

Abstract
Highly repetitive satellite DNA (satDNA) repeats are found in most eukaryotic genomes. SatDNAs are rapidly evolving and have roles in genome stability and chromosome segregation. Their repetitive nature poses a challenge for genome assembly and makes progress on the detailed study of satDNA structure difficult. Here, we use single-molecule sequencing long reads from Pacific Biosciences (PacBio) to determine the detailed structure of all major autosomal complex satDNA loci in Drosophila melanogaster, with a particular focus on the 260-bp and Responder satellites. We determine the optimal de novo assembly methods and parameter combinations required to produce a high-quality assembly of these previously unassembled satDNA loci and validate this assembly using molecular and computational approaches. We determined that the computationally intensive PBcR-BLASR assembly pipeline yielded better assemblies than the faster and more efficient pipelines based on the MHAP hashing algorithm, and it is essential to validate assemblies of repetitive loci. The assemblies reveal that satDNA repeats are organized into large arrays interrupted by transposable elements. The repeats in the center of the array tend to be homogenized in sequence, suggesting that gene conversion and unequal crossovers lead to repeat homogenization through concerted evolution, although the degree of unequal crossing over may differ among complex satellite loci. We find evidence for higher-order structure within satDNA arrays that suggest recent structural rearrangements. These assemblies provide a platform for the evolutionary and functional genomics of satDNAs in pericentric heterochromatin.
Satellite DNAs (satDNAs) (Kit 1961; Sueoka 1961; Szybalski 1968) are tandemly repeated DNAs frequently found in regions of low recombination (Charlesworth et al. 1994), e.g., centromeres, telomeres, and Y Chromosomes that can make up a large fraction of eukaryotic genomes (Britten and Kohne 1968). SatDNA families are classified according to their repeat unit size and composition—simple satellites generally correspond to uniform clusters of small (e.g., 1–10 bp) repeat units, and complex satellites correspond to more variable clusters of larger (e.g., >100 bp) repeat units (Rosenberg et al. 1978; Charlesworth et al. 1994). SatDNAs are highly dynamic in copy number and chromosomal location over short evolutionary time scales (Lohe and Brutlag 1987b; Plohl et al. 2012; Larracuente 2014). Changes in satDNA composition and abundance contribute to the evolution of genome structure (Charlesworth et al. 1994), speciation (Yunis and Yasmineh 1971; Ferree and Barbash 2009), and meiotic drive (Henikoff et al. 2001; Fishman and Saunders 2008). Early studies on satDNA (correctly) assumed that it must have some function in protecting against nondisjunction during chromosome segregation (Walker 1971) or a structural role in the nucleus (Yunis and Yasmineh 1971). However, subsequent studies suggested that satDNAs were inert “junk” (Ohno 1972) that expand in genomes due to selfish replication (Doolittle and Sapienza 1980; Orgel and Crick 1980; Orgel et al. 1980). In the last 15 yr, researchers across the fields of evolutionary, cell, and molecular biology have accumulated evidence that some satDNAs have important roles in centromere function heterochromatin formation and maintenance (Dernburg et al. 1996; Sun et al. 1997; Csink and Henikoff 1998; Ferree and Barbash 2009; Hughes et al. 2009; Zhu et al. 2011; He et al. 2012). However, the highly repetitive nature of satDNA makes the detailed study of their loci difficult.
Gross-scale techniques, such as density-gradient centrifugation and in situ hybridization, demonstrate that satDNAs are organized into large contiguous blocks of repeats (Peacock et al. 1974; Lohe and Brutlag 1986). Molecular assays based on restriction digest mapping indicate that satDNA blocks may be interrupted by smaller “islands” of more complex repeats such as transposable elements in Drosophila melanogaster mini-chromosomes (Le et al. 1995; Sun et al. 1997). Although these methods have been useful in detailing the overall structure of satDNA loci, detailed sequence-level analysis of these arrays is stymied by the shortcomings of traditional sequencing methods. Highly repetitive arrays are unstable in BACs and cloning vectors (Brutlag et al. 1977; Lohe and Brutlag 1986, 1987a)—in some cases they are even toxic to E. coli and thus are underrepresented in BAC libraries and among Sanger sequence reads (Hoskins et al. 2002). Next-generation short-read sequencing methods, such as Illumina or Roche 454, circumvent bacterial-based cloning related issues. These methods still pose difficulties for repeat assembly because of PCR biases and short-read lengths that result in the collapse of, or assembly gaps in, repetitive regions (Hoskins et al. 2002; Schatz et al. 2010). However, recent developments in single-molecule real-time (SMRT) sequencing (e.g., from Pacific Biosciences; PacBio) (Eid et al. 2009) address some of these issues (Koren et al. 2012; Chin et al. 2013; Berlin et al. 2015). With current sequencing chemistries, PacBio read lengths are ∼16 kb on average but reach ∼50 kb, which can bridge repetitive regions not easily resolved with short read technology. Although PacBio reads have a high error rate (∼15%), because these errors are randomly distributed, several approaches can correct the reads for use in de novo assembly (Koren et al. 2012; Ross et al. 2013; Chaisson et al. 2014; Lam et al. 2014). Hybrid approaches use deep coverage from Illumina reads for error correction of the raw PacBio reads (Koren et al. 2012). However, hybrid assemblies have difficulty dealing with regions that have large dips in short-read coverage, which can be caused by GC or sequence context biases known to affect Illumina data, resulting in breaks in the final assembly (Koren et al. 2012; Chin et al. 2013). More promising for the de novo assembly of repetitive regions are algorithms that use the PacBio reads themselves for self-correction (Koren et al. 2012; Chin et al. 2013). With sufficiently high read coverage (>50×), the longest subset of reads are corrected by overlapping the shorter reads, and the corrected long reads are then used for contig assembly, which can produce assemblies that are more contiguous than hybrid assemblies (Berlin et al. 2015; Chakraborty et al. 2016). One popular package for de novo PacBio assembly is the PBcR pipeline included in the Celera assembler. Earlier versions of the assembler (Celera 8.1) used a time-intensive all-by-all alignment step called BLASR to compute overlaps among the uncorrected reads, which accounts for >95% of runtime and is a significant bottleneck for larger genomes (Berlin et al. 2015). Newer versions of the PBcR pipeline (Celera 8.2 and later) use the recently developed MinHash Alignment Process (MHAP) algorithm to overlap and correct the reads, which is several orders of magnitude faster than BLASR (Berlin et al. 2015). Recently, development of the Celera assembler was forked to create Canu, which specializes in assembling large genomes using noisy, error-prone long reads (e.g., PacBio reads) (Berlin et al. 2015; Koren et al. 2017). Similar to Celera's PBcR pipeline, Canu uses the MHAP algorithm for fast read alignment and assembly but has been completely redesigned to improve on the Celera assembler, requiring lower read depth, faster runtime, and improved repeat and haplotype separation (Koren et al. 2017). Specifically, Canu modifies the MHAP algorithm to better recognize true overlaps in repetitive reads, improving both runtime and contiguity (Koren et al. 2017).
Assembly quality is most often evaluated based on increased overall contiguity and ability to close gaps in the euchromatin. Here, we assess the utility of long-read SMRT sequencing approaches for the accurate assembly of repetitive regions near centromeres. We experiment with three different PacBio de novo assembly pipelines—Celera 8.1 (PBcR-BLASR), Celera 8.3 (PBcR-MHAP), and Canu—to assemble satDNA regions in the pericentric heterochromatin of the D. melanogaster genome. We focus on two families of complex satDNA loci—Responder (Rsp) and 1.688 gm/cm3—and assess assembly quality through computational and molecular validation. Rsp is a satDNA that primarily exists as a dimer of two related 120-bp repeats, referred to as Rsp Left and Rsp Right, on Chromosome 2R (Wu et al. 1988; Pimpinelli and Dimitri 1989; Houtchens and Lyttle 2003; Larracuente 2014). Rsp is well-known for being a target of the selfish male meiotic drive system Segregation Distorter (for review, see Larracuente and Presgraves 2012). 1.688 gm/cm3 satDNA repeats (hereafter 1.688) are a family of related repeats, which include the 353-bp and 356-bp repeats (3L pericentromere), 260-bp (2L pericentromere), and the 359-bp (X pericentromere; Losada and Villasante 1996; Abad et al. 2000). Previous estimates show ∼80% similarity between repeat family members, with 5%–11% sequence divergence within arrays (Losada and Villasante 1996; Kuhn et al. 2012). Together, this family makes up the most abundant tandemly repeated complex satDNA in the D. melanogaster genome (Lohe and Roberts 1988). Due to the lower read coverage on the X Chromosome, we focus on the autosomal members of the 1.688 family (353-bp, 356-bp, and 260-bp), with particular focus given to 260-bp because it is more easily distinguishable from other family members. Using high-coverage (∼90×) PacBio data for D. melanogaster (Kim et al. 2014), we determine the optimal assembly protocols for complex satDNA loci and provide a detailed, base pair–level analysis of the Rsp and 1.688 family complex satDNAs.
Results
Rsp and 1.688 FISH
To confirm the gross-scale genomic distribution of Rsp and 1.688 satellites in the sequenced strain (ISO1), we performed multicolor fluorescence in situ hybridization (FISH) on mitotic chromosomes. Rsp is located in the pericentric heterochromatin on Chromosome 2R (Fig. 1; Supplemental Fig. S1), proximal to clusters of Bari-1 repeats (Supplemental Fig. S1A), in agreement with previous studies (Caizzi et al. 1993) and the PacBio assemblies. Rsp is flanked by AAGAG repeats at the cytological level (Supplemental Fig. S1B). Our 260-bp probe cross-hybridizes with 353-bp, 356-bp, and 359-bp. The 260-bp satellite is in 2L heterochromatin, whereas 353-bp/356-bp are located on Chromosome 3L at two close (but distinct) loci, and 359-bp is a large block of satellite on Chromosome X (Fig. 1).
Figure 1.
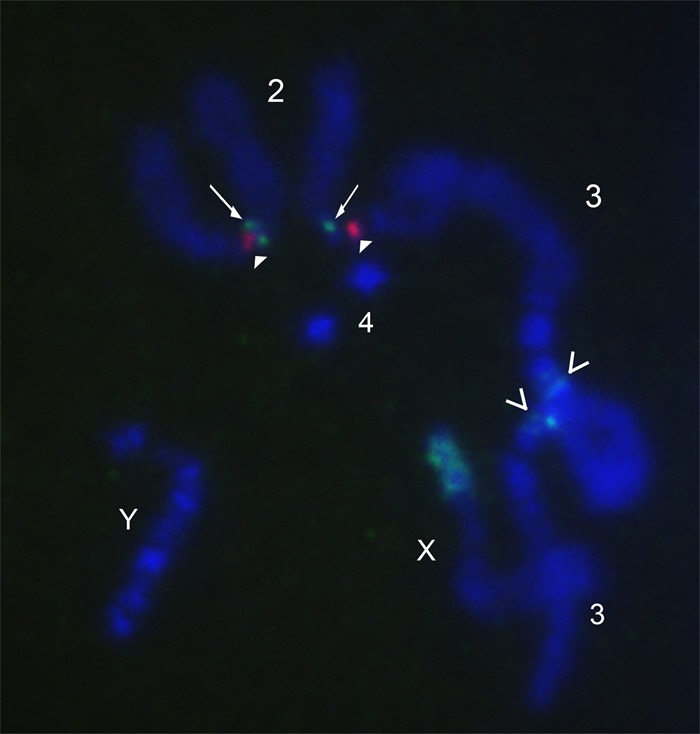
FISH image of D. melanogaster mitotic chromosomes showing Rsp and 1.688 satellites. DNA is stained with DAPI (blue). Rsp is in red (closed arrowheads) and 1.688 family satellites are in green. The 260-bp array is located on Chromosome 2L (arrows), and 353/356-bp arrays are located on Chromosome 3L (open arrowheads).
Optimal approaches to complex satellite DNA assembly
Our goal was to determine the best pipelines for assembling arrays of complex satellites. We compared de novo PacBio assemblies generated using different methods and parameters (both our own and existing assemblies) and evaluated them based on the contiguity of complex satellite sequences. We generated de novo PacBio-only assemblies using the Celera 8.3 and 8.2 PBcR pipelines (referred to as “PBcR-MHAP”) using a range of parameters (Supplemental Table S1). We generated assemblies with the experimental FALCON diploid assembler that yielded highly fragmented assemblies that we will not discuss further (Supplemental Table S2). We also experimented with the recently developed Canu 1.2 assembler using a range of error rates for the overlapping steps (referred to as “Canu” followed by the error rate) (Supplemental Table S3). Last, to determine which step is most important for proper assembly, we also generated two assemblies using the Canu and Celera 8.3 assemblers but with precorrected reads from the computationally intensive BLASR method (referred to as Canu-corr and BLASR-corr Cel8.3, respectively).
For the 260-bp locus, all PBcR-MHAP and Canu assemblies that we built using the diploid/large genome parameters, as well as the PBcR-BLASR assembly, recovered a 1.3-Mb contig that contains 284 260-bp repeats spanning ∼75 kb (Table 1). The other contigs containing 260-bp have fewer than 10 copies or are short contigs made up of only satellite sequence. Our PBcR-MHAP assemblies tended to produce these short contigs comprised entirely of Rsp or 1.688 family satellites, which were not present in the Canu, PBcR-BLASR, and the BLASR-corr Cel8.3 assemblies. In contrast to the 260-bp locus, the Rsp locus on Chromosome 2R was more variable among the different assembly methods (F-test; F = 49.09; P < 10−16). PBcR-MHAP assemblies that lacked the diploid/large genome parameters and Canu assemblies with more stringent error rates produced a fragmentary locus consisting of several contigs with approximately 200–300 Rsp repeats per contig. The PBcR-BLASR and BLASR-corr Cel8.3 assemblies each contained a single contig with approximately 1000 Rsp repeats, and whose distal end matched the Rsp locus in the latest release of the D. melanogaster reference genome (Release 6.03, which contains only about 340 copies) and is supported by a BAC. Our genomic Southern results are consistent with this number, although our pulse field gel analysis suggested a somewhat higher number of repeats (Fig. 2; Supplemental Fig. S2). The latter could be due to the different conditions under which we ran these gels (Supplemental Fig. S2). Our slot blot analysis estimating the relative abundance of Rsp in ISO1 compared to three genotypes with previously published estimates of Rsp copy number (cn bw, lt pk cn bw and SD) (Supplemental Methods; Wu et al. 1988) is also consistent with our assembly. It is important to note that although we can estimate relative abundances accurately, we believe that the precise quantification of Rsp copy number using any hybridization-based method is not feasible due to sequence variability in the repeats at the Rsp locus (e.g., Houtchens and Lyttle 2003). Rsp-containing BACs mapping to 2R heterochromatin align with 99% identity to the distal portion of the locus.
Table 1.
Summary of Rsp and 260-bp repeat counts for a subset of assemblies
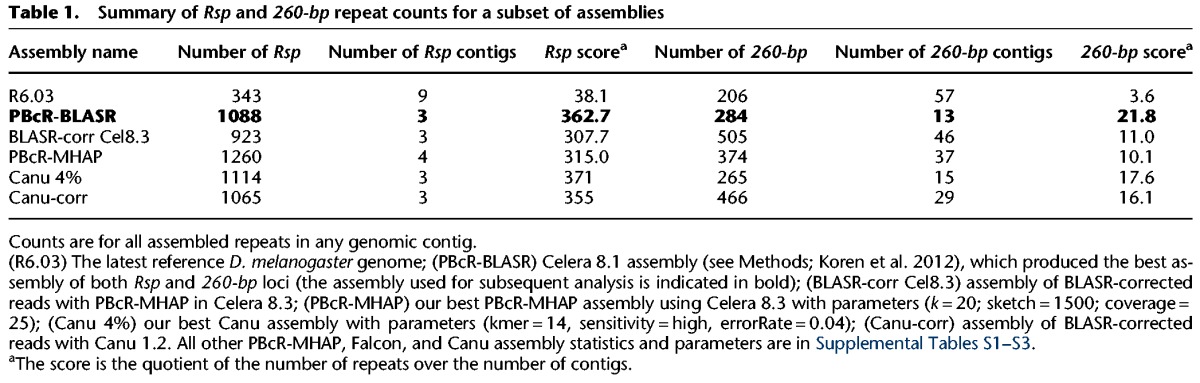
Figure 2.

(A) Southern blot of a 15-kb PCR amplicon (primer pair 3) (Fig. 3, see below) from the distal region of the Rsp locus digested with EagI, HindIII, SstI, and XmaI. We detected bands for all predicted fragments (predictions in boxes). The location of the15-kb PCR amplicon and the predicted restriction sites are shown in Supplemental Figure S2. (B) The left side shows a genomic Southern blot used to determine the assembly with the correct Rsp locus organization. Only fragments <10 kb in size were resolved. The right side shows a schematic representation of the results. Fragment sizes consistent with the PBcR-BLASR and BLASR-corr Cel8.3 are indicated with red and blue bars, respectively. Thick bars indicate double bands and dashed bars indicate fragments with few predicted Rsp repeats and thus, a comparatively weak signal. Empty boxes represent detected bands from fragments proximal to the assembled Rsp array and/or partially digested DNA. Boxes with an asterisk represent predicted fragments from Rsp repeats on Chromosome 3L. The actual banding pattern is consistent with the PBcR-BLASR assembly (red). Results from the pulse field gel confirming the overall size of the locus are in Supplemental Figure S2. (C) PCR results confirming the presence of two G5 clusters flanking the major Rsp array (primer pairs 1, 2, and 4). Primer pairs 1 and 2 yield a product for a genomic DNA template and not the 15-kb amplicon; primer pair 4 yields a product for both template types, as expected: (*) a product from elsewhere in the genome. The size (in kb) of the predicted band is below each lane: (–) no predicted product.
Several of our PBcR-MHAP assembly parameter combinations (e.g., PBcR-MHAP with k = 20; sketch = 1500; coverage = 25) and Canu assemblies with a more permissive error rate (e.g., Canu 4%) also produced a Rsp locus with about 1000 repeats, similar to PBcR-BLASR and BLASR-corr Cel8.3 (Table 1). Notably, the Canu 4% assembly resulted in a contig with about 1100 Rsp repeats but also extending another ∼250 kb distal to the Bari1 repeats (total contig length ∼587 kb) (Supplemental Fig. S3). However, although the total locus size and number of repeats were roughly consistent between the PBcR-BLASR, BLASR-corr Cel 8.3, Canu, and PBcR-MHAP assemblies, we detected rearrangements in the central Rsp repeats between these assemblies. Specifically, the Rsp locus in the Canu-corr, BLASR-corr Cel 8.3, and several of our PBcR-MHAP assemblies had an inversion relative to the PBcR-BLASR assembly. There were also various medium-to-long indels over the center of the locus between the different assemblies. Although these assemblies had contiguous satDNA loci, the disagreements over the Rsp locus indicate that some must be misassembled. It is essential to validate assemblies: those with the highest contiguity (e.g., largest contigs containing the most repeats) are not necessarily correct.
Molecular and computational validation of the Rsp locus
To distinguish between the possible configurations of the major Rsp locus, we mapped high coverage Illumina and raw PacBio reads to the assemblies containing about 1000 Rsp copies (PBcR-BLASR, BLASR-corr Cel 8.3, Canu 4%, and our PBcR-MHAP assemblies). Each PBcR-MHAP assembly, including those with highest contiguity of Rsp, had dips in coverage across the Rsp locus, suggesting that they might be misassembled (e.g., Supplemental Fig. S4). Similarly for the Canu 4% assembly, both the PacBio and Illumina mapped reads have sharp dips in coverage near the center of the Rsp locus (Supplemental Fig. S5). Several regions that have zero Illumina read coverage also have very low PacBio coverage (fewer than 10 reads), suggesting that these are misassembled regions that may also be underrepresented due to bias in DNA extraction, library construction, or representation of genomic DNA in the tissues used for library preparation. In contrast, the PBcR-BLASR and BLASR-corr Cel8.3 assemblies had uniform coverage across the contig for both the Illumina and PacBio reads (e.g., Supplemental Figs. S6–S8). We therefore focus on these two assemblies. Although both were well supported by read mapping, our alignment of the PBcR-BLASR assembly against the BLASR-corr Cel8.3 assembly showed an inversion in the central segment of the major Rsp locus (Supplemental Fig. S9). To determine the correct orientation, we designed long PCR primers that should amplify a 15-kb product based on the PBcR-BLASR assembly or no product based on the BLASR-corr Cel8.3 assembly (Figs. 2, 3A, primer pair 3). We obtained a 15-kb fragment, which we excised and digested with several restriction enzymes; Southern analysis of these digests matched the predictions from the PBcR-BLASR assembly (Fig. 2; Supplemental Fig. S2A). In addition, we performed Southern blot analysis of restriction enzyme–digested genomic DNA to look at large segments across the entire major Rsp locus; these results supported the PBcR-BLASR assembly and were inconsistent with other assemblies (Fig. 2B; Supplemental Fig. S2B,C). It is unclear why the post-error-correction assembly steps implemented in Celera 8.1 (Myers et al. 2000) produce a better assembly of the Rsp locus than subsequent versions of the assembler.
Figure 3.

Maps of complex satDNAs contigs. Counts for each repetitive element family in our custom Repbase library were plotted in 3-kb windows across each contig. (A) Rsp locus on Chromosome 2R. Blue bars correspond to Left, Right, variant or truncated repeats, whereas other colors correspond to various TE families as indicated to the right of each contig. Rsp spans ∼170 kb of the 300-kb contig (thick blue line below the x-axis). Above the plot is a schematic showing the orientation of two G5 clusters flanking the Rsp locus and a separate contig containing Rsp and the Jockey element G2, which is directly adjacent to AAGAG satellite repeats. The colors of the chevron outlines indicate the G5 elements with the highest degree of similarity with one another. Solid and dashed lines within the insertions show the approximate locations of shared insertions or deletions, respectively. Several configurations of indels are unique, such as the two in G5_5 or the deletion in G5_1, which allows verification of the cluster. The G2 contig may contain the most centromere-proximal repeats (black circle; see text). (B) Minor Rsp locus on Chromosome 2R. The inset shows the detailed orientation of the two clusters (five Rsp repeats per cluster, ∼100 kb apart); the direction of arrows indicates the relative orientation of the elements. The Rsp repeats (blue chevrons) are nested within Doc5 (orange chevrons) insertions, which are in turn nested within insertions of a transposon known as ProtoP (purple chevrons). The clusters of Rsp+Doc5+ProtoP share ∼96% sequence identity with one another, and are in an inverted orientation. (C) 260-bp locus on Chromosome 2L. Only the area surrounding the 260-bp array is shown (300 kb of ∼1.1-Mb contig). The 260-bp locus spans ∼70 kb of the 1.1-Mb contig (green below the x-axis) and is interrupted with Copia transposable elements.
To evaluate the bias in error rate, we used Pilon with high-coverage Illumina data to compare the rate of single nucleotide substitutions and indels over the contigs containing Rsp and 260-bp to the rest of the assembly. The nucleotide substitution error rate for both satDNA loci are close to the median of the empirical cumulative distribution function (ECDF) of the rate for all contigs (Supplemental Fig. S10). The indel error rates are in the first quartile of the ECDF, but still not significantly different (Supplemental Fig. S11).
Structure of Rsp loci
We find that a single 300-kb contig contains most of the major Rsp locus, and a 150-kb contig contains a minor locus directly distal to the major locus. The minor locus contains Bari1 repeats and two small clusters of variant and Left Rsp repeats (five repeats per cluster, 10 total) separated by ∼100 kb (Fig. 3B). In the Canu 4% assembly, both the major and minor Rsp loci are contained within a single contig. Interestingly, these small Rsp clusters are each inserted in the middle of a Doc5 transposon (which is itself inserted in a ProtoP element). This Rsp-Doc5-ProtoP feature is duplicated in inverted orientation ∼100 kb away (Fig. 3B), and these two units are ∼96% identical to one another.
The major Rsp locus is ∼170 kb and contains ∼1050 Rsp repeats, which are interrupted by transposable element sequences at the centromere proximal (left) and distal (right) ends of the array (Fig. 3A). The PBcR-BLASR assembly contains an additional ∼70 kb of Rsp sequence compared to the R6 (version 6.03) reference assembly, whose 2R contig terminates in a 54-kb array of Rsp and scattered TEs. Our alignment of the PBcR-BLASR and R6 assemblies shows that the Rsp locus is collapsed in R6, and that the proximal-most sequences in R6 are actually from the center of the locus; precise breakpoints are shown in Supplemental Fig. S12A and Supplemental Table S4. The presence of the Bari1 repeats at the distal end of the contig agrees with our FISH analysis (Supplemental Fig. S1) and previous studies (Wu et al. 1988; Caizzi et al. 1993). However, the PBcR-BLASR and R6 assemblies disagree over the distal-most Bari1 repeats. A Bari1-containing BAC supports the R6 configuration, suggesting that these terminal Bari1 repeats are misassembled in the PBcR-BLASR assembly. The proximal end of the contig terminates in Rsp, meaning it may be missing the most centromere-proximal repeats. We searched the raw uncorrected PacBio reads for missing Rsp repeats and found seven reads containing large (up to 6-kb) blocks of both tandem Rsp repeats and the AAGAG simple satellite, a widely distributed repeat that also localizes cytologically to the Chromosome 2 centromere and distal to Rsp on Chromosome 2R (Supplemental Fig. S1B; Lohe et al. 1993). The AAGAG+Rsp reads were not present in the error-corrected PacBio reads, and due to the high error rate of the uncorrected reads, we could not compare the AAGAG-adjacent Rsp repeats to our contig. However, the AAGAG+Rsp reads also contain a single Jockey element insertion called G2, which we used to identify 11 error-corrected reads containing Rsp and the G2 insertion. These Rsp repeats have the highest similarity to the most centromere-proximal repeats in the major Rsp locus, suggesting that they may be derived from the centromere-proximal region (Fig. 3A). We created a contig from the 11 error-corrected reads (Supplemental Fig. S13) that, when combined with the AAGAG+Rsp raw reads, suggests that our 300-kb contig is missing ∼22 kb of sequence containing about 200 Rsp repeats.
Satellites tend to undergo concerted evolution—unequal crossing over and gene conversion homogenize repeat sequences within arrays (Dover 1982, 1994; Charlesworth et al. 1994). To test the hypothesis that Rsp undergoes concerted evolution, we examined the relationship between genetic and physical distance within the 2R array. We built neighbor-joining trees for each satellite family using each full-length repeat monomer (Fig. 4). We find a pattern consistent with concerted evolution: two large clades of nearly identical repeats corresponding to the Rsp Right and Rsp Left repeats consist mainly of repeats from the center of the array. In contrast, the variant Rsp repeats have longer branch lengths and tend to occur toward the proximal and distal ends of the array (Fig. 4A). To examine the higher-order structure of the array, we studied the distribution of all unique repeat sequences across the locus according to their abundance (Fig. 5A). The approximate 1050 Rsp repeats on the main contig correspond to roughly 480 unique variants. Consistent with our phylogenetic analysis, low copy number Rsp repeats tend to dominate the ends of the array, whereas higher copy number variants dominate the center of the array (Fig. 5A).
Figure 4.

Neighbor-joining tree of complex satDNA monomers. (A) Rsp repeats in the Chromosome 2R locus. Repeats were divided into bins each of which contains one-sixth of the locus, or about 180 repeats/bin. Tip color corresponds to position in the array (red is most centromere-proximal; blue is most distal). The tip symbol indicates if the repeat is Rsp Left (square), Rsp Right (triangle), or variant/truncated (circle). (*) Repeats corresponding to the G2 contig suspected of being centromere-proximal are indicated in pink. Note that these repeats cluster with the repeats on the proximal end of the Rsp contig (red), although it is possible that these are actually distal to the locus. (B) 260-bp repeats in the Chromosome 2L locus. Repeats were divided into bins each of which contains one-fourth of the locus, or about 57 repeats/bin. Tip color corresponds to position in the array (green is most centromere proximal; red is most centromere distal). (C) Aligned consensus Rsp Left and Rsp Right repeat sequences with PCR primers (arrows) used to amplify across Left/Right Rsp dimers. (D) Consensus 260-bp repeat sequence.
Figure 5.

Distribution of satDNA sequence variants across loci. Each row corresponds to a unique monomer, and the x-axis shows the position of that monomer sequence in the array. The color of the point indicates the copy number of each monomer in the array. (A) The Rsp locus on Chromosome 2R. Several high copy number Rsp variants dominate the center of the array (purple and blue), with the low frequency and unique sequences found more toward the proximal and distal ends (gray and green). One cluster of repeats is duplicated on either side of the array (boxed). (B) The 260-bp locus on Chromosome 2L. The majority of repeats occur only once, although a few variants have intermediate copy number.
There are several TE insertions within the major Rsp array located toward the proximal and distal ends of the locus. The homogenized Rsp repeats in the center of the array are flanked by two nearly identical clusters of G5 Jockey elements (Fig. 3A). These G5 repeats form their own clade with respect to the other G5 insertions in the genome and have a high degree of similarity to one another (Supplemental Fig. S14). They have a complicated orientation, with each repeat having a near 99% identical (albeit inverted) match on the opposite side of the locus ∼100 kb away (Fig. 3A). Despite the similarity between the two clusters, there are several unique configurations of indels in each that distinguish them. We examined the pileup of raw PacBio reads over sets of long indels found in the G5 clusters and identified eight and 20 individual long reads that spanned the unique configuration of indels in the proximal G5 cluster (G5-5 and G5-6) (Fig. 3A) and distal G5 cluster (G5-3 and G5-2) (Fig. 3A), respectively. This suggests that the proximal cluster actually exists and is not an error in the assembly of the distal cluster. For further confirmation, we designed PCR primers complementary to the unique indels in the proximal cluster (primer pairs 1 and 2) and controls (primer pairs 3 and 4). In all cases, we obtain products of the expected sizes, supporting the existence of the two G5 clusters (Fig. 2C). The Rsp elements surrounding the G5 elements also show a mirrored structure (Fig. 5A,C). Interestingly, one 1.7-kb stretch of inter-G5 Rsp repeats is repeated three times, which suggests a complex series of duplication and inversion within the G5 cluster. The Rsp repeats are oriented on the same strand across most of the array, but they flip orientation at the fragmentary G5 element, mirroring what we see with the orientation of the G5 elements (Fig. 3A). Thus, the inversion did not occur only in the local area around the G5s, but across the entire proximal end of the contig.
Structure of 1.688 loci
Our FISH showed three different autosomal pericentromeric loci corresponding to members of the 1.688 family (Fig. 1; Abad et al. 2000): the 260-bp locus on 2L, and two 353/356-bp loci on 3L that are located ∼2 Mb apart in the R6 reference. The 260-bp satellite is most easily distinguishable from other family members due to a large internal deletion. The 260-bp locus is fully contained within a 1.2-Mb contig and contains 284 repeats interrupted by identical Copia transposable elements (Fig. 3C). Similar to Rsp, the 260-bp locus is collapsed in the R6 assembly relative to PBcR-BLASR (about 100 versus 284 repeats) (Table 1; Supplemental Fig. S12A). Specifically, although the proximal and distal ends of the 260-bp locus in R6 align to the PBcR-BLASR assembly, the center of the locus is absent in R6, resulting in a 39-kb gap in the alignment corresponding to the additional repeats in our assembly. We confirmed that the R6 assembly is missing 260-bp repeats using long PCR and restriction digests to verify the orientation in the PBcR-BLASR assembly (Supplemental Fig. S12B,C). The 353-bp and 356-bp monomers are more difficult to distinguish from one another and are spread across multiple contigs, all of which terminate in satellite sequence, making it difficult to assess their contiguity. We identified five contigs with large arrays of 353/356-bp, three of which align to the 3L pericentromeric heterochromatin in R6, whereas two (comprised mostly of satDNA sequence) we could not definitively place. As with the other satellites we examined, these loci are either contracted in R6 or extend into long runs of “N”s (Supplemental Fig. S12D). Like Rsp and 260-bp, the 353/356-bp arrays are organized into blocks of mostly uninterrupted satDNA sequence with TE insertions clustered toward the terminal edges of each array (Supplemental Fig. S15). Unfortunately, the similarity between 1.688 family members and the tendency for probes to cross-hybridize prevented us from using extensive molecular methods to verify the structure of these loci. However, we find uniform PacBio and Illumina read coverage over the 260-bp and 353/356-bp arrays (except contig utg 564, which has a dip in read coverage), supporting the genomic structure of these assemblies (Supplemental Fig. S15).
To detect patterns of concerted evolution, we constructed a neighbor-joining tree using monomers from the 260-bp locus, the five contigs containing 353/356-bp repeats, an X-linked 1.688 repeat (359-bp), and a related repeat (360-bp) from Drosophila simulans (Fig. 6). We find four major clades of repeats that cluster by locus, suggesting that concerted evolution is occurring within each array. Repeats from the more distal 353/356-bp locus on 3L (contig utg 43) form a well-defined cluster and are comprised almost entirely of the 356-bp sequence, whereas repeats from the more proximal locus (contigs utg 47 and utg 93) are a mix of 353-bp and 356-bp. Repeats in one of the contigs that did not align to the reference (utg 564) clustered with repeats from the proximal 1.688 locus, suggesting that they might correspond to the center of the proximal locus marked as “N”s in the reference assembly (Fig. 6). The other 1.688 contig that did not align to the reference (utg 565) formed its own group of repeats (Fig. 6), suggesting that it is distinct from the other 1.688 arrays. To determine the extent of localized gene conversion and unequal exchange, we constructed neighbor-joining trees within each contig. Unlike Rsp, we do not see positional differences in the degree of homogenization of the 1.688 satellite loci. For example, the 260-bp satellite array lacks the homogenized center and has more variant sequences (Fig. 4B). The 260-bp satellite has relatively more unique variants than Rsp: the 284 monomers correspond to 170 unique variants, and there are fewer high copy number variants (Fig. 5B). The 353/356-bp contigs are similar to the 260-bp locus: there are more variant sequences and the contigs lack a homogenized center.
Figure 6.

Neighbor-joining tree of D. melanogaster 1.688 family satellites: the 353/356-bp satellites on Chromosome 3L, the 260-bp locus on Chromosome 2L and a single consensus repeat of 359-bp from the X Chromosome, and a related repeat from D. simulans (360-bp). Tips are colored according to the contig from which they originate. The tip symbol refers to monomer repeat type: 353-bp, 356-bp, or 260-bp. The inset shows a schematic of the Chromosome 3L 1.688 loci and the organization of their respective contigs (the two clusters are ∼2 Mb apart). Contig utg 564 does not align to the reference genome, but we infer its location (*) based on repeats clustering with those in utg 47 and a gap in the reference at this genomic location. Contig utg 565 is unmapped.
Discussion
Assembly methods for complex satellites
For large complex centromeric repeats, such as human centromeres, the complete assembly of a contiguous stretch of repeats has not been possible with current technologies (Miga 2015). Instead, researchers have inferred human centromere composition using graph-based modeling strategies (Miga et al. 2014). In contrast, single-molecule sequencing has produced assemblies of more tractable, but still challenging, highly repetitive genomic regions (Chaisson et al. 2014; Carvalho et al. 2015; Krsticevic et al. 2015), including some plant centromeres (VanBuren et al. 2015; Wolfgruber et al. 2016). However, validation of these assemblies is difficult. Here, we annotate accurate de novo assemblies of two complex satDNA families in D. melanogaster using SMRT PacBio sequencing reads, allowing us to examine the detailed spatial distribution of elements within these arrays for the first time. We found that assemblers differed in their ability to produce a complete assembly for the two satellites we focused on. Although the 260-bp locus assembly was consistent between almost all PacBio assembly methods, the larger Rsp locus required the time-intensive BLASR correction algorithm for an accurate assembly. We validated the major features of the PBcR-BLASR Rsp assembly through extensive molecular and computational approaches. There are four features of the Rsp locus that could present a particular challenge for de novo assembly, especially for MHAP- and FALCON-based methods: (1) it is large (more than twice the size of the autosomal 1.688 loci); (2) it is close to the centromere (Pimpinelli and Dimitri 1989); (3) the array center is occupied by a contiguous stretch of nearly identical repeat variants, which could pose a problem when creating overlaps in the assembly process; and (4) these repeats are flanked by nearly identical TEs in inverted orientation. In contrast, the autosomal 1.688 loci are smaller, lack large runs of totally identical sequence, and do not have a complex higher-order organization. In addition to struggling with the major Rsp satDNA locus, we found that even our most contiguous PBcR-MHAP assemblies produced short contigs consisting entirely of what we believe are extraneous repeats. Despite these caveats, we recover the gross-scale organization of each of the complex satellite loci with our best PBcR-MHAP and Canu parameter combinations. We found that the PBcR-MHAP assembler requires the “diploid/large genome” parameters to produce a contiguous major Rsp locus. These parameters allow for a greater fraction of errors in the overlapping steps during error-correction, consensus calling, and unitig construction/assembly. Similarly, we found that a more strict (lower) error rate with Canu resulted in more fractured satDNA loci, whereas allowing a more lenient (higher) error rate produced more contiguous loci, but at the cost of increased assembly time. In addition, the Canu 4% error rate assembly produced a major Rsp locus in an orientation matching the validated PBcR-BLASR locus. However, read mapping indicates that the center of the locus is misassembled. Notably, areas with no support from the Illumina reads have very low PacBio coverage, suggesting that variant or error-prone reads can create spurious overlaps in ambiguous regions. Thus, the PBcR-MHAP and Canu assemblies show that a more permissive error threshold allows for better assembly of complex satDNA loci, but there is a trade-off with accuracy. The faster PBcR-MHAP and Canu approaches may offer a reasonable starting point for determining the structure of difficult repetitive loci.
One explanation for why reads corrected with BLASR generate superior assemblies of certain satDNA loci compared with PBcR-MHAP and Canu is the difference in how the two methods calculate overlaps. The MHAP algorithm used in both PBcR-MHAP and Canu converts the k-mers for each read into an integer “fingerprint,” which are collected into a set (sketch) representing the whole sequence that can be easily compared to other reads to generate an overlap; in other words, it does not actually perform an alignment (Berlin et al. 2015). A known limitation of algorithms such as MHAP is that separating true overlaps from false ones in highly repetitive regions becomes extremely computationally intensive, so in the case of the default MHAP algorithm, many overlaps are discarded; Canu attempts to address this problem by weighting repetitive overlaps (Koren et al. 2017). In contrast, BLASR generates a computationally expensive but sensitive all-by-all alignment of all the reads, which may imply full alignment of the reads is necessary for these regions. The fractured assemblies generated by PBcR-MHAP suggest that, even when using large hash sizes, this method is not sensitive enough for complex satDNAs. This finding is in contrast with recent work evaluating the repeat-rich Mst77Y region on the D. melanogaster Y Chromosome, which found that the region is misassembled in the PBcR assembly but improved with PBcR-MHAP (Krsticevic et al. 2015). Another study showed hybrid PacBio assembly combined with PBcR-MHAP assembly produced the most contiguous assembly for D. melanogaster (Chakraborty et al. 2016). Thus, optimal PacBio assembly methods seem to be dependent on the region analyzed, and careful, independent verification of the assembly is important. We find that slower but more sensitive overlapping is required for base pair–level resolution of large complex satDNA loci like Rsp, whereas PBcR-MHAP and Canu are sufficient for smaller, less homogeneous complex satDNA loci (such as 260-bp). Although the latest reference genome (R6) (Hoskins et al. 2015) offered an impressive improvement in the assembly of pericentric regions over previous releases, the de novo PacBio assembly methods evaluated here (PBcR-MHAP, Canu, and PBcR-BLASR) produced more complete and contiguous assemblies of these complex satDNAs.
Distribution of simple satellites
Despite these improvements in complex satDNA regions, no assembly method allowed us to resolve centric heterochromatin, which is enriched for simple satellite sequences. We find that in the case of two simple satellites we examined, the reads containing the satellites were progressively lost through the process of error-correction and assembly (Supplemental Table S5). Furthermore, although the raw reads did have a higher proportion of simple satellites than the error-corrected reads and the finished assembly, there still seems to be a reduced representation of simple satellite-rich raw reads. AAGAG is known to be the most abundant simple satellite in the D. melanogaster genome (∼5.6% of the genome) (Lohe and Brutlag 1986), yet it only makes up ∼0.69% of the bases in the raw reads (Supplemental Table S5). This apparent bias against raw reads derived from simple repeats has two potential explanations that are not mutually exclusive. First, PacBio sequencing may be subject to a bias that is difficult to measure because it occurs in the most highly repetitive regions of the genome. Second, the inherent structural properties of some highly repetitive DNAs may lead to a misrepresentation of these sequences in library preparation (e.g., nonrandom chromosome breakage during DNA isolation or library preparation; underreplication in tissues with endoreplicated cells). Therefore, the assembly of some simple tandem repeats still pose a significant challenge for PacBio-based assembly methods.
Structure of complex satDNA loci
Consistent with gross-scale structural analyses of satellite DNA (Brutlag et al. 1977; Lohe and Brutlag 1987a; Lohe et al. 1993; Le et al. 1995; Sun et al. 1997), we find that Rsp and 1.688 loci have uninterrupted blocks of homogeneous repeats alternating with “islands” of complex DNA. For both of these complex satDNAs, TE insertions cluster toward the array ends. The TEs in and around these loci tend to be full-length and similar to euchromatic copies, suggesting recent insertion. What gives rise to this structure? Repetitive tandem arrays are thought to expand and contract via unequal crossing over (Smith 1976), which along with gene conversion, can homogenize the array and lead to a pattern of concerted evolution (Dover 1982, 1994; Charlesworth et al. 1994). The localization of the TEs in islands near the proximal and distal ends of the locus is consistent with the “accretion model,” which predicts that repeated unequal exchange over the center of the array cause TEs to accumulate at the ends of an array (McAllister and Werren 1999). The organization of the sequence variants across the locus and the degree of homogeneity differs between Rsp and 1.688 satellites. The center of the Rsp locus is highly homogeneous and dominated by a few high-copy number variants, whereas the other loci show little homogenization over the centers of their arrays and are comprised of more variant monomers. The 260-bp locus, although showing more homogenization than the 353/356-bp loci, is comprised mostly of low-copy number or unique repeats, and the homogenized repeats are spread across the entire array rather than localized (Fig. 4B). However, in agreement with previous studies (Kuhn et al. 2012), we observed that repeats from each 1.688 locus are more similar to other repeats within their array than between arrays (Fig. 6), indicating that they are undergoing concerted evolution. It may be that gene conversion and unequal exchange occurred more frequently or more recently (e.g., recent expansion) at the Rsp locus than the 1.688 loci.
Unequal exchange breakpoints are more likely to occur within repeats rather than perfectly at the junction between two repeats, resulting in truncated repeats. For the Rsp locus, the lack of truncated repeats within the array center suggests that any unequal exchange event involved a large part of the array. The nearly identical G5 elements flanking the major Rsp array suggest a complicated rearrangement, likely involving duplication and an inversion. The high degree of similarity between the clusters is unlikely a result of gene conversion: the clusters are ∼100 kb apart, and studies in rice have shown that rates of gene conversion decrease as a function of distance between elements (Xu et al. 2008). Instead, the intervening Rsp locus may have recently expanded. Interestingly, we see a similar structure at the minor Rsp locus directly distal to the main locus: two small clusters of repeats are located 100 kb apart in an inverted orientation. Both clusters have five Rsp repeats inserted in the middle of a Doc5 Jockey element, which itself interrupts a ProtoP element. In each case, the Doc5 and Rsp elements are inserted in the same site, making it unlikely that the insertions occurred independently; instead, the entire Rsp-Doc5-ProtoP unit duplicated and inverted, and the pair are now separated by 100 kb. Because the Rsp repeats clearly interrupt the Doc5 elements, it does not appear that the movement of Rsp was mediated by TE activity. We speculate that intra-sister chromatid exchange events in the major Rsp locus—in this case, the most centromere-proximal repeats (Fig. 4A)—may have generated an extrachromosomal circular DNA, perhaps amplified through rolling circle replication, and reintegrated distal of the main cluster, in the middle of Doc5. Analogous events may seed the movement and expansion of satDNAs to new genomic regions (Cohen and Segal 2009). With some manual scaffolding, we were able to extend the assembly at the junction between Rsp and the AAGAG satDNA. Although the AAGAG satellite is known to localize to the Chromosome 2 centromeric region, it is widespread in the D. melanogaster genome and also occurs distal to the Rsp locus on Chromosome 2R (Supplemental Fig. S1B). We suspect that these reads come from the most centromere-proximal region for four reasons: (1) Rsp and AAGAG both appear 2R centromere-proximal at the cytological level; (2) the distal end of the Rsp locus is supported by BACs, and it is unlikely (but possible) that ∼20 kb of Rsp from this region would be undetected at the cytological level; (3) the major locus contig terminates in Rsp repeats proximal to the centromere; and (4) the Rsp-AAGAG repeats cluster phylogenetically with the proximal-most repeats in the main Rsp locus (Fig. 4A). Therefore, although we cannot extend the 2R assembly to the centromere, we verify that most of the pericentric Rsp satellite is assembled.
The difficulty in linking pericentric satellite arrays to centromere-adjacent repeats demonstrates that we have not entirely overcome problems assembling satellite DNA. Nevertheless, de novo PacBio assembly methods allow for exciting progress in studying the structure of previously inaccessible regions of the genome in unprecedented detail. We show here that some complex satDNA loci are tractable models for determining tandem repeat organization in pericentric heterochromatin. These assemblies provide a platform for evolutionary and functional genomic studies of satDNA in Drosophila.
Methods
Assemblies
We downloaded raw and error-corrected SMRT PacBio sequence reads from the ISO1 strain of D. melanogaster (raw read SRA accession SRX499318) (Kim et al. 2014). We also downloaded an assembly made using Celera 8.1 (Myers et al. 2000) with reads corrected using a computationally intensive all-by-all alignment by BLASR, which we refer to as “PBcR-BLASR” (http://cbcb.umd.edu/software/PBcR/dmel) (Koren et al. 2012).
We generated new assemblies using the PBcR pipeline from Celera 8.2 and 8.3 (“PBcR-MHAP”) to explore the parameter space that produces the best assembly of repetitive loci (Table 1; Supplemental Table S1). We tested 39 combinations of k-mer size, sketch size, and coverage, without the large/diploid genome parameters (http://wgs-assembler.sourceforge.net/wiki/index.php/PBcR#Assembly_of_Corrected_Sequences) or with the large/diploid parameters, which allows a more permissive error rate (e.g., Supplemental Files S1, S2, respectively). Not all parameter combinations resulted in finished assemblies, as numerous parameter combinations exceeded their allotted memory and failed, and others resulted in impractically long assembly times. For those that did finish, we evaluated assemblies for their ability to generate large contiguous blocks of the Rsp and 260-bp satellites. We chose the assembly with the most contiguous satellite arrays (“PBcR-MHAP,” k = 20, sketch = 1500, coverage = 25) as our example PBcR-MHAP assembly, although other parameter combinations produced assemblies that were very similar (Supplemental Tables S1–S3). We also created assemblies using the recently developed Canu 1.2 pipeline (Supplemental File S3). Because Canu also uses the MHAP algorithm to overlap reads similar to the Celera 8.2+ pipeline, we attempted to use parameter settings that we had optimized for MHAP (k-mer = 14, sensitivity = high). We used a range of values for the master errorRate parameter, which implicitly sets other error rates (Supplemental Table S3). The assembly with errorRate = 0.04 (“Canu 4%”) gave the most contiguous satellite arrays. In addition to the Celera and Canu assemblers, we tested different parameter combinations in the experimental diploid PacBio assembler Falcon (https://github.com/PacificBiosciences/FALCON). We tested a range of -min_cov lengths, which controls the minimum coverage when overlapping reads in the preassembly error correction step, and a range of -min_len sizes, which sets the minimum length of a read to be used in assembly. Overall, we tested 19 different combinations (example spec file in Supplemental File S4). All combinations of FALCON parameters produced a highly fragmented Rsp locus (Supplemental Table S2), and thus were excluded from further analysis.
To determine the step in the assembly process that leads to the most contiguous assembly of repeats, we assembled reads corrected with the Celera 8.1 pipeline by BLASR (http://cbcb.umd.edu/software/PBcR/dmel) (Koren et al. 2012) using the MHAP algorithm implemented in Celera 8.3 and the Canu 1.2 pipelines (Supplemental Files S5–S7). In each case, we sampled the longest 25× subset of the BLASR-corrected reads, which we then converted to an .frg file and assembled using Celera 8.3 (“BLASR-corr Cel8.3”) or Canu 1.2 (“Canu-corr”).
We ran all assemblies on a node with a pair of Intel Xeon E5-2695 v2 processors (24 cores) and 124 GB on a Linux computing cluster (Center for Integrated Research Computing, University of Rochester) using the SLURM job management system (http://slurm.schedmd.com/) (e.g., Supplemental File S8). The PBcR-BLASR assembly is available for download on NCBI (BioSample ID: SAMN02614627; PBcR-BLASR assembly accession: GCA_ 002050065.1). All other assemblies are available on the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.c0g33).
Assembly evaluation
We used custom repeat libraries that we compiled from Repbase (Supplemental File S9) and updated with consensus sequences of 1.688 family and Responder (Rsp) satellites as BLAST (blast/2.2.29+) queries against all assemblies. We created a custom Perl script to annotate contigs containing repetitive elements based on the BLAST output (Supplemental File S10). The GFF files containing our repeat annotations for the PBcR-BLASR assembly are in Supplemental Files S11–S13. For Rsp, we categorized repeats as either Left, Right, variant, or truncated based on their length and BLAST score. Our cutoff value to categorize Rsp repeats as Left or Right corresponds to the 90th percentile of the BLAST score distribution in reciprocal BLAST searches. We categorized Rsp repeats with a score below this cutoff as variant and partial repeats <90 bp as truncated. We evaluated PacBio assemblies based on the copy number and contiguity of Rsp and 260-bp repeats (Table 1; Supplemental Table S1). For both the Rsp and 260-bp loci, we imported our custom GFF files into the Geneious genome analysis tool (http://www.geneious.com) (Kearse et al. 2012) and manually annotated repeats that were still ambiguous. We also compared these assemblies to the D. melanogaster reference genome v6.03 (Hoskins et al. 2015).
Cytological validation
We confirmed the higher-order genomic organization of Rsp and 1.688 with fluorescence in situ hybridization (FISH). We designed a Cy5-labeled oligo probe to the Bari1 repeats distal to the Rsp locus (Bari1: 5′-/Cy-5/ATGGTTGTTTAAGATAAGAAGGTATCCGTTCTGAT-3′) and a FM6-labeled probe to the AAGAG repeats (of five AAGAG repeats) found both distal and proximal to Rsp on Chromosome 2R (Supplemental Fig. S1B). We generated biotin- and digoxigenin-labeled probes using nick translation on gel-extracted PCR products from the Rsp and 260-bp repeats, respectively—260F: 5′-TGGAAATTTAATTACGAGCT-3′; 260R: 5′-ATGAAACTGTGTTCAACAAT-3′ (Abad et al. 2000); RspF: 5′-CCGATTTCAAGTACCAGAC-3′; RspR: 5′-GGAAAATCACCCATTTTGACCGC-3′ (Larracuente 2014). We conducted FISH according to Larracuente and Ferree (2015) (Fig. 1; Supplemental Fig. S1). Briefly, larval brains were dissected in 1× PBS, treated with a hypotonic solution (0.5% sodium citrate) and fixed in 1.8% paraformaldehyde, 45% acetic acid, and dehydrated in ethanol. Probes were hybridized overnight at 30°C, washed in 4× SSCT and 0.1× SSC, blocked in a BSA solution, and treated with 1:100 Rhodamine-avadin (Roche) and 1:100 anti-dig fluorescein (Roche), with final washes in 4× SSCT and 0.1× SSC. Slides were mounted in VectaShield with DAPI (Vector Laboratories), visualized on a Leica DM5500 upright fluorescence microscope at 100×, imaged with a Hamamatsu Orca R2 CCD camera, and analyzed using Leica's LAX software.
Computational validation
Because we only use a subset of error-corrected PacBio reads to create de novo assemblies, we assessed the computational support for each assembly using independently derived short Illumina reads, Sanger-sequenced BACs, and the entire set of raw PacBio reads. We mapped high-coverage Illumina reads from the ISO1 strain (Gutzwiller et al. 2015) to each assembly using “–very-sensitive” settings in Bowtie 2 (Langmead and Salzberg 2012) to identify regions of low coverage that could indicate misassemblies (e.g., Supplemental Figs. S4, S5). We quantified error rate with these Illumina reads using Pilon 1.2 (Walker et al. 2014) and BCFtools 0.1.19. We calculated the number of nucleotide substitutions/contig length and the number of indels/contig length for each contig in the assembly and plotted the distribution (Supplemental Figs. S10, S11). We mapped raw PacBio reads to the PBcR-BLASR, BLASR-corr Cel 8.3, PBcR-MHAP, and Canu 4% error rate assemblies using the default parameters in the PacBio-specific BLASR aligner in the SMRT Analysis 2.3 software package available from Pacific Biosciences (e.g., Supplemental Figs. S7, S8). For the BLASR-corr Cel 8.3, PBcR-MHAP, and Canu 4% error rate assemblies, we provided the mapped PacBio reads to the Quiver genomic consensus caller to correct remaining SNPs/indels (https://github.com/PacificBiosciences/GenomicConsensus). We also mapped available BACs sequences (BACN05C06, BACR32B23, CH221-04O17) that localize to the Rsp locus (Larracuente 2014) to our assemblies.
Molecular validation
We confirmed the presence of two distinct G5 clusters using PCR analysis with primers designed in and around informative indels (Figs. 2, 3A). We used the following primers to verify the G5 clusters: primer pair 1 (5′-GGGAGCAAATGAAAAAGATTC-3′ and 5′-GTGGTATGCCTAATGGGAG-3′), primer pair 2 (5′-GGGAGCAAATGAAAAAGATTC-3′ and 5′-AGCTGATCGCCATGTGAG-3′), and primer pair 4 (5′-TCGATGAAGCTAATTGCTGG-3′ and 5′-GTGGTATGCCTAATGGGAG-3′). To confirm the locus orientation, we designed PCR primers that could only amplify an ∼15-kb segment of the distal part of the locus found in the PBcR-BLASR assembly (primer pair 3, 5′-CATGTGTGAACAGTGTATTCTG-3′ and 5′-GGCAGGAGTATTAAATCGATCTTC-3′) (Fig. 3A; Supplemental Fig. S2A) and confirmed its organization using restriction enzyme digestion with HindIII, EagI, SstI, and XmaI, and Southern blot analysis using a biotinylated Rsp probe and the North2South kit (ThermoFisher #17175) (Fig. 2A). We validated the distal and proximal ends of the locus with a Southern blot analysis (see below) on genomic DNA digested with AccI, EcoRI, FspI, and SstI (Fig. 2B; Supplemental Fig. S2B,C). For the 260-bp locus, we designed long PCR primers that span two Copia transposable element insertions in the locus that are not present in the R6 reference. We obtained a product of the expected size, which we gel extracted and digested using HinfI to confirm our PBcR-BLASR assembly (Supplemental Fig. S12B,C).
Composition and structure of satellite loci
Using maps of the locus based on our BLAST output, we extracted individual repeat units and created alignments using Geneious 8.05, which we inspected and manually adjusted (Kearse et al. 2012). We then examined the relationship between genetic distance and physical distance between repeats. We used the APE phylogenetics package in R (Paradis et al. 2004) to construct neighbor-joining trees for all monomers of each repeat family, using the “indelblock” model of substitution (Fig. 4). We then collapsed the repeats down to individual unique variants and plotted their distribution across the locus using a custom Perl script to examine any higher-order structures (Fig. 5). To evaluate how the assembly process affects simple satellite sequences, we used RepeatMasker 4.0.5 to identify simple polynucleotide repeats (using the –no-int option) in the raw (uncorrected) PacBio reads, the BLASR-corrected reads, and the final PBcR-BLASR assembly. For the raw reads, we allowed for extra divergence (--div 85) to accommodate the ∼15% error rate of the reads. We then parsed the GFF files using custom scripts to calculate the percentage of each data set comprised of the simple satellites AAGAG and AATAT (Supplemental Table S5).
Southern blot analyses
Spooled genomic DNA was obtained from approximately 60 adult females in standard phenol-chloroform extractions and resuspended in TE buffer. We performed Southern blot analyses on ∼10 ng of the 15-kb PCR amplicon and 10 µg of genomic DNA. In short, restriction enzyme digested DNA was fractionated on a 1% agarose/TAE gel and then depurinated, denatured, and neutralized before being transferred for 16 h in high salt (20 × SSC/1 M ammonium acetate) to a nylon membrane (Genescreen PlusR). DNA was UV crosslinked and hybridizations were done overnight at 55°C in North2South hybridization buffer (ThermoScientific). To make the biotinylated RNA probe, we transcribed a 240-bp Rsp gel extracted PCR amplicon (primers: T7_rsp1 5′-TAATACGACTCACTATAGGGGAAAATCACCCATTTTGATCGC-3′ and rsp2 5′-CCGAATTCAAGTACCAGAC-3′) using the Biotin RNA Labeling Mix (Roche) and T7 polymerase (Promega). The hybridized membrane was processed as recommended for the Chemiluminescent Nucleic Acid Detection Module (ThermoScientific), and the signal recorded on a ChemiDoc XR+ (BioRad). For the slot blot protocol, see Supplemental Methods.
Nuclei isolation and pulse-field gel analysis
Nuclei isolation was performed as described in Kuhn et al. (2008) with some modification. Approximately 100 flies were ground in liquid nitrogen. The powder was suspended in 0.9 µL of nuclei isolation buffer with 5 mM DTT, filtered first through a 50-µm and then through a 20-µm nitex nylon membrane (03-50/31 and 03-20/14, Sefar America) and pelleted by centrifugation at 3500 rpm for 10 min. Nuclei were resuspended in 0.2 µL of 30 mM Tris, pH 8.0, 100 mM NaCl, 50 mM EDTA, 0.5% Triton X-100, combined with an equal volume of 1% agarose, and set using a block maker (BioRad). The agarose blocks were incubated in 0.5 M EDTA (pH 8.0), 1% sodium lauryl sarcosine, and 0.1 mg/mL proteinase K overnight at 50°C and then washed in TE and restriction enzyme buffer. The blocks were digested overnight in fresh buffer with BSA and 100 units of EcoRI and AccI at 37°C. The digested blocks were run in a 1% agarose/TBE gel using a pulse field apparatus for 21 h at 8°C (4.5 V/cm; 0.5–50 sec pulses). Southern analysis was performed as above using the biotinylated Rsp probe.
Data access
Newly drawn consensus sequences for satellite sequences from this study have been submitted to NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers KY575278 (Rsp Left), KY575279 (Rsp Right), KY575280 (260-bp), KY575281 (353-bp), and KY575282 (356-bp). For 353-bp and 356-bp, the lengths of the consensus sequences do not match the expected lengths (e.g., the 353-bp consensus is not 353 bp long)—there is considerable heterogeneity in lengths of the monomers in the arrays, which was also noted when the satellites were initially characterized (Carlson and Brutlag 1979; Lohe and Brutlag 1986; Losada and Villasante 1996; Abad et al. 2000). Thus, we have kept their original names for clarity. The PBcR-BLASR assembly validated for Rsp and 260-bp satellites is available at NCBI (https://www.ncbi.nlm.nih.gov/biosample and https://www.ncbi.nlm.nih.gov/assembly/) under BioSample ID SAMN02614627 and PBcR-BLASR assembly accession GCA_002050065.1. All other assemblies used in the main text that each contain some rearrangement of the satellite loci have been submitted to the Dryad Digital Repository (http://datadryad.org/) (http://dx.doi.org/10.5061/dryad.c0g33). The Release 6.03 D. melanogaster assembly can be downloaded from FlyBase. Specification files and SLURM scripts used to construct assemblies are located in Supplemental Files S1–S7. The custom Perl script used to annotate repetitive elements using BLAST output is found in Supplemental File S10 and on the Larracuente laboratory's GitHub site (https://github.com/LarracuenteLab/Khost_Eickbush_Larracuente2017).
Supplementary Material
Acknowledgments
We would like to thank Casey Bergman, Adam Phillippy, and Sergey Koren for helpful conversations about PacBio assembly methods and for sharing assemblies, reads or protocols, and three anonymous reviewers for constructive comments. We would like the thank the staff of the Center for Integrated Research Computing at the University of Rochester for maintenance of the computing cluster and access to computational resources and Tom Eickbush for discussion. This work was supported by the University of Rochester and the National Institutes of Health–National Institute of General Medical Sciences R35-GM119515-01 to A.M.L.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.213512.116.
Freely available online through the Genome Research Open Access option.
References
- Abad JP, Agudo M, Molina I, Losada A, Ripoll P, Villasante A. 2000. Pericentromeric regions containing 1.688 satellite DNA sequences show anti-kinetochore antibody staining in prometaphase chromosomes of Drosophila melanogaster. Mol Gen Genet 264: 371–377. [DOI] [PubMed] [Google Scholar]
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. [DOI] [PubMed] [Google Scholar]
- Britten RJ, Kohne DE. 1968. Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science 161: 529–540. [DOI] [PubMed] [Google Scholar]
- Brutlag D, Carlson M, Fry K, Hsieh TS. 1977. DNA-sequence organization in Drosophila heterochromatin. Cold Spring Harb Sym 42: 1137–1146. [DOI] [PubMed] [Google Scholar]
- Caizzi R, Caggese C, Pimpinelli S. 1993. Bari-1, a new transposon-like family in Drosophila melanogaster with a unique heterochromatic organization. Genetics 133: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, Brutlag D. 1979. Different regions of a complex statellite DNA vary in size and sequence of the repeating unit. J Mol Biol 135: 483–500. [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Vicoso B, Russo CA, Swenor B, Clark AG. 2015. Birth of a new gene on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci 112: 12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2014. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ. 2016. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res 44: e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Sniegowski P, Stephan W. 1994. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371: 215–220. [DOI] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Cohen S, Segal D. 2009. Extrachromosomal circular DNA in eukaryotes: possible involvement in the plasticity of tandem repeats. Cytogenet Genome Res 124: 327–338. [DOI] [PubMed] [Google Scholar]
- Csink AK, Henikoff S. 1998. Something from nothing: the evolution and utility of satellite repeats. Trends Genet 14: 200–204. [DOI] [PubMed] [Google Scholar]
- Dernburg AF, Sedat JW, Hawley RS. 1996. Direct evidence of a role for heterochromatin in meiotic chromosome segregation. Cell 86: 135–146. [DOI] [PubMed] [Google Scholar]
- Doolittle WF, Sapienza C. 1980. Selfish genes, the phenotype paradigm and genome evolution. Nature 284: 601–603. [DOI] [PubMed] [Google Scholar]
- Dover G. 1982. A molecular drive through evolution. Bioscience 32: 526–533. [Google Scholar]
- Dover G. 1994. Concerted evolution, molecular drive and natural selection. Curr Biol 4: 1165–1166. [DOI] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138. [DOI] [PubMed] [Google Scholar]
- Ferree PM, Barbash DA. 2009. Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol 7: e1000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman L, Saunders A. 2008. Centromere-associated female meiotic drive entails male fitness costs in monkeyflowers. Science 322: 1559–1562. [DOI] [PubMed] [Google Scholar]
- Gutzwiller F, Carmo CR, Miller DE, Rice DW, Newton IL, Hawley RS, Teixeira L, Bergman CM. 2015. Dynamics of Wolbachia pipientis gene expression across the Drosophila melanogaster life cycle. G3 (Bethesda) 5: 2843–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Caudy A, Parsons L, Rosebrock A, Pane A, Raj S, Wieschaus E. 2012. Mapping the pericentric heterochromatin by comparative genomic hybridization analysis and chromosome deletions in Drosophila melanogaster. Genome Res 22: 2507–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293: 1098–1102. [DOI] [PubMed] [Google Scholar]
- Hoskins RA, Smith CD, Carlson JW, Carvalho AB, Halpern A, Kaminker JS, Kennedy C, Mungall CJ, Sullivan BA, Sutton GG, et al. 2002. Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol 3: RESEARCH0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, Booth BW, Pfeiffer BD, George RA, Svirskas R, et al. 2015. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res 25: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtchens K, Lyttle TW. 2003. Responder (Rsp) alleles in the segregation distorter (SD) system of meiotic drive in Drosophila may represent a complex family of satellite repeat sequences. Genetica 117: 291–302. [DOI] [PubMed] [Google Scholar]
- Hughes SE, Gilliland WD, Cotitta JL, Takeo S, Collins KA, Hawley RS. 2009. Heterochromatic threads connect oscillating chromosomes during prometaphase I in Drosophila oocytes. PLoS Genet 5: e1000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KE, Peluso P, Babayan P, Yeadon PJ, Yu C, Fisher WW, Chin CS, Rapicavoli NA, Rank DR, Li J, et al. 2014. Long-read, whole-genome shotgun sequence data for five model organisms. Sci Data 1: 140045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kit S. 1961. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J Mol Biol 3: 711–716. [DOI] [PubMed] [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res (this issue). 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krsticevic FJ, Schrago CG, Carvalho AB. 2015. Long-read single molecule sequencing to resolve tandem gene copies: the Mst77Y region on the Drosophila melanogaster Y chromosome. G3 (Bethesda) 5: 1145–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn GC, Sene FM, Moreira-Filho O, Schwarzacher T, Heslop-Harrison JS. 2008. Sequence analysis, chromosomal distribution and long-range organization show that rapid turnover of new and old pBuM satellite DNA repeats leads to different patterns of variation in seven species of the Drosophila buzzatii cluster. Chromosome Res 16: 307–324. [DOI] [PubMed] [Google Scholar]
- Kuhn GC, Küttler H, Moreira-Filho O, Heslop-Harrison JS. 2012. The 1.688 repetitive DNA of Drosophila: concerted evolution at different genomic scales and association with genes. Mol Biol Evol 29: 7–11. [DOI] [PubMed] [Google Scholar]
- Lam KK, Khalak A, Tse D. 2014. Near-optimal assembly for shotgun sequencing with noisy reads. BMC Bioinformatics 15(Suppl 9): S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM. 2014. The organization and evolution of the Responder satellite in species of the Drosophila melanogaster group: dynamic evolution of a target of meiotic drive. BMC Evol Biol 14: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, Ferree PM. 2015. Simple method for fluorescence DNA in situ hybridization to squashed chromosomes. JoVE 95: e52288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, Presgraves DC. 2012. The selfish Segregation Distorter gene complex of Drosophila melanogaster. Genetics 192: 33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le MH, Duricka D, Karpen GH. 1995. Islands of complex DNA are widespread in Drosophila centric heterochromatin. Genetics 141: 283–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohe AR, Brutlag DL. 1986. Multiplicity of satellite DNA sequences in Drosophila melanogaster. Proc Natl Acad Sci 83: 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohe AR, Brutlag DL. 1987a. Adjacent satellite DNA segments in Drosophila structure of junctions. J Mol Biol 194: 171–179. [DOI] [PubMed] [Google Scholar]
- Lohe AR, Brutlag DL. 1987b. Identical satellite DNA sequences in sibling species of Drosophila. J Mol Biol 194: 161–170. [DOI] [PubMed] [Google Scholar]
- Lohe AR, Roberts PA. 1988. Evolution of satellite DNA sequences in Drosophila. In Heterochromatin: molecular and structural aspects (ed. Verma RS). Cambridge University Press, Cambridge, UK. [Google Scholar]
- Lohe AR, Hilliker AJ, Roberts PA. 1993. Mapping simple repeated DNA sequences in heterochromatin of Drosophila melanogaster. Genetics 134: 1149–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Villasante A. 1996. Autosomal location of a new subtype of 1.688 satellite DNA of Drosophila melanogaster. Chromosome Res 4: 372–383. [DOI] [PubMed] [Google Scholar]
- McAllister BF, Werren JH. 1999. Evolution of tandemly repeated sequences: what happens at the end of an array? J Mol Evol 48: 469–481. [DOI] [PubMed] [Google Scholar]
- Miga KH. 2015. Completing the human genome: the progress and challenge of satellite DNA assembly. Chromosome Res 23: 421–426. [DOI] [PubMed] [Google Scholar]
- Miga KH, Newton Y, Jain M, Altemose N, Willard HF, Kent WJ. 2014. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res 24: 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196–2204. [DOI] [PubMed] [Google Scholar]
- Ohno S. 1972. So much “junk” DNA in our genome. Brookhaven Symp Biol 23: 366–370. [PubMed] [Google Scholar]
- Orgel LE, Crick FH. 1980. Selfish DNA: the ultimate parasite. Nature 284: 604–607. [DOI] [PubMed] [Google Scholar]
- Orgel LE, Crick FH, Sapienza C. 1980. Selfish DNA. Nature 288: 645–646. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289–290. [DOI] [PubMed] [Google Scholar]
- Peacock WJ, Brutlag D, Goldring E, Appels R, Hinton CW, Lindsley DL. 1974. The organization of highly repeated DNA sequences in Drosophila melanogaster chromosomes. Cold Spring Harb Symp Quant Biol 38: 405–416. [DOI] [PubMed] [Google Scholar]
- Pimpinelli S, Dimitri P. 1989. Cytogenetic analysis of segregation distortion in Drosophila melanogaster: the cytological organization of the Responder (Rsp) locus. Genetics 121: 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plohl M, Mestrovic N, Mravinac B. 2012. Satellite DNA evolution. Genome Dyn 7: 126–152. [DOI] [PubMed] [Google Scholar]
- Rosenberg H, Singer M, Rosenberg M. 1978. Highly reiterated sequences of SIMIANSIMIANSIMIANSIMIANSIMIAN. Science 200: 394–402. [DOI] [PubMed] [Google Scholar]
- Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. 2013. Characterizing and measuring bias in sequence data. Genome Biol 14: R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz MC, Delcher AL, Salzberg SL. 2010. Assembly of large genomes using second-generation sequencing. Genome Res 20: 1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP. 1976. Evolution of repeated DNA sequences by unequal crossover. Science 191: 528–535. [DOI] [PubMed] [Google Scholar]
- Sueoka N. 1961. Variation and heterogeneity of base composition of deoxyribonucleic acids: a compilation of old and new data. J Mol Biol 3: 31–40. [Google Scholar]
- Sun X, Wahlstrom J, Karpen G. 1997. Molecular structure of a functional Drosophila centromere. Cell 91: 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szybalski W. 1968. Use of cesium sulfate for equilibrium density gradient centrifugation. Methods Enzymol 12B: 330–360. [Google Scholar]
- VanBuren R, Bryant D, Edger PP, Tang H, Burgess D, Challabathula D, Spittle K, Hall R, Gu J, Lyons E, et al. 2015. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527: 508–511. [DOI] [PubMed] [Google Scholar]
- Walker PM. 1971. Origin of satellite DNA. Nature 229: 306–308. [DOI] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgruber TK, Nakashima MM, Schneider KL, Sharma A, Xie Z, Albert PS, Xu R, Bilinski P, Dawe RK, Ross-Ibarra J, et al. 2016. High quality maize centromere 10 sequence reveals evidence of frequent recombination events. Front Plant Sci 7: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI, Lyttle TW, Wu ML, Lin GF. 1988. Association between a satellite DNA sequence and the Responder of Segregation Distorter in D. melanogaster. Cell 54: 179–189. [DOI] [PubMed] [Google Scholar]
- Xu S, Clark T, Zheng H, Vang S, Li R, Wong GK, Wang J, Zheng X. 2008. Gene conversion in the rice genome. BMC Genomics 9: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunis JJ, Yasmineh WG. 1971. Heterochromatin, satellite DNA, and cell function. Structural DNA of eucaryotes may support and protect genes and aid in speciation. Science 174: 1200–1209. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM. 2011. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.