while 22% of indels in Illumina-based call sets were not detected by SMRT-SV.
In addition to SVs and indels, we also identified 3,885,137 SNVs in the theoretical diploid with support from two or more local assemblies. Of these total SNVs, 1,431,052 sites (37%) were homozygotes, while 1,253,422 (32%) were present only in CHM1 and 1,200,663 (31%) only in CHM13. We similarly estimated the false-discovery rate (FDR) of SMRT SNVs using 3,761,923 joint-called SNVs for CHM1 and CHM13 Illumina data from both FreeBayes and GATK. We observed 3,413,913 SNVs shared between SMRT and Illumina calls, corresponding to 88% of SMRT SNVs and 91% of Illumina SNVs (Supplemental Fig. S17). De novo assemblies of CHM1 and CHM13 SMRT WGS supported 75% of SNVs (350,976 of 470,268) from SMRT local assemblies that were not reported in Illumina call sets. By comparing the different forms of genetic variation in the total call set for the theoretical diploid, we estimate the ratio of SNVs to indels to be 9.0 (3,885,137/431,488) by total events and 1.2 (3,885,137/3,162,795 bp) by base-pair events. SNVs far outpace SVs in terms of the number of events (ratio, 129 SNVs to one SV event) but are dwarfed with respect to their effect on base pairs (3.4 SV base pairs to one SNV base pair).
Of all 477,776 insertions and deletions detected as SVs or indels, only 1.8% of events occurred within a GENCODE or RefSeq coding exon, noncoding exon, or untranslated region (UTR) (Table 2). An additional 4.2% of events occurred in predicted noncoding regulatory regions, including DNase hypersensitivity sites, promoters (H3K27ac), and enhancers (H3K4me3), while 35.8% of events occurred in introns (Harrow et al. 2012). The proportion of these putatively functional variants was consistent for different variant classes (46% for SVs and 43% for indels). Of particular interest are SVs or indels that occur within coding exons (RefSeq or GENCODE), do not map within segmental duplications or tandem repeats, and were not observed in previous studies. We identified 39 such variants (eight SVs and 31 indels) that affected 16 distinct genes and potentially warrant future investigation and in-depth genotyping in existing genome cohorts (Supplemental Table S5).
Table 2.
Summary of SVs and indels in the theoretical diploid CHM1/CHM13 by putative functional effect
Pseudodiploid variant detection
We modeled the accuracy of SMRT-SV for calling in diploid human samples. To establish the accuracy of SMRT-SV in diploid samples with reasonable sequencing statistics, we matched the read-length distributions of CHM1 and CHM13 genomes, downsampled both genomes to 30-fold sequence coverage each, and called variants. We combined calls from the downsampled CHM1 and CHM13 genomes to determine how many total variants to expect in a diploid sample sequenced to 60-fold total coverage. The 9754 SVs shared between the downsampled CHM1 and CHM13 represented idealized homozygotes, while the 11,477 SVs specific to CHM1 and 10,120 SVs specific to CHM13 represented heterozygous SVs where the haplotype of origin is known. Given these baseline expectations, we generated an effectively 60-fold “pseudodiploid” genome in silico by combining the 30-fold coverage SMRT sequence reads from each sample to assess sensitivity for the detection of SVs and indels when allelic variation was present and no haplotype phasing of reads was performed prior to assembly.
In the pseudodiploid experiment, we recovered 87% of “homozygous alternate” SVs shared by both CHM1 and CHM13 haplotypes (Table 3; Supplemental Fig. S18). In contrast, only 41% of heterozygous CHM1 or CHM13 SVs could be recovered. Together, the SVs recovered from the pseudodiploid correspond to an overall 44% FNR (false-negative rate) with a 13% FNR for homozygous variants and a 59% FNR for heterozygous variants. Similarly, we recovered 91% of homozygous alternate indels, 36% of heterozygous CHM1 indels, and 37% of heterozygous CHM13 indels for an overall indel FNR of 49%. Differences in sequence coverage seemed to have minimal effect on sensitivity. For example, of the 30,065 SVs we discovered with 60-fold coverage per haplotype with CHM1/CHM13, we recovered 26,211 variants (87%) with 30-fold coverage per haplotype. Of the SVs that could not be recovered at lower coverage, 93% were found only in CHM1 or CHM13 and were thus “heterozygous” in the context of the theoretical diploid. These results highlight the limitation of calling heterozygous variants in diploid genomes and the importance of methods that can effectively phase long reads into correct haplotype bins prior to variant discovery.
Table 3.
SVs and indels observed in a downsampled theoretical diploid (CHM1/CHM13) and an in silico pseudodiploid of reads from both genomes
Genotyping of SVs in Illumina genomes
It is difficult to maintain both high specificity and sensitivity when detecting SVs from short-read sequence data (Mills et al. 2011; Chaisson et al. 2015b; Sudmant et al. 2015a,b; Huddleston and Eichler 2016). To maintain a low FDR, most callers apply stringent criteria that essentially eliminate significant fractions of true SVs from further genotyping. The availability of short- and long-read data from the same source material and the fact that the majority of SVs had been resolved at the base-pair level allowed us to uncouple discovery from genotyping. We reasoned that the sequence-resolved alternate allele of the SV would facilitate more accurate genotyping of those variants in short-read data. To this end, we developed a short-read genotyper (SMRT-SV Genotyper) to assay the allele frequency of SMRT-SV variants, taking advantage of the sequence-resolved alternate alleles from each of the haploid genomes.
We first applied SMRT-SV Genotyper to PCR-free paired-end Illumina reads (151 bp) generated from the hydatidiform moles using SVs (insertions and deletions) that had been sequence-resolved from the CHM1 and CHM13 PacBio local assemblies. The moles are effectively haploid samples, so all CHM1 Illumina genotypes for CHM1 SVs are expected to be homozygous for the alternate allele, while all CHM13 Illumina genotypes for CHM1 SVs should be homozygous for either allele but never heterozygous. CHM1 homozygous reference and heterozygous genotypes and heterozygous CHM13 genotypes thus indicate potential genotyping error or invalid SV calls. From the union of 40,979 CHM1 and CHM13 insertions and deletions, we found that 77% (31,371) had sufficient coverage across the breakpoints to be genotyped by their respective sample's short reads (Supplemental Table S6). The majority of these genotypes from each mole's Illumina WGS against its respective SVs had the expected homozygous alternate genotype (29,570 or 94%), while 630 (2%) were classified as heterozygous and 1171 (4%) as homozygous reference. Similarly, 28,829 (91%) of the 31,633 SVs genotyped from each mole's Illumina WGS against the other mole's SVs were homozygous for one of the alleles. When we assessed only nonredundant calls that could be correctly assayed in the CHM1 and CHM13 Illumina WGS (see Methods), we found that 61% (18,211 of 29,992) could be concordantly genotyped in human genomes sequenced by Illumina. Of the 9608 (23% of 40,979) ungenotyped SVs, 9255 (96%) occurred within tandem repeats or segmental duplications, where it is difficult or impossible for Illumina reads to map uniquely in the human reference (Alkan et al. 2009; Li 2014). These results suggest an initial FDR of 6%–9% for those regions that can be assayed in short-read sequence data.
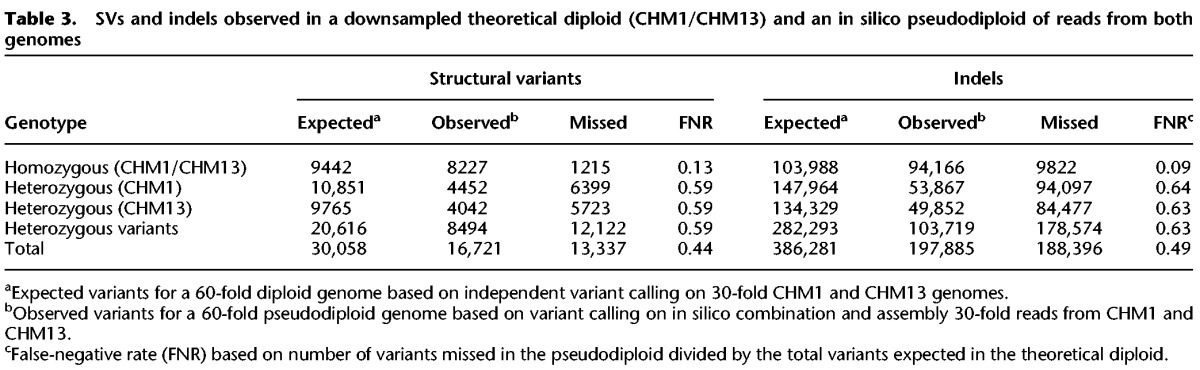
We next applied the SMRT-SV Genotyper to paired-end Illumina WGS data generated from both moles and a human diversity panel of 30 high-coverage genomes, including the NA19240 and NA12878 trios sequenced as part of the 1000 Genomes Project Consortium (The 1000 Genomes Project Consortium 2015). SVs from CHM1 and CHM13 were genotyped independently and then merged into a nonredundant set of 29,992 SVs. In keeping with the CHM1 and CHM13 genotype results, 23,613 SVs (79%) had sufficient breakpoint coverage to be genotyped in at least one sample. Of these genotyped SVs, 21,861 (93%) were present in at least one haplotype from the 30 diploid samples, corresponding to an allele frequency of 1.7% (Fig. 3A; Supplemental Fig. S11). Additionally, 1161 SVs (5%) were homozygous for the alternate allele in all 30 diploid samples, suggesting that the human reference is in error or represents the minor allele for these variants. In contrast, 1752 genotyped SVs (7%) were only observed in CHM1 or CHM13, indicating that these were rare variants or false positives from SMRT-SV. As expected, SVs mapping to GC-rich regions of the genome were less likely to be genotyped by Illumina WGS (Fig. 3B). Overall, these results confirm that the majority of SVs detected by SMRT-SV not only were previously unreported but also are polymorphic in human populations.
Figure 3.

SMRT-SV genotyping with Illumina sequence data. (A) The heatmap depicts genotypes for 18,211 of 29,992 (61%) nonredundant CHM1 and CHM13 SVs that could be concordantly genotyped in both moles by their respective Illumina WGS. Each row is a sample (two moles and 30 PCR-free samples from the 1000 Genomes Project), each column is an SV, and each cell is colored by genotype: homozygous alternate (dark blue), heterozygous (light blue), and homozygous reference (white). The number of heterozygous and homozygous alternate genotypes for each sample is indicated (parentheses). Columns are ordered by presence/absence of the SV in CHM1, CHM1/CHM13, and CHM13 and then by allele count and genomic coordinate. Specifically highlighted are 1161 SVs present in both CHM1/CHM13 and fixed (homozygous alternate) in all 30 diploid human genomes, suggesting minor alleles or sequencing errors in GRCh38. (B) The density plot compares the GC composition (x-axis) of CHM1 and CHM13 SVs that could be successfully genotyped by their respective PCR-free Illumina WGS data (77%) versus those that could not. Density plots do not represent relative proportion between the two SV categories. SVs that failed to genotype were particularly biased for GC-rich regions of the genome.
As an orthogonal assessment of genotype accuracy of the SMRT-SV Genotyper on diploid samples, we selected 20 insertions and 20 deletions each from CHM1 and CHM13 calls for validation by PCR amplification and sequencing across five DNA samples from the 1000 Genomes Project diversity panel. Specifically, we considered SVs <500 bp in size with support from 5–8 local assemblies and excluded SVs mapping to mobile element insertions (MEIs), segmental duplications, tandem repeats, or calls from previous studies. Of the 80 sites, 75% (n = 60) were successfully PCR amplified and Sanger sequenced in at least one sample. Across these 60 SVs and five samples, we successfully genotyped 264 SV/sample pairs of which 90% (n = 237) were concordant between SMRT-SV Genotyper and PCR (Table 4; Supplemental Table S7). Accuracy for MEI genotypes was slightly less than non-MEI accuracy, with 79% concordance between SMRT-SV Genotyper and PCR for MEIs shared between CHM1 and CHM13 and four samples in Stewart et al. (2011; Supplemental Table S8).
Table 4.
Genotype concordance between SMRT-SV Genotyper and PCR genotypes for five samples across 56 non-MEI SVs from CHM1 and CHM13
Discussion
We find that the theoretical amount of genetic variation in a single human diploid genome far exceeds expectations established by previous whole-genome studies (Conrad et al. 2010; Kidd et al. 2010a; Mills et al. 2011; Sudmant et al. 2015a,b). We estimate a fivefold increase in discovery from indels >7 bp and SVs <1 kbp. Although this represents only a fraction of variant sites between two haplotypes, this missing variation accounts for most of the variant base pairs between two human genomes. This increase in sensitivity stems from the improved mappability of long-read sequence data to repeat-rich regions (especially STRs and variable number tandem repeats), GC-rich DNA, and low-complexity DNA. These represent regions of the genome where short-read sequence data and variant callers are less able to discover and genotype with certainty (Gymrek et al. 2012; Willems et al. 2014; The 1000 Genomes Project Consortium 2015; Carlson et al. 2015; Sudmant et al. 2015a,b), but long-read sequence technology can access these regions because alignments are sufficiently anchored within the flanks. Although the discovery of these intermediate-sized variants is likely to remain challenging for short-read-sequencing data sets, once the alternate SV allele is resolved at the breakpoint level, we show that short reads can be used to genotype the majority of SVs relatively accurately.
Cost and throughput significantly limit the number human genomes that can be sequenced with current long-read sequencing technologies (Chaisson et al. 2015b). Sequence coverage, thus, becomes a critical consideration. For the purposes of SV discovery, we show that accurate haplotype phasing of long reads provides significantly greater yield as opposed to simply doubling the sequence coverage once a haploid sequence coverage of 30-fold is achieved. Undercalling of heterozygous SVs remains the most significant challenge for comprehensive assessment of SVs, and this limitation can be resolved if haplotypes are resolved first. The continued development of genome haplotype-phasing methods (Chaisson et al. 2015b; Chin et al. 2016; Garg et al. 2016) should facilitate partitioning long reads. This should, in turn, dramatically increase the yield of SVs.
Despite the dramatic increase in long-read variant discovery and the subsequent power of genotyping these variants in short-read data, large and highly identical repetitive regions remain effectively inaccessible to this technology. Specifically, recent segmental duplications confound most modern alignment and de novo assembly (Berlin et al. 2015; Chaisson et al. 2015b; Gordon et al. 2016). Similarly, STRs, especially longer ones, cannot be genotyped by short reads by naive assessment of genome alignments (Gymrek et al. 2012; Chaisson et al. 2015a). These regions of our genome are simultaneously the hardest to inspect, show elevated rates of mutation, and are frequently associated with human disease and evolution. As such, they warrant the continued development of duplication-aware de novo assembly methods and application of more sophisticated genotyping tools (Sudmant et al. 2010; Handsaker et al. 2011, 2015; Gymrek et al. 2012; Chin et al. 2016).
One possible scenario going forward would be to comprehensively discover SVs in a relatively small number of diverse human genomes using long-read sequence data in order to understand the full spectrum of human genetic variation, including indels. Once sequence-resolved, extensive genotyping of these variants across a larger set of human genomes sequenced deeply with short-read data would likely dramatically increase the number of SVs as part of Phase 3 of the 1000 Genomes Project (The 1000 Genomes Project Consortium 2015; Sudmant et al. 2015b). If such data sets were generated using 1000 Genomes Project samples, the efficacy of SV imputation from flanking single-nucleotide polymorphisms could also be readily assessed. Moreover, this resource would facilitate more robust genotyping of SVs in disease and population cohorts, potentially leading to novel disease associations using tools such as those developed here.
Methods
Genome sequencing and assembly
We resequenced CHM1 with PacBio long reads using a P6/C4 chemistry to produce 62.4-fold coverage (9.4-kbp median subread length; SRA accession: SRP044331). Additionally, we sequenced CHM13 with a combination of P5/C3 (60%) and P6/C4 (40%) chemistries to generate 66.3-fold coverage (7.4-kbp median subread length; SRA accessions: SRX818607, SRX825542, and SRX825575-SRX825579). Each genome's SMRT WGS was de novo assembled by FALCON v0.4 (Chin et al. 2016) and refined to high-quality consensus by Quiver (Chin et al. 2013). CHM13 was sequenced with a PCR-based Illumina library to approximately 30-fold coverage of paired 101-bp reads. PCR-free Illumina WGS for CHM1 and CHM13 were previously provided by the Broad Institute through SRA accessions ERX1413366 and ERX1413367. PCR-based Illumina WGS from CHM1 was previously published under the SRA accession SRX652547 (Chaisson et al. 2015a).
Variant discovery and validation
SMRT WGS reads were aligned to the human reference (GRCh38/hg38) with BLASR (Chaisson and Tesler 2012) and parsed to identify genomic regions with “signatures” of SVs as previously described (Chaisson et al. 2015a). Additionally, 60-kbp genomic windows with a 20-kbp slide were created across the genome. We assembled raw reads aligned to each signature region and tiled window with the PBcR de novo assembler (Berlin et al. 2015), applied Quiver (Chin et al. 2013) to generate high-quality consensus, and aligned the consensus for each region back to the corresponding reference sequence. Inversions were detected by scanning local assembly alignments for contiguous subsequences that mapped with higher identity to the reference after being reverse complemented (see Supplemental Methods). Insertions and deletions were detected by parsing alignments of local assemblies to the reference as previously described for SVs (Chaisson et al. 2015a) and with more sensitive parser settings for 2- to 49-bp indels and SNVs (see Supplemental Methods). SV insertions were further assessed for their status as nontemplate insertions (Supplemental Table S10) and as duplications of existing reference sequence (Supplemental Table S11). Indels and SNVs were called from Illumina WGS from CHM1 and CHM13 with FreeBayes 0.9.21-19-gc003c1e (Garrison and Marth 2012) and GATK HaplotypeCaller (McKenna et al. 2010). SNVs and indels from the theoretical diploid of CHM1/CHM13 were compared with variants from the Venter genome, which contained 3,213,401 SNVs and 181,730 indels totaling 1,037,246 bp (Levy et al. 2007). SVs were called from the same Illumina WGS with WHAM (Kronenberg et al. 2015) and LUMPY (Layer et al. 2014). Variants were validated by targeted sequencing of 47 BAC clones (∼9 Mbp) from CHM1 (CHORI17) and CHM13 (VMRC59), de novo assemblies of SMRT WGS by FALCON, and targeted PCR and Sanger sequencing for SVs <500 bp (Supplemental Table S4).
Genotyping
We aligned paired-end Illumina reads with BWA-MEM (v. 0.7.12-r1039) (Li 2013) in alt-aware mode to a custom reference assembly consisting of the human reference assembly (GRCh38) and either CHM1 or CHM13 local assemblies containing SV calls. Only SV-associated reads were used for genotyping (see Supplemental Methods), a heuristic used to reduce analysis time 10-fold at the expense of a 1%–5% genotyping error resulting from under-supported alternate alleles (Supplemental Table S9). For each pair of breakpoints associated with a variant (reference and alternate haplotypes), we determined the median and standard error of read depth across 25-bp windows on either side of both deletion breakpoints or the single insertion breakpoint requiring mapping quality greater than 20 and base quality greater than 20. We calculated genotype probabilities for all three biallelic genotypes using the binomial probability mass function parameterized by the median depth minus the standard error for both reference and alternate haplotypes. The final genotype for each variant was the genotype with the highest binomial probability (Supplemental Fig. S19). The number of SVs that could be genotyped was strictly defined based on the haploid nature of the hydatidiform moles as the number of SVs with homozygous alternate genotypes for one mole's SVs based on that mole's Illumina WGS and homozygous reference or alternate genotypes from the other mole's Illumina WGS. Genotype concordance was measured by targeted PCR and Sanger sequencing of CHM1 and CHM13 non-MEI SVs in five 1000 Genomes Project samples (Table 4; Supplemental Table S7) and comparison with previously PCR-validated MEI genotypes from Stewart et al. (2011) (Supplemental Table S8). Prior to visualization by heatmap, variants were filtered to include SVs where CHM1 and CHM13 had a homozygous alternate genotype in calls from their respective SMRT WGS and nonheterozygous or missing genotypes from each other's Illumina WGS. Missing genotypes were imputed using BEAGLE v4.1 (Browning and Browning 2016). Rows were clustered by UPGMA using the Euclidean distance metric.
Software
SMRT-SV provides an official software package for previously described tools (Chaisson et al. 2015a) and adds several key features, including a unified variant calling user interface with built-in cluster compute support, small indel calling (2–49 bp), improved inversion calling (screenInversions), a quality metric for SV calls based on the number of local assemblies supporting each call, higher sensitivity for SV calls using tiled local assemblies across the entire genome instead of “signature” regions, and genotyping of SVs with Illumina paired-end reads from WGS samples.
Data access
SMRT WGS for CHM1 and CHM13 from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP044331 for CHM1 and SRX818607, SRX825542, and SRX825575-SRX825579 for CHM13. Illumina WGS for CHM13, BAC assemblies from CHM13's VMRC59 BAC library, variants from CHM1/CHM13, and genotypes for SVs from this study have been submitted to the NCBI BioProject database (BioProject; https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA335618. Raw and genotyped SVs for both CHM1 and CHM13 from this study have been submitted to NCBI's database of genomic structural variation (dbVar; https://www.ncbi.nlm.nih.gov/dbvar) under accession number nstd137. De novo assemblies of CHM1 and CHM13 SMRT WGS with FALCON from this study have been submitted to the NCBI Assemblies database (Assembly; https://www.ncbi.nlm.nih.gov/assembly/) under accession numbers GCA_001297185.1 and GCA_000983455.2, respectively. SMRT-SV and screenInversions are available in the Supplemental Material (see Supplemental Code) and at https://github.com/EichlerLab/pacbio_variant_caller. Sanger sequences have been submitted to SRA under accession numbers SRR5398681-SRR5398805, and SRR5398854-SRR5398945.
Supplementary Material
Acknowledgments
We thank M.W. Hunkapiller for helpful discussions; L. Harshman, M. Scofield, C. Hill, and P. Audano for technical assistance; U. Surti for providing access to CHM1 and CHM13 cell lines for DNA isolation; and T. Brown for assistance in manuscript preparation. This work was supported, in part, by National Institutes of Health (NIH) grants HG002385 to E.E.E. and HG007635 to E.E.E. and R.K.W. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Author contributions: E.E.E., J.H., M.J.P.C., K.M.S., W.W., R.K.W., C-S.C., and J.K. designed experiments. P.P., M.B., L.V., and T.A.G. prepared libraries and generated sequence data; K.H. performed PCR validations and genotyping; J.H., M.J.P.C., D.G., Z.N.K., and C-S.C. performed bioinformatics analyses; K.M.M. performed targeted sequencing of clones; and J.H. and E.E.E. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.214007.116.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. 2009. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 41: 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. [DOI] [PubMed] [Google Scholar]
- Bhangale TR, Rieder MJ, Livingston RJ, Nickerson DA. 2005. Comprehensive identification and characterization of diallelic insertion-deletion polymorphisms in 330 human candidate genes. Hum Mol Genet 14: 59–69. [DOI] [PubMed] [Google Scholar]
- Browning BL, Browning SR. 2016. Genotype imputation with millions of reference samples. Am J Hum Genet 98: 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson KD, Sudmant PH, Press MO, Eichler EE, Shendure J, Queitsch C. 2015. MIPSTR: a method for multiplex genotyping of germline and somatic STR variation across many individuals. Genome Res 25: 750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015a. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Wilson RK, Eichler EE. 2015b. Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16: 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Chin C-S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. 2016. Phased diploid genome assembly with single molecule real-time sequencing. Nat Methods 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. 2010. Origins and functional impact of copy number variation in the human genome. Nature 464: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destouni A, Zamani Esteki M, Catteeuw M, Tsuiko O, Dimitriadou E, Smits K, Kurg A, Salumets A, Van Soom A, Voet T, et al. 2016. Zygotes segregate entire parental genomes in distinct blastomere lineages causing cleavage-stage chimerism and mixoploidy. Genome Res 26: 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Salerno WJ, Hampton OA, Gonzaga-Jauregui C, Ambreth S, Ritter DI, Beck CR, Davis CF, Dahdouli M, Ma S, et al. 2015. Assessing structural variation in a personal genome-towards a human reference diploid genome. BMC Genomics 16: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Martin M, Marschall T. 2016. Read-based phasing of related individuals. Bioinformatics 32: i234–i242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. arXiv:p1207.3907 [q-bio.GN]. [Google Scholar]
- Gordon D, Huddleston J, Chaisson MJ, Hill CM, Kronenberg ZN, Munson KM, Malig M, Raja A, Fiddes I, Hillier LW, et al. 2016. Long-read sequence assembly of the gorilla genome. Science 352: aae0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gymrek M, Golan D, Rosset S, Erlich Y. 2012. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res 22: 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Korn JM, Nemesh J, McCarroll SA. 2011. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet 43: 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA. 2015. Large multiallelic copy number variations in humans. Nat Genet 47: 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston J, Eichler EE. 2016. An incomplete understanding of human genetic variation. Genetics 202: 1251–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs PA, Wilson CM, Sprenkle JA, Rosenshein NB, Migeon BR. 1980. Mechanism of origin of complete hydatidiform moles. Nature 286: 714–716. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE. 2010a. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143: 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Sampas N, Antonacci F, Graves T, Fulton R, Hayden HS, Alkan C, Malig M, Ventura M, Giannuzzi G, et al. 2010b. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Methods 7: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. 2007. Paired-end mapping reveals extensive structural variation in the human genome. Science 318: 420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg ZN, Osborne EJ, Cone KR, Kennedy BJ, Domyan ET, Shapiro MD, Elde NC, Yandell M. 2015. Wham: identifying structural variants of biological consequence. PLoS Comput Biol 11: e1004572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Nickerson DA. 2001. Variation is the spice of life. Nat Genet 27: 234–236. [DOI] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, Hall IM. 2014. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN]. [Google Scholar]
- Li H. 2014. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30: 2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Goode DL, Kvikstad E, Albers CA, Zhang ZD, Mu XJ, Ananda G, Howie B, Karczewski KJ, Smith KS, et al. 2013. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res 23: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Guo Y, Dong C, Huddleston J, Yang H, Han X, Fu A, Li Q, Li N, Gong S, et al. 2016. Long-read sequencing and de novo assembly of a Chinese genome. Nat Commun 7: 12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C, Kural D, Stromberg MP, Walker JA, Konkel MK, Stutz AM, Urban AE, Grubert F, Lam HY, Lee WP, et al. 2011. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet 7: e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. 2010. Diversity of human copy number variation and multicopy genes. Science 330: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, et al. 2015a. Global diversity, population stratification, and selection of human copy-number variation. Science 349: aab3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. 2015b. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. 2001. The sequence of the human genome. Science 291: 1304–1351. [DOI] [PubMed] [Google Scholar]
- Weber JL, David D, Heil J, Fan Y, Zhao C, Marth G. 2002. Human diallelic insertion/deletion polymorphisms. Am J Hum Genet 71: 854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems T, Gymrek M, Highnam G, Mittelman D, Erlich Y. 2014. The landscape of human STR variation. Genome Res 24: 1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, Salit M. 2014. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol 32: 246–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.