

Abstract
Long sequencing reads generated by single-molecule sequencing technology offer the possibility of dramatically improving the contiguity of genome assemblies. The biggest challenge today is that long reads have relatively high error rates, currently around 15%. The high error rates make it difficult to use this data alone, particularly with highly repetitive plant genomes. Errors in the raw data can lead to insertion or deletion errors (indels) in the consensus genome sequence, which in turn create significant problems for downstream analysis; for example, a single indel may shift the reading frame and incorrectly truncate a protein sequence. Here, we describe an algorithm that solves the high error rate problem by combining long, high-error reads with shorter but much more accurate Illumina sequencing reads, whose error rates average <1%. Our hybrid assembly algorithm combines these two types of reads to construct mega-reads, which are both long and accurate, and then assembles the mega-reads using the CABOG assembler, which was designed for long reads. We apply this technique to a large data set of Illumina and PacBio sequences from the species Aegilops tauschii, a large and extremely repetitive plant genome that has resisted previous attempts at assembly. We show that the resulting assembled contigs are far larger than in any previous assembly, with an N50 contig size of 486,807 nucleotides. We compare the contigs to independently produced optical maps to evaluate their large-scale accuracy, and to a set of high-quality bacterial artificial chromosome (BAC)-based assemblies to evaluate base-level accuracy.
Long-read sequencing technologies have made significant advances in the past few years, with read lengths rapidly increasing while costs steadily dropped. Current technology can yield reads with average lengths of 5–10 kilobases (kb) and a throughput that can reach a gigabase (Gb) from a single Pacific Biosciences (PacBio) SMRT cell. Although this technology remains more expensive and has lower throughput than Illumina sequencing, it is now feasible to generate deep coverage of a large plant or animal genome at a modest cost. The long read lengths are extremely valuable for de novo genome assembly, allowing assemblers to overcome many of the problems caused by repeated sequences that are longer than Illumina reads. This is particularly true for plant genomes, in which transposable elements with lengths >1 kb are pervasive, often occupying over half of the genome. In the absence of other linking information, any near-exact repeat longer than a read will create a break in an assembly.
Traditionally, the primary strategy for spanning long repeats has been to create paired-end libraries from long DNA fragments, ranging in size from 2–10 kb, or from even longer fosmids (∼40 kb) or BACs (∼125–150 kb). These strategies yield valuable long-range linking information, but they require more complex and more expensive methods of preparing DNA so that both ends can be sequenced. In contrast, when a single read spans a repeat and contains unique flanking sequences, the repeat can be directly incorporated into the assembly without the need to use paired-end information.
Recently, several assembly techniques have been developed for de novo assembly of a large genome from high-coverage (50× or greater) PacBio reads. These include: the PBcR assembler, which employs the MHAP algorithm (Berlin et al. 2015) together with the CABOG assembly system; the HGAP assembler (Chin et al. 2013); the Canu assembler (Koren et al. 2017), which also uses MHAP; and the FALCON assembler developed at Pacific Biosciences (https://github.com/PacificBiosciences/FALCON). Other methods employ a hybrid assembly strategy, in which short Illumina reads are used to correct errors in longer PacBio reads (Koren et al. 2012; Hackl et al. 2014; Salmela and Rivals 2014).
In this paper, we describe a new hybrid assembly technique that can produce highly contiguous assemblies of large genomes using a combination of PacBio and Illumina reads. The new method extends Illumina reads into super-reads (Zimin et al. 2013) and then combines these with the PacBio data to create mega-reads, essentially converting each PacBio read into one or more very long, highly accurate reads. The mega-reads software, which is now incorporated into the MaSuRCA assembler, can handle hybrid assemblies of almost any plant or animal genome, including genomes as large as the 22-Gbp loblolly pine. The memory usage of the hybrid assembly algorithm scales linearly with the size of the genome, and its execution time scales linearly with the depth of coverage in PacBio reads. One terabyte of memory is sufficient for most genomes under 10 Gbp in length. Here, we use this method to produce an assembly of the large and complex genome of Aegilops tauschii, one of the three diploid progenitors of bread wheat. The Ae. tauschii genome is unusually repetitive and has proven extremely difficult to assemble using short-read data.
Ae. tauschii is a self-pollinating inbred grass species whose genome is nearly homozygous, making it an excellent asset for evaluation of error rates in assembly. To this end, we have generated an optical BioNano genome (BNG) map for the Ae. tauschii genome, which provided a sequence-independent means of evaluating the large-scale accuracy of the assembly, as we discuss below. We have also independently sequenced the Ae. tauschii genome using an ordered BAC-clone sequencing approach, which provides a means of evaluating the base-level accuracy of the assembly.
Methods
Sequencing data requirements
Our hybrid assembly algorithm expects at least 100× genome coverage by paired Illumina reads of 100–250 bp, combined with at least 10× coverage in PacBio reads. Based on preliminary data, we expect that generating deeper PacBio coverage, up to 60×, is likely to improve the final results. Deeper Illumina coverage may also be beneficial. The mega-reads algorithm has the following main steps.
Super-read construction
We first transform Illumina paired-end reads into super-reads, as described previously (Zimin et al. 2013). The super-reads algorithm builds a database of all sequences of a user-specified length k, and then extends these k-mers in both directions as long as the extensions are unambiguous. In most cases, the super-reads will be much longer than the original Illumina reads, typically averaging 400 bp or more, depending on the repetitiveness of the genome. Subsequent steps of our algorithm use the longer but much lower coverage (usually 2–4×) super-reads, thus providing a very substantial degree of data compression. Super-reads are longer and have fewer errors than Illumina reads; thus, they can be mapped to high-error PacBio reads more reliably and therefore provide a better vehicle for error correction.
For the next two steps, we treat each PacBio read as a template to which super-reads can be attached, as illustrated in Figure 1.
Figure 1.
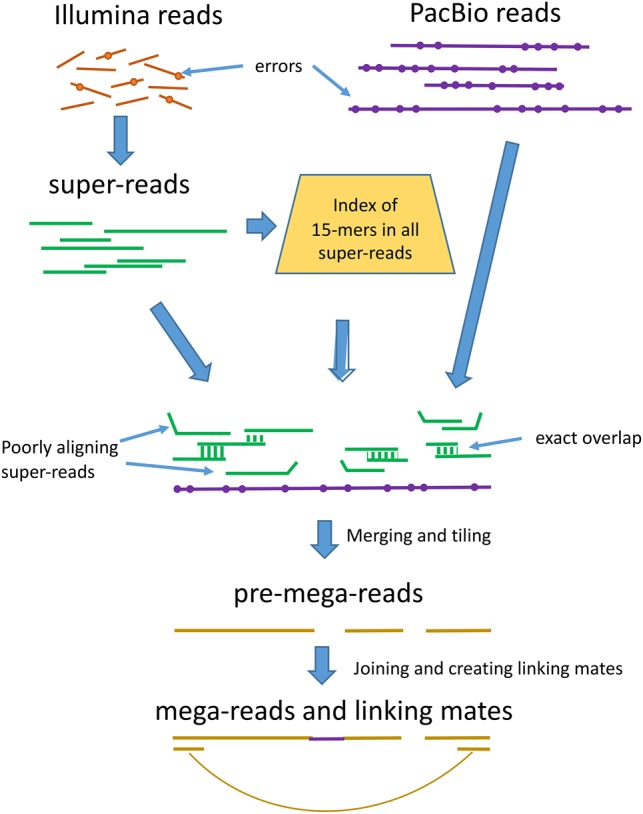
Overview of the mega-reads algorithm. Low-error rate Illumina reads (top left) are used to build longer super-reads (green lines), which in turn are used to construct a database of all 15-mers in those reads. PacBio reads (purple lines) and super-reads are then aligned, using the 15-mer index. Inconsistent super-reads are shown as kinked lines; these are discarded, and the remaining super-reads are merged, using the PacBio read as a template, to produce pre-mega-reads (yellow). These are further merged to produce the final mega-reads and to generate linking mates across gaps.
Approximate alignment along a PacBio read
We create approximate alignments of the super-reads to each PacBio read using 15-mers that the PacBio read has in common with super-reads. We used 15-mers so that we would have a sufficient number of “seeds” along most PacBio reads; smaller k-mers might also work but could increase the spurious match rate. Note that the choice of k = 15 for this step can be changed for different data sets. We first build a database of all 15-mers in the super-reads and use this database to compute, for each super-read, its approximate start and end positions on each PacBio read. This approach is similar (although different in many details) to both MHAP (Berlin et al. 2015) and minimap (Li 2016), in that both these other algorithms find chains of “seed” alignments in long PacBio reads. Our method does not compute a full alignment.
For each PacBio read P, we walk down the read looking at each 15-mer. We use the 15-mer database to determine (in constant time for each 15-mer) which 15-mers are found in super-reads. Once we have the super-reads that match P, for each such super-read S we look for ordered subsequences of the 15-mers that both P and S have in common. (The 15-mers can be overlapping.) We then assign a score to each super-read S, where the score is the number of 15-mers in the longest common subsequence (LCS) of 15-mers in the two reads. We label an alignment as plausible if the score of S exceeds some specified minimum. For each plausible alignment, we compute an approximate position of S along P based on the positions of the LCS 15-mers in P and S.
Note that a super-read can align to many different PacBio reads; the number will depend on the depth of coverage of the PacBio data.
Graph traversal for a PacBio read
Let K denote the k-mer size that was used to generate the super-reads. After super-reads are constructed, we record all exact overlaps of pairs of super-reads for which the length of the overlap is at least K.
Using all super-read positions on a PacBio read P, we create possible paths of (plausible) super-reads along P. Each path consists of a sequence of super-reads where two adjacent super-reads must have an exact overlap of at least K bases and also must have positions on P that make it possible for them to overlap. We compute an LCS score for each path.
A path might span only part of P, and conversely, subsequences of P might not be covered by any path. We then form a graph consisting of the paths along P, where super-reads are the nodes and K-overlaps are the edges. For each connected component of paths (or more precisely, a connected graph of super-reads), we compute the LCS score and we choose the path with the highest score. We call each such path a pre-mega-read. The sequence of a mega-read is essentially a long, high-quality “read” that covers part or all of the original PacBio read P.
At this point, each connected component is a directed acyclic graph (DAG) of super-reads that overlap by at least K bases and that align to P. The approximate positions of the super-reads on P impose a topological order on the DAG. We impose an overall direction on the DAG from the 5′ end toward the 3′ end of P.
Tiling
We tile the PacBio read P with the pre-mega-reads in a greedy fashion, beginning with the longest pre-mega-read, and disallowing overlaps longer than K bases (Fig. 1). We choose the pre-mega-reads for P by maximizing the total of all LCS scores in the tiling.
Many PacBio reads will be tiled by more than one pre-mega-read; i.e., the tiling has gaps. Gaps might be caused by lack of Illumina read coverage for parts of the genome, or by long stretches of poor-quality sequence in a PacBio read, or (rarely) by chimeric PacBio reads. Even though we have PacBio sequence spanning these gaps, we choose not to simply merge the pre-mega-reads using raw PacBio read sequence because that might create stretches of low-quality sequence in the mega-reads. However, if multiple PacBio reads overlap one another for the sequence in one of these gaps, we can sometimes fill the gap between pre-mega-reads. We only use raw PacBio read sequence to join neighboring pre-mega-reads if (1) the tilings for at least three PacBio reads have nearly identical gaps, (2) the pre-mega-reads surrounding the gap have identical sequence, and (3) the gap lengths are nearly identical. Here, gap length is determined by the length of the PacBio sequence between the aligned pre-mega-reads. If these conditions are met, we compute the gap-filling sequence from the original PacBio reads that span the gap. The consensus step in the CABOG assembler then creates the final version of the sequence. For the Ae. tauschii assembly, the average length of filled-in gaps was 206 bp, and the final assembly contained a total of 14 Mbp (0.03%) that resulted from filling gaps, of which 5.8 Mb (0.014%) was covered by the minimum of three PacBio reads.
It is also possible that a gap in the tiling is not a gap at all but instead is an erroneous insertion in the PacBio read. In these cases, the pre-mega-reads flanking the gap may overlap one another. If the pre-mega-reads overlap by at least 37 bp, then we merge them to close the gap.
Note that the user can set the maximum gap size for the gap-filling procedure, and the algorithm will not attempt to fill gaps larger than this maximum. If one sets the maximum gap size to zero, the mega-reads assembler will not use raw PacBio sequence at all and will only join pre-mega-reads when they have an exact overlap of 37 bases or more. For Ae. tauschii, the maximum gap size was 750 bp.
The result of this tiling and gap-filling process is the final set of mega-reads.
Creating linking pairs
When mega-reads cannot be merged and a gap remains, we create a linked pair of “reads” that spans the gap. We extract two 500-bp sequences from the mega-reads flanking the gap and link them together as mates (Fig. 1). (If either mega-read is <500 bp, we create a shorter linking read.) The assembler uses these sequences in its scaffolding step to ensure that all mega-reads from the same PacBio read are kept adjacent in the assembly; i.e., they are placed into the same scaffold. We call these artificial mates the linking pairs.
Assembly
Finally, we assemble the mega-reads along with the linking pairs into contigs and scaffolds using the CABOG assembler (Miller et al. 2008). For this step, we can also use other linking information, if available, for scaffolding.
Results
Data sets
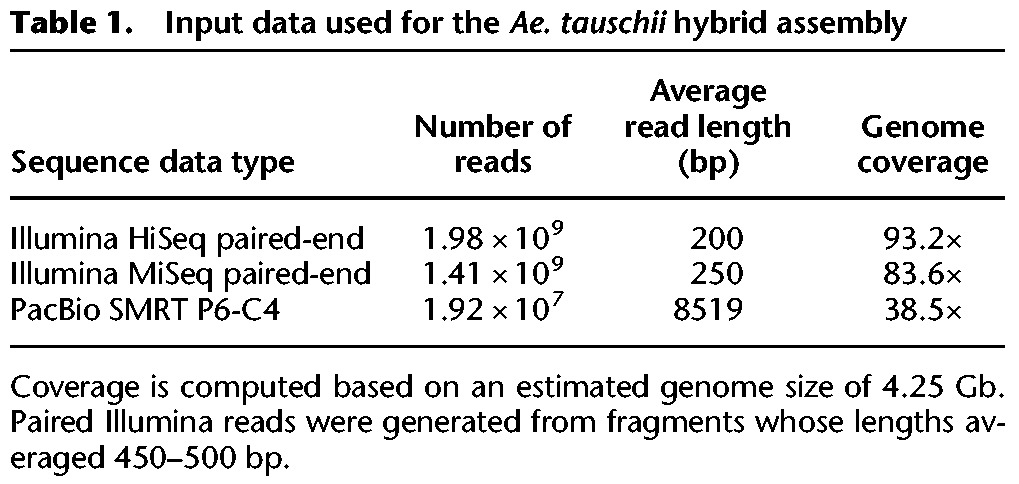
We generated over 19 million PacBio reads, equivalent to ∼38× genome coverage, using the SMRT P6-C4 chemistry. We also generated a total of 177× coverage on an Illumina HiSeq 2500 in paired 200-bp reads and an Illumina MiSeq with paired 250-bp reads (Table 1). These data sets were the only input used for our hybrid assembly of Ae. tauschii.
Table 1.
Input data used for the Ae. tauschii hybrid assembly
Ae. tauschii assembly
The methods described above produced 16.7 M super-reads (using a k-mer length of 127) from the Illumina data, and 18.7 M mega-reads from the super-reads and PacBio reads (Table 2). We used the CABOG assembler (version wgs-8.3rc2) in the MaSuRCA mega-reads pipeline to produce an initial assembly of the mega-reads. This assembly contained 128,898 scaffolds totaling 4.778 Gb in length. We then manually ran a post-processing step that included four rounds of aligning each scaffold to all others, in order to remove scaffolds that were either duplicated or that were completely contained within other scaffolds. For this alignment step, we ran BWA-MEM (Li 2013) with parameters –k127 –e and then used NUCmer (Delcher et al. 2002) to find and remove duplicate alignments. This procedure identified a total of 75,338 scaffolds (most of them very small) that were contained in other scaffolds and could be safely removed. Total computational time for all steps of the assembly was approximately 110,000 CPU hours, with about 72,000 CPU hours for computing super-reads and mega-reads, and the remaining time spent in assembly of contigs and scaffolds. The code is highly parallelized so that most procedures were run in parallel on large computing grids.
Table 2.
Statistics for super-reads and mega-reads
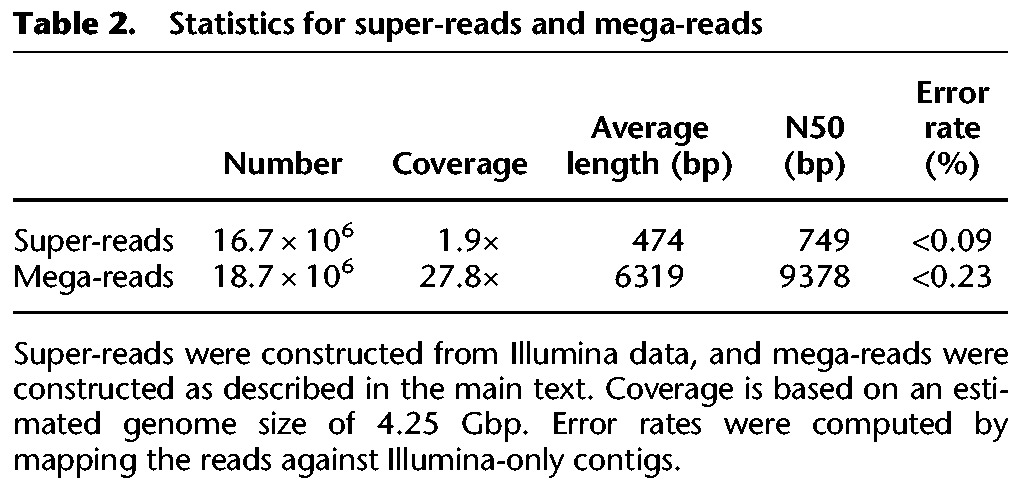
The resulting Ae. tauschii assembly, version Aet_MR.1.0, contains 53,560 scaffolds with a total span of 4.338 Gb, a contig N50 size of 486,807 bp, and a scaffold N50 size of 521,653 bp (Table 3). As described above, scaffolding was minimal because it used only the linking pairs created from mega-reads that flanked gaps in the original PacBio reads. Thus, for every gap internal to a scaffold, we have at least one PacBio read spanning the gap. The principal benefit of PacBio reads and of the mega-reads algorithm is the much larger contigs that result, ∼30 times larger than the contigs from an Illumina-only assembly (Table 3).
Table 3.
Assembly statistics for Ae. tauschii Aet_MR.1.0 compared to other assemblies

We also created a whole-genome assembly with the PacBio reads only, using the Canu assembler (Berlin et al. 2015). For this assembly, we generated additional data to bring the PacBio coverage up to 55×. The Canu assembly had a total length of 4.06 Gb and an N50 size (using 4.25 Gb as the genome size) of 311,860 bp (Table 3). It is worth noting here that the Aet_MR.1.0 assembly has many more contigs than the Canu assembly, but this is due to a large number of small contigs in the tail of the distribution and to the fact that the Aet_MR.1.0 assembly is ∼264 Mb larger. If we select contigs from the Aet_MR.1.0 assembly whose sizes total 4.06 Gb (the same total as the Canu assembly), we need only 24,309 contigs, almost exactly the same number as in the Canu assembly, and the smallest such contig is 15,911 bp. The total computation time for the Canu assembly was 38,000 CPU hours, while the mega-reads assembly took 110,000 hours. However, expected improvements in these algorithms suggest that the two assemblers would take less computational time today and will likely continue to improve.
Evaluation of assembly quality
We evaluated the quality of the output contigs using two metrics: large-scale contiguity and consensus sequence accuracy. To evaluate large-scale accuracy, we used an independently constructed BioNano Genomics (BNG) map. This technology, developed by BioNano Genomics, allows the construction of accurate maps based on restriction enzymes, in which DNA molecules are passed through a nanochannel and fluorescently tagged restriction sites are detected (Lam et al. 2012). This process creates many small restriction-mapped regions that can span several megabases each. BNG maps have recently been used to improve the assembly of portions of several highly repetitive plant genomes, including one arm of the bread wheat genome (Stankova et al. 2016), a 2-Mb fragment of Ae. tauschii (Hastie et al. 2013), and six small but complex regions of the maize genome (Dong et al. 2016). To use these maps to assess quality of a sequence assembly, the distances and positions of the same restriction sites along the BNG map are compared with a restriction map constructed computationally.
Here, we used the Ae. tauschii BNG map only to evaluate the correctness of Aet_MR.1.0; it was not used to construct or modify the assembly. We aligned our contigs to the BNG map using the restriction sites in the contigs, and searched for regions of disagreement, which indicate either an error in the BNG map or a misassembled contig. We only considered alignments that shared at least 15 restriction sites, which in most cases meant the aligned contigs were longer than 140 kb. Note that the BNG map does not contain sequence, and the only errors this procedure can detect are relatively large-scale rearrangements, insertions, or deletions.
We found 572 locations where a contig disagreed with the BNG map. All disagreements were apparent chimeric joins in the contigs with respect to the BNG map. A typical signature of a misassembly is a sharp drop in coverage, indicating a possible “weak” overlap holding the contig together. Given that the average coverage in mega-reads was ∼28× (Table 2), we flagged as a possible misassembly any disagreement with the BNG map where the coverage was <4; this analysis flagged 342 locations. We examined a small set of the higher-coverage discrepancies manually, and in each case the assembly appeared correct (based on underlying support from paired Illumina reads); thus, we concluded that these are likely to represent errors in the BNG map rather than in Aet_MR.1.0. Therefore, for the full assembly of 4.28 Gb, we estimate approximately one assembly error per 12.5 Mb.
To estimate base-level accuracy, we aligned a subset of the PacBio reads (50,000 randomly sampled reads), the super-reads, the mega-reads, and the final set of assembled contigs to the previously released Ae. tauschii assembly, version 0.4 (ftp://ftp.ccb.jhu.edu/pub/data/Aegilops_tauschii). That assembly was built from 5216 pools of bacterial artificial chromosomes (BACs), with each pool containing eight overlapping BACs spanning approximately 1 Mb. (Each BAC was a haploid sequence averaging ∼120 kb in length.) These pools were independently assembled from 250-bp Illumina MiSeq reads using SOAPdenovo2 (Luo et al. 2012). The reads used for the BAC pool assemblies were a subset of reads used for creating the mega-reads. For alignment, we used the NUCmer program from the MUMmer package (Delcher et al. 2002; Kurtz et al. 2004) with a minimum match length of 127 bp to anchor each alignment.
The average identity of the alignments was 85.1%, 99.91%, and 99.77% for the PacBio reads, super-reads, and mega-reads, respectively. From this, we estimate that the error rates for super-reads and mega-reads were <0.09% and 0.23%, respectively (Table 2). The mega-reads have a higher error rate because some of them include small patches of sequence computed from alignments of PacBio-only data. The PacBio error rate of 14.9% was consistent with previous reports on this technology.
For the final assembly, we computed the average identity for all alignments longer than 10 kb, which is long enough to span almost all repeats in the genome. This yielded 10,597 alignments covering 1,629,700,561 bp, with an average identity of 99.96% (one mismatch every 2500 bases), with values ranging from 99.24% to 100%. Thus, the base-level accuracy of the assembly appears very high: note that some differences between the haploid BAC assemblies and the diploid whole-genome assembly are likely due to haplotype differences rather than errors. In support of this hypothesis, if we consider only the alignments at 99.99% identity, which presumably come from regions of the whole-genome assembly whose haplotype matches the BAC sequence, these cover 650,572,225 bp (40%) of the aligned regions. Note that we also evaluated the base-level accuracy of the Canu assembly using the same procedure, which yielded a rate of 99.32% identity between the contigs and the BAC assemblies.
Assembly quality as a function of sequencing depth
To evaluate how the mega-reads assembly method performs with varying amounts of PacBio data, we performed a series of experiments on a data set from Arabidopsis thaliana Ler-0. We used a hybrid data set for this genome generated previously (Lee et al. 2014), which includes over 100× coverage by PacBio reads using P5-C4 chemistry and 110× coverage by paired, 300 bp Illumina MiSeq reads. We sampled the PacBio data to create six data sets ranging from 10× to 60× coverage and combined each of these with 100× coverage in MiSeq reads. Figure 2 shows how the resulting assembly N50 size varies with PacBio coverage. The N50 size was greatest at a depth of 60×. Note that because this genome is far less repetitive than Ae. tauschii, the assembly was much more contiguous; also note that with newer P6-C4 chemistry, which produces longer reads, the results with lower PacBio coverage would very likely improve.
Figure 2.

Change in the N50 contig size of genome assemblies using the mega-reads algorithm with varying PacBio coverage and 100× Illumina coverage for the Arabidopsis thaliana genome. At 60×, the N50 size of 9.15 Mb approaches the maximum possible N50 contig size for this genome, which is determined by the sizes of the chromosome arms.
Discussion
Both Ae. tauschii and its close relative, hexaploid wheat (Triticum aestivum), have proven difficult to assemble because of their unusually high proportion of repetitive sequences. A previously published version of Ae. tauschii (Jia et al. 2013) yielded only 2.69 Gbp (∼63% of the genome) spread across 1.1 million contigs. Attempts to assemble T. aestivum have met with similar problems: a massive effort to sequence T. aestivum chromosome-by-chromosome yielded only 61% of the genome in very small contigs with N50 sizes from 1.7 to 8.9 kb (International Wheat Genome Sequencing 2014). Most of the repeats in Ae. tauschii and in other plants consist of transposons (Lisch 2013), which occur in thousands of copies, many of them nearly identical, throughout the genome. Assembly algorithms can find the correct location for these elements if the input data include reads that are long enough to contain the entire span of a repeat plus unique flanking regions on either side.
The PacBio reads generated in this study, with an average read length of 8520 bp, are easily long enough to span most transposable elements, which are usually 2–3 kb in length (though some can be longer). However, the high error rate of PacBio reads requires some form of error correction before these sequences can be used in a final assembly. The mega-reads introduced here solve both these problems: with an average length of 6319 bp, they are long enough to contain the ubiquitous 2–3 kb repeats in the Ae. tauschii genome, and they are accurate enough—much more accurate than raw Illumina reads—to be used to generate a high-quality assembly. Using these mega-reads, we have generated a whole-genome assembly of Ae. tauschii with an N50 contig size of 486,807 bp, more than 20 times longer than any previous assembly. The unprecedented contiguity of this assembly provides a strong foundation for additional mapping and assembly work to create a far more complete picture of this important plant genome. The strategy described here, using deep coverage Illumina sequencing with moderate coverage PacBio sequencing, demonstrates a cost-effective approach to generating highly contiguous, accurate assemblies of large genomes, even when those genomes contain large numbers of long, near-identical repeats.
Software availability
The MaSuRCA mega-reads software is freely available from http://genome.umd.edu/masurca.html and as Supplementary Material.
Data access
The Ae. tauschii assembly (Aet_MR.1.0) as well as the Illumina and PacBio sequencing data from this study have been submitted to NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA329335.
Supplementary Material
Acknowledgments
This work was supported in part by National Science Foundation (NSF) grant IOS-1238231 to J.D. and by National Institutes of Health (NIH) grant R01 HG006677 to S.L.S. S.K. was supported by the Intramural Research Program of the National Human Genome Research Institute, NIH. This study utilized the computational resources of the Biowulf system (http://biowulf.nih.gov) at NIH and the MARCC system (http://marcc.jhu.edu) at JHU.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.213405.116.
References
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. [DOI] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Phillippy A, Carlton J, Salzberg SL. 2002. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 30: 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Feng Y, Kumar D, Zhang W, Zhu T, Luo MC, Messing J. 2016. Analysis of tandem gene copies in maize chromosomal regions reconstructed from long sequence reads. Proc Natl Acad Sci 113: 7949–7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackl T, Hedrich R, Schultz J, Forster F. 2014. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30: 3004–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie AR, Dong L, Smith A, Finklestein J, Lam ET, Huo N, Cao H, Kwok PY, Deal KR, Dvorak J, et al. 2013. Rapid genome mapping in nanochannel arrays for highly complete and accurate de novo sequence assembly of the complex Aegilops tauschii genome. PLoS One 8: e55864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Wheat Genome Sequencing Consortium. 2014. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345: 1251788. [DOI] [PubMed] [Google Scholar]
- Jia J, Zhao S, Kong X, Li Y, Zhao G, He W, Appels R, Pfeifer M, Tao Y, Zhang X, et al. 2013. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496: 91–95. [DOI] [PubMed] [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res (this issue). 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al. 2012. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol 30: 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Gurtowski J, Yoo S, Marcus S, McCombie WR, Schatz M. 2014. Error correction and assembly complexity of single molecule sequencing reads. bioRxiv 10.1101/006395. [DOI] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v1. [Google Scholar]
- Li H. 2016. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32: 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D. 2013. How important are transposons for plant evolution? Nat Rev Genet 14: 49–61. [DOI] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. 2008. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24: 2818–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmela L, Rivals E. 2014. LoRDEC: accurate and efficient long read error correction. Bioinformatics 30: 3506–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankova H, Hastie AR, Chan S, Vrana J, Tulpova Z, Kubalakova M, Visendi P, Hayashi S, Luo M, Batley J, et al. 2016. BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes. Plant Biotechnol J 14: 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Marcais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. 2013. The MaSuRCA genome assembler. Bioinformatics 29: 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.