

Abstract
The most polymorphic part of the human genome, the MHC, encodes over 160 proteins of diverse function. Half of them, including the HLA class I and II genes, are directly involved in immune responses. Consequently, the MHC region strongly associates with numerous diseases and clinical therapies. Notoriously, the MHC region has been intractable to high-throughput analysis at complete sequence resolution, and current reference haplotypes are inadequate for large-scale studies. To address these challenges, we developed a method that specifically captures and sequences the 4.8-Mbp MHC region from genomic DNA. For 95 MHC homozygous cell lines we assembled, de novo, a set of high-fidelity contigs and a sequence scaffold, representing a mean 98% of the target region. Included are six alternative MHC reference sequences of the human genome that we completed and refined. Characterization of the sequence and structural diversity of the MHC region shows the approach accurately determines the sequences of the highly polymorphic HLA class I and HLA class II genes and the complex structural diversity of complement factor C4A/C4B. It has also uncovered extensive and unexpected diversity in other MHC genes; an example is MUC22, which encodes a lung mucin and exhibits more coding sequence alleles than any HLA class I or II gene studied here. More than 60% of the coding sequence alleles analyzed were previously uncharacterized. We have created a substantial database of robust reference MHC haplotype sequences that will enable future population scale studies of this complicated and clinically important region of the human genome.
In studying the genetics of human disease, the major histocompatibility complex (MHC) region is arguably the most important part of the genome (Lechler and Warrens 2000; Chapman and Hill 2012). The MHC region on Chromosome 6 is ∼5 Mbp in length and contains ∼165 protein-encoding genes (Horton et al. 2004). Almost half of these proteins are directly involved in immune defense against pathogens, including the highly polymorphic HLA class I and II (Horton et al. 2004; Fairfax et al. 2014). Among the first and best examples of personalized medicine, hundreds of thousands of solid organ and bone marrow transplants have involved HLA-matched donors and recipients (Terasaki 1969; Petersdorf et al. 2013). The most striking correlation of the MHC region with disease remains that of HLA-B*27 with ankylosing spondylitis, one of the first discovered (Brewerton et al. 1973; Schlosstein et al. 1973). Since that time, hundreds of diseases have been associated with HLA class I or II alleles, and for many of them this remains the strongest genetic correlation (Trowsdale and Knight 2013; Li et al. 2015). In general, the causative mechanisms that underlie these disease associations are poorly understood. Notable exceptions are the effects of HLA-B*57 in slowing the progress of HIV infections to AIDS and in causing life-threatening hypersensitivity reactions to abacavir, a drug used to treat HIV infections (Illing et al. 2013; McLaren and Carrington 2015).
HLA molecules are cell-surface proteins that bind peptide antigens, engage T cell receptors, and stimulate the T cell arm of adaptive immunity. This, in turn, stimulates the B cell arm and production of pathogen-specific antibodies. The highly polymorphic class I molecules are HLA-A, HLA-B, and HLA-C, and their nomenclature is described in the Supplemental Information. In addition to their role in adaptive immunity, HLA class I molecules contribute to innate immunity by serving as ligands for the killer-cell immunoglobulin-like receptors (KIR) of natural killer (NK) cells (Moretta et al. 1996; Parham and Moffett 2013). Contrasting with HLA class I, the highly polymorphic HLA class II molecules, HLA-DR, HLA-DQ, and HLA-DP are dedicated to adaptive immunity (Germain 1994). Also encoded by MHC region genes are components of the machinery that produces peptide antigens and delivers them to HLA class I and II. Other MHC region genes encode complement components, cytokines, transcription factors, structural and developmental proteins, olfactory receptors, and numerous tRNAs (Horton et al. 2004). Extensive linkage disequilibrium (LD) characterizes the MHC region, creating long-range haplotypes (Price et al. 1999; Larsen et al. 2014). This feature complicates the analysis of HLA-associated diseases, because polymorphisms in genes having no known participation in disease causation can be disease-associated (Trowsdale and Knight 2013; O'Donovan 2014).
The high polymorphism, sequence divergence, and structural variation of the MHC region has made it intractable to high-throughput and automated genome-wide analysis (Horton et al. 2004). Previous studies to characterize the region and identify causative alleles required combinations of strategies and lengthy procedures (Horton et al. 2004; Nair et al. 2006; Yang et al. 2007). To surmount these difficulties, we developed a method to sequence the entire MHC region from genomic DNA, and in large numbers. We applied the method to a panel of 95 MHC region homozygous cell lines and devised a de novo-based approach to assemble their complete MHC haplotypes. Application of the method, in conjunction with the reference haplotypes we characterized, to population genetics and disease studies will refine genetic associations and facilitate discovery of the causative mechanisms.
Results
Enrichment method captures divergent sequences
A set of oligonucleotide probes was used to capture the ∼5-Mbp MHC region from sheared genomic DNA libraries. The targeted region is located at Chromosome 6p21 and encompasses all genes from GPX5 to ZBTB9 (Fig. 1A). The method for sequencing the MHC was optimized using DNA from the COX cell line, which is homozygous throughout the entire genomic region (Stewart et al. 2004) and is the source of one of the two complete MHC haplotype sequences included in the human genome reference (COX_hap2, GenBank: NT_113891). The method yielded 99.96% coverage of the COX MHC haplotype, with a mean read-depth of 59.4 (SD 18.4) and with 99.6% of the coverage being >10× (Fig. 1B). A total of 25 gaps in the sequence represent 0.04% (2414 bp) of the targeted region; they have a size range of 1–509 bp with a median of 49 bp, and 80% of them are outside the bounds of a coding region (Supplemental Fig. S1). These measurements show the method produced over 5 Mbp of high coverage, depth, and fidelity of nucleotide sequence from the target cell line.
Figure 1.

The target MHC region is >99.96% covered by the sequence data. (A) SNP and gene density throughout the target MHC region. Blue line: The number of dbSNP markers in discrete windows of 10 kbp. Orange diamonds: Genes that are involved in immunity. Black diamonds: Other genes. Red text: Classical HLA class I and II genes. Areas of significant structural diversity are indicated with green brackets; these are HLA-DR and C4A/C4B. Build 142 of dbSNP was used, which has 225,302 SNP sites mapped to the target region. The full list of genes in the target region is shown in Supplemental Table S3A. (B) Depth of sequence reads (duplicates removed) derived from the COX cell line following stringent alignment to the COX reference MHC haplotype. (C) Read depth after sequence reads derived from chimpanzee Clint had been mapped to the reference sequence (also derived from Clint) and PCR duplicates removed. Stringent alignment criteria were used to map the sequence reads back to the chimpanzee genomic segment that is equivalent to the human MHC region (the panTro4 MHC, Chr 6: 28774516-33956232).
The MHC region has the highest sequence diversity of the human genome (Sachidanandam et al. 2001). To establish a comprehensive set of reference haplotypes, it was therefore important to determine if our probe set could cope with the extent of this diversity. As a test, we applied our method to genomic DNA from the subject of the chimpanzee genome project (The Chimpanzee Sequencing and Analysis Consortium 2005) and obtained 97.66% coverage of the previously characterized target region (Fig. 1C). This high coverage included the highly polymorphic chimpanzee Patr- class I and II genes, which have equivalent or greater pairwise nucleotide distance from the target sequences as their most divergent human counterparts (Supplemental Fig. S2). When applied to a panel of 95 MHC region homozygous cells (described in Methods and obtained from the International Histocompatibility Working Group [IHWG]), the method obtained an estimated mean 97.2% coverage of the genomic region (Supplemental Table S1). Again, this included the highly polymorphic HLA class I and II genes, which have been extensively genotyped from these cells (Mickelson et al. 2006). That we could confirm and extend the resolution of these genotypes (Supplemental Table S2) shows that the method captures the most polymorphic portions of the MHC region (the peaks of Fig. 1A). The full extent of sequence diversity in the MHC region is unknown, but together, these results suggest our method achieved equivalent success for the ‘unknown’ sequence from these cell lines as it did for the previously sequenced controls.
Generation of high-fidelity haplotypes spanning the MHC region
We targeted cell lines that are MHC region homozygous so that their haplotypes could be assembled without bias or ambiguity of phase. The assembly pipeline, a composite of established and in-house bioinformatics programs, consists of three stages that are fully described in the section titled “Methods.” Briefly, in the first stage, we generated and filtered multiple contigs, and in the second, we used read-pair information to join these contigs together to form scaffolds. In the third stage, we determined the relative orientation of the scaffolds and joined them where possible with read-pairs to form ‘super-scaffolds.’ The process was designed to be conservative in order to limit or eliminate false joins. The assembly pipeline was optimized on sequence data from the COX cell line. From the COX data, 16 ‘super scaffolds’ were derived, with a mean length of 306.1 kbp and representing 98.97% of the target sequence. A pairwise comparison with the original Sanger-sequenced haplotype showed that the assembly process created a correctly characterized and ordered final sequence (Fig. 2A).
Figure 2.

In silico reference-guided scaffold orientation correctly assembles complex structural variation. (A) Results obtained from dot plot comparisons of the original COX haplotype (hap_2: left) and our newly-sequenced and assembled COX haplotype (right) against the human reference MHC region sequence (huref). This shows the haplotypes were assembled in the correct order. (B) The human reference MHC region sequence (huref) was derived from the PGF cell line. Here, the PGF haplotype was assembled using a modified COX haplotype sequence as a guide to orient the scaffolds obtained from the PGF sequence data. Because PGF had been sequenced previously, we were able to compare dot plots of the original sequence against itself (left) with plots of our newly sequenced and assembled version against the original (right). Identical results were obtained. (C) Complex structural variation observed from comparison of the COX and PGF haplotypes. The colored segment from COX is inverted (blue/yellow) and partially deleted (red) in PGF. The assembly process for COX (upper box) and PGF (lower box) is denoted from the top down in each case. (1) Shows the de novo generated contigs, (2) shows the scaffolds that were built from the contigs, and (3) shows the final sequence scaffolds generated.
Although the methods we used to generate scaffolds were entirely de novo, the final stage of scaffold orientation was guided by a reference sequence based on the published COX haplotype (described in Methods). To demonstrate that our approach could capture unknown structural variation, we targeted the PGF cell line. The haplotype sequence from PGF was also characterized previously, forming the human reference from build hg18 onwards, and differs in sequence and structure from COX by ∼16,000 SNPs and 2400 indels (Stewart et al. 2004; Horton et al. 2008). The complement component 4 genes C4A and C4B are located in a region of modular structural diversity (Yang et al. 2007). Importantly, PGF has a longer C4A/C4B region than COX (the result of a duplication) and a different set of genes in the HLA class II region (HLA-DRB1*15) than COX (HLA-DRB1*03) (respectively representing the DR2 and DR3 haplotypes structures shown in Supplemental Fig. S3). From PGF, we assembled 5.04 Mbp of haplotype sequence, which includes the C4A/C4B and HLA class II regions (Fig. 2B). As a further check for the scaffold orientation step of the assembly, we analyzed a known complex structural rearrangement that distinguishes PGF from COX and occurs in the MUC22 gene, located 234 kbp telomeric of the HLA-C gene (Fig. 1). The variant consists of a 5-kbp inversion coupled with a 4-kbp deletion (Fig. 2C). Here, we show the variant junctions had been included in contigs made in stage one of our assembly process. These contigs were then incorporated into a larger de novo-assembled scaffold of 30 kbp. Finally, this scaffold was assembled in stage three as part of a correctly ordered super-scaffold of 120 kbp. Thus, our analysis showed this segment of the PGF haplotype assembled correctly when the COX haplotype was used as the reference guide. Our approach is therefore able to capture highly complex structural and sequence diversity of the sort that characterizes the MHC region.
A database of MHC haplotype sequences
The complete sequencing and assembly method was then applied to the remaining MHC homozygous cell lines. Sixty of the IHWG cell lines are homozygous through the entire target of ∼5 Mbp (Norman et al. 2015). From these cells, we obtained a mean of 4.86 Mbp (±71.2 kbp) final sequence, consisting of ∼100 ‘super-scaffolds’ per cell line (Fig. 3A; Supplemental Table S1). These data therefore represent unambiguously phased MHC region haplotypes with an estimated mean completion of 98.3%. These final sequences contain short gaps (a mean of 192 ± 42 gaps per full haplotype) representing contigs that were identified to be adjacent but not completely joined. Many of these gaps could be closed using less stringent scaffold assembly or by simple manual intervention, but we chose to retain an approach that limits the potential for false scaffold joining. Included in the final full-length sequences are six haplotypes, not completed previously, that form the current alternative reference sequences of the human genome assembly (Horton et al. 2004). The most significant of these is the haplotype from APD, which we have raised from an estimated completion of 45% to 98% (Fig. 3B). Consistent with expectations, the longest final sequence, of 5.2 Mbp, was obtained from the DEU cell line, which has an HLA-DRB1*04 (DR4) haplotype. The shortest final sequence, of 4.74 Mbp, was obtained from the SPL cell line, which has an HLA-DRB1*08 (DR8) haplotype (Supplemental Fig. S3). Data from the 35 cell lines that are not completely MHC homozygous were trimmed to the coordinates previously defined (Norman et al. 2015) to give a mean of 3.02 Mbp of phased MHC region haplotype sequence. All of the haplotype sequences have been deposited in public databases as a resource for future studies. Their accession numbers are listed in Supplemental Table S1.
Figure 3.

Characterizing 95 MHC region haplotypes. (A) Summary statistics for 60 full-length MHC haplotypes. (B) Dot plot comparisons of the deposited APD alt_ref haplotype (left) and the final haplotype we generated (right) vs. the PGF haplotype of the human reference.
High sequence and structural diversity of MHC region coding sequences
The human MHC reference sequence contains 165 expressed genes (Supplemental Table S3). We generated unambiguous full-length coding DNA sequence (CDS) for each of these genes. In analyzing these data, our emphasis was on the predicted polypeptide sequences. This was partly because phased CDS diversity has rarely been investigated in genome studies but mainly because HLA class I and II gene polymorphism involves nonsynonymous substitutions at sites that modulate protein function and are implicated in the mechanisms that underlie the associations of HLA class I and II with disease (de Bakker and Raychaudhuri 2012; Illing et al. 2013; McLaren and Carrington 2015).
For 23 of the MHC region genes, the allelic variation is curated by the Immuno Polymorphism Database (IPD) (Robinson et al. 2015). In the IHWG cell lines, we observed 304 unique CDSs of these 23 genes (encoding 271 unique polypeptides), and they are given in Supplemental Table S2. From the remaining 142 genes, we characterized 11,571 CDS. For these 95 homozygous cell lines, the CDSs represent 1.82 Gbp of DNA, comprising 800 unique CDS. BLAST searches showed that 140/800 CDS matched the RefSeq allele sequence and that 131 of the remaining 660 CDS are present in sequence databases. Thus, 66% of the (non-IPD) CDSs we characterized have not been described previously (Supplemental Table S3). Although a majority of variant sites are known and have dbSNP IDs, their contribution in the context of genomic CDS diversity has not been examined previously. The IHWG cells are monomorphic for 32 of the expressed polypeptides (Fig. 4A; Supplemental Table S3). Several genes, such as HLA-F, CCHCR1, and the heat-shock protein genes HSPA1L, HSPA1A, and HSPA1B, combine low polypeptide diversity with higher nucleotide diversity in the CDS. This observation raises the possibility that low frequency protein-coding variants of these genes might influence disease. Because the cell panel represents a variety of common HLA class I and II haplotypes, many of which are associated with diseases mediated by immune mechanisms that are poorly understood, we also compiled a database of the CDS alleles for use in future studies (Supplemental Table S4).
Figure 4.

Coding DNA sequence (CDS) and polypeptide diversity of the MHC region. (A) Combined incidence (%) of non-reference CDS alleles through the MHC region of the 95 cell lines. The values are calculated from the homozygous segments only. (B,C) Number of unique coding-sequences (CDS alleles) for each of the expressed genes in the target MHC region detected from analysis of cell lines derived from homozygous individuals. (B) In red are the CDS alleles that encode unique polypeptide sequences, and in blue are additional alleles having synonymous changes. HSPA1 represents three heat-shock protein genes: HSPA1L, HSPA1A, and HSPA1B. MUC22 is the only gene to have more alleles than HLA-B. (C) Total number of CDS alleles corrected for the size of the transcript. Classical HLA genes are indicated in red text. Other polymorphic genes mentioned in the main text are indicated in B; full list of the CDS and alleles is given in Supplemental Table S3. (D) Mean pairwise difference between polypeptide sequences of the unique allotypes expressed by each of the genes in the MHC genomic region.
MUC22 has the greatest number of alleles of any gene in the MHC region
The CDSs have from 1 to 43 alleles (Fig. 4B). The highly polymorphic genes localize to three main segments: one containing the HLA-A gene, a second containing HLA-B and -C, and a third containing HLA-DRB1. These same regions contain the major peaks of SNP diversity (Fig. 1A). Unexpectedly, the gene with 43 alleles is not an HLA class I or II gene, but MUC22. These alleles encode 40 allotypes of the MUC22 protein (Supplemental Table S5). MUC22 (also known as PBMUCL1), a transmembrane mucin expressed in the bronchi of the lungs (Hijikata et al. 2011), is up-regulated on infection with respiratory syncytial virus (Del Rocío Baños-Lara et al. 2015). Polymorphism of MUC22 is associated with diffuse panbronchiolitis (Hijikata et al. 2011) and possibly with asthma (Galanter et al. 2014). Exon 3 of MUC22 is particularly large (∼5 kbp) and exhibits VNTR polymorphism (Hijikata et al. 2011). The human reference CDS consists of 124 repeats of a 90-nt sequence, with ∼70% of the repeats being unique (Hijikata et al. 2011). A similar level of structural variation to what we observed was present in a Japanese cohort, including six MUC22 indel variants (Hijikata et al. 2011). The 1860-bp MUC22 deletion we identified in the HLA-B*44:03 homozygous HOR cell line is likely the same as that identified by Hijikata et al. In the MUC22 alleles of the cell panel, we identified 10 different indel variants of exon 3, ranging in length from 1 to 1860 bp (Supplemental Table S5). Two of the 10 are frameshift mutations leading to significantly truncated polypeptide fragments (340 and 674 amino acids, respectively, are deleted from the full-length reference CDS of 1773 residues). Both of these frame shifts are rare in the cell panel: one being specific to the BM09 cell line and the other to the RSH cell line. RSH is Sub-Saharan African, so this allele could be more frequent in Africa. The other six deletions and the one insertion are all in-frame, leading to putative transcripts encoding polypeptides from 1133 to 1183 residues in length.
Following MUC22 in allele number are HLA-B and NOTCH4, each with 34 alleles. MUC22 and NOTCH4 are much larger genes than HLA class I and II. When allele number is normalized to gene length, HLA class I and II emerge as the most polymorphic genes (Fig. 4C). With this normalization, MUC22 is still seen to be a highly polymorphic gene, but NOTCH4 is not. As a measure of difference between a gene's protein allotypes, we plotted the mean-pairwise difference of their amino acid sequences (Fig. 4D). For almost all genes, the values are low, the variants differing by only one or a few amino acid substitutions. MUC22 also scored low, because the structural variants (insertions and deletions) that characterize this protein were considered equivalent to single amino acid substitutions. In striking contrast are the high values exhibited by the HLA-A, -B and -C class I molecules and the HLA-DRB1, -DQA1, -DQB1, -DPA1, and -DPB1 class II molecules. For these highly polymorphic HLA proteins, the allotypes differ by multiple amino acid substitutions. Apart from these antigen-presenting molecules, the only other protein encoded in the MHC region that has a high mean pairwise difference is another mucin, MUC21, for which the basis of the diversity is two divergent allotype groups.
Structural diversity in the MHC exemplified by the genes encoding NOTCH4 and C4
NOTCH4, encoding Notch homolog 4, and C4A and C4B, encoding the C4 complement protein, are highly polymorphic by virtue of complex structural diversity combined with SNP diversity. Polymorphism of NOTCH4, an important transmembrane receptor for cell development, correlates with several diseases, notably schizophrenia (Wei and Hemmings 2000). Characterizing NOTCH4 diversity is a short tandem repeat (STR) polymorphism in exon 1, involving 6–13 copies of a GCT sequence (Dorak et al. 2006). These in-frame indels vary the number of leucine residues in the leader peptide (Supplemental Table S5), variation that likely affects protein expression, as do some SNP variants of the NOTCH4 gene (Shayevitz et al. 2012). The structural variation in the 34 NOTCH4 allotypes identified here has considerable potential for functional diversity and differential disease associations. The results of a previous investigation of NOTCH4 STR polymorphism in 63 IHWG cell lines (Dorak et al. 2006) are fully consistent with our analysis, increasing confidence in our method (Supplemental Table S5).
The C4 component of the complement system cooperates with specific antibodies to neutralize pathogens (Horton et al. 2004; Yang et al. 2007). Variable gene content and allelic polymorphism of C4A and C4B are associated with complement deficiency and the autoimmune disease systemic lupus erythematosus (SLE) (Yang et al. 2007). To date, no single typing method has encompassed the diversity of these genes, and in whole-genome analysis, the C4A/C4B region is poorly covered by SNP probes (Fig. 1). Forty of the IHWG cell lines were analyzed previously using conventional methods (Wu et al. 2007). There is good concordance between those data and ours (Methods; Supplemental Table S5D,E). Our method identified between one and four C4A or C4B genes per MHC haplotype. It also discriminated the functionally important antigenic determinants of the C4A and C4B isotypes, as well as the presence and absence of a HERV insertion that reduces gene expression (Supplemental Table S5). In addition to these complex markers, we identified 34 single nucleotide differences in the CDS of complement C4, including 20 not described previously. We identified 53 different C4A/C4B haplotypes in the cell panel (Supplemental Table S5). The four cell lines having the same HLA class I and II haplotype as COX also had the COX C4A/C4B genotype. Likewise, the four cell lines with the PGF HLA class I and II haplotype also had the PGF C4A/C4B genotype. Contrasting with these similarities, the cell panel included 74 different HLA-B, C4A/C4B, HLA-DRB1 haplotypes and six examples of haplotypes having identical HLA-B and -DRB1 alleles but different C4A/C4B haplotypes. The latter includes three of four examples of the HLA-B*52/-DRB1*15:02 haplotype. Such high variability among haplotypes considered identical by standard methods suggests that further investigation of this variability in the context of disease would be fruitful. It would be difficult and laborious to assess all the functionally important variants we describe without targeted analysis of these genomic regions.
Discussion
We set out to characterize haplotypic sequence diversity in the highly polymorphic and structurally complex MHC region. This goal was achieved by exploiting a panel of 95 cell lines that were selected for being homozygous for HLA class I and II genes (Yang et al. 1987; Marsh et al. 1996; Mickelson et al. 2006). HLA homozygous individuals are infrequently identified, usually in consanguineous families, and the cell lines were collected over a period of >30 yr. Targeting these homozygous individuals reduced the complexity of resolving haplotype phase and permitted assembly of 95 haplotypes spanning the 4.8-Mbp MHC region. These haplotypes will form reference sequences essential for future studies that aim to understand the numerous roles of MHC factors in immune-mediated disease.
For this project, we designed and implemented a bioinformatics pipeline that involved no manual input or decision making for much of the process. Because the contigs were generated entirely de novo and the haplotypes were automatically assembled with only limited external data to map their relative order, we could capture information on the structure and sequence of the targeted genes. This was critical because many of the MHC region genes exhibit both structural and sequence variation, as was clearly evident from the many CDS allele variants we uncovered that were not previously observed. A second reason for adopting this approach was because it increased the efficiency of the process, an important consideration given the large number of haplotypes analyzed. We chose short-read sequencing technology for this study because of its high fidelity. We were thus able to generate essentially contiguous sequence runs of equivalent or greater length than those achieved with most long-read or cloning technologies.
The bioinformatics methods we used to assemble the MHC region haplotypes were designed specifically for application to the HLA class I and II homozygous individuals that have driven the advance of HLA research for the last 25 yr. They are not suitable for the analysis of HLA heterozygous samples. We have shown, however, through family studies of heterozygous samples that the capture and sequencing method can generate accurate data from HLA and other highly polymorphic genes (Norman et al. 2016). For large-scale population studies, any manner of formal haplotype assembly will be impractical, and alternative methods that rely on population information will be required. We believe the most promising of these involves mapping sequence reads to a population reference graph (Dilthey et al. 2015). To this end, the data we describe here will be valuable toward populating these graphs.
Eighty percent of the cell panel we studied is of European origin. We estimate that these cells represent 60%–80% of HLA class I and II alleles worldwide (Norman et al. 2015). For future studies, it will be critical to target other population groups in order to represent fully human MHC diversity. Our previous analyses of HLA class I genes show that the method can be applied to Sub-Saharan African and Asian populations and detects the most divergent human HLA class I allele, HLA-B*73 (Ashouri et al. 2016; Norman et al. 2016). To establish if the probes used in our capture method can cope with the complete range of human MHC diversity, we targeted the chimpanzee whose genome was sequenced. Although trans-species polymorphism of HLA class I and II alleles is apparent, with longer coalescence times between specific human allele pairs than some human-chimpanzee pairs (Lawlor et al. 1988; Mayer et al. 1988), we demonstrate the method does capture sequences, such as the chimpanzee-specific Patr-AL MHC class I gene, that are more divergent from the probes than any known HLA allele (Supplemental Fig. S2). This specific finding, combined with the good overall coverage of the chimpanzee MHC haplotype we achieved, make it likely that most human MHC haplotypes will be sequenced successfully with our method. Furthermore, the method is flexible and further probes can be added should new regions of the human MHC be discovered.
We identified high levels of polymorphism at several genes that are not HLA class I or II genes. The most striking of these is MUC22, which had the most alleles of any MHC region gene in our cell panel. All MUC22 variants we describe have potential to influence disease, yet they have been largely invisible to whole-genome SNP analysis. These findings have significant implications for fine-mapping of disease associations. Since the MHC region haplotypes we studied were not selected randomly, the data are unsuitable for formal linkage disequilibrium analyses. We note, however, that there is clear evidence for correlation among specific pairs of MUC22 and HLA class I alleles, forming haplotypes being shared by unrelated individuals. In contrast, for other pairs of alleles there is little sign of LD. These extremes in background haplotype diversity likely result from HLA class I and II alleles that have different demographic and evolutionary histories (Ahmad et al. 2003; Barreiro and Quintana-Murci 2010; Abi-Rached et al. 2011). For example, the five HLA-B*08:01 haplotypes sequenced all have the same MUC22 allele (Supplemental Table S4). In contrast, there are four MUC22 alleles in the five HLA-B*51:01 haplotypes. These differences show that the evolutionary stability of an MHC haplotype will determine the depth and breadth of analysis required to correlate disease with functionally important genetic variants.
Our method for complete sequencing of MHC haplotypes has the capacity to analyze the large cohorts necessary for defining population genetics and disease associations. Moreover, we demonstrate that data generation and analysis for this complex genomic region are robust, versatile, and consistent. The method enables an extensive characterization of sequence and structural variation not possible with any other single method. This sets the stage for future studies that will take the analysis of HLA and disease to the highest resolution. This should distinguish genetic variation that functionally contributes to disease from genetic variation that is carried with it by hitch-hiking, the latter being a consequence of the linkage disequilibrium that pervades the MHC and varies between haplotypes. The approach we describe permits comprehensive assessment of all candidate genetic markers, including previously uncharacterized variation. The procedure is designed to be cost-effective and run on a large scale (for example, 96 individuals possible per HiSeq instrument run). With these qualities, it can form the basis for significant, broad-based advancement in understanding the strongest genetic associations with human disease.
Methods
DNA samples
DNA was extracted from a panel of 95 EBV-transformed human B cell lines. These cells, which derive from the 10th, 12th, and 13th International Histocompatibility Workshops (Yang et al. 1987; Marsh et al. 1996; Mickelson et al. 2006), are homozygous for some or all of the highly polymorphic HLA class I and II genes (Dorak et al. 2006; Mickelson et al. 2006). The coordinates of their homozygous tracts were defined using a high-density SNP array (Norman et al. 2015). The panel represents 90%–95% of the European and 60%–80% of the world population, as assessed by HLA class I and II allele frequency (Norman et al. 2015). Also included in the analysis was one chimpanzee B cell line, derived from Clint (Yerkes pedigree number C0471), a chimpanzee of the Pan troglodytes verus (Western chimpanzee) subspecies, who was the subject of the chimpanzee genome sequencing project (The Chimpanzee Sequencing and Analysis Consortium 2005). Low-passage samples of the B cell lines were grown from archival material, available at Stanford University, or purchased from the International Histocompatibility Working Group (http://www.ihwg.org/). Included in our panel were the eight HLA class I and II homozygous cell lines studied by the MHC Sequencing Consortium (PGF, COX, DBB, QBL, MANN, SSTO, MCF, APD) and for which the MHC haplotypes were sequenced to varying extent (Horton et al. 2004). Data from these ‘Sanger 8 reference’ cells have formed the reference, and alternative references for the MHC region of the human genome, since build NCBI36/hg18.
Assessment of the capture/sequence method and construction of reference haplotypes
Oligonucleotide probes targeting the genomic region of (GRCh38/hg38) Chr 6:28510120-33532223 were designed and manufactured as described in the Supplemental Information. These coordinates encompass all eight of the alternative reference haplotypes for the MHC region, which were used as templates for the probe design. Of these, the two completed sequences (Stewart et al. 2004), of PGF and COX, overlap through 4.5 Mbp and form the telomeric (COX) and centromeric (PGF) boundaries of our ∼5-Mbp target. The enrichment process we used will tolerate up to 18% difference in nucleotide sequence between the probe and the target (InanlooRahatloo et al. 2014), which is greater than the difference between any known pair of HLA alleles (Supplemental Fig. S2). The library preparation, enrichment process, and sequencing methodologies are all described in the Supplemental Information. Except where indicated otherwise, we used 800-bp DNA fragments and 2 × 100-bp paired-end reads.
While optimizing methods using the COX cell line, we identified and confirmed 19 sequence differences—18 SNPs and one 4-kbp insertion—from the published COX haplotype (Supplemental Information; Supplemental Fig. S4). A modified reference haplotype (COX_IIb; 4,799,503 bp), containing these differences, was used to harvest and map reads having no nucleotide mismatches in order to generate a final set of coverage statistics for the data derived from COX. We optimized the methods using COX because this cell line is homozygous through the entire MHC region, while PGF is only homozygous through ∼4 Mbp (Norman et al. 2015). A second reference haplotype (superCOX_IIb; 4,947,983 bp) that included the additional non-overlapping portion of sequence originating from PGF was generated for the main analyses.
Characterizing phased full-length MHC region haplotypes
The three stages of haplotype building, which are contig assembly, scaffold building, and final sequence construction, are summarized in Figures 5 and 6 and Supplemental Figure S5.
Figure 5.

Pipeline for (A) generating and (B) filtering de novo contigs. Shown is an overview of the protocol for selecting reads, making de novo contigs, and filtering them in preparation for haplotype assembly. Yellow boxes denote data, red boxes denote existing computer programs that were used (these are named at the left), and blue boxes denote in-house scripts. Gray hexagons are used to show data that are not used further in the pipeline.
Figure 6.
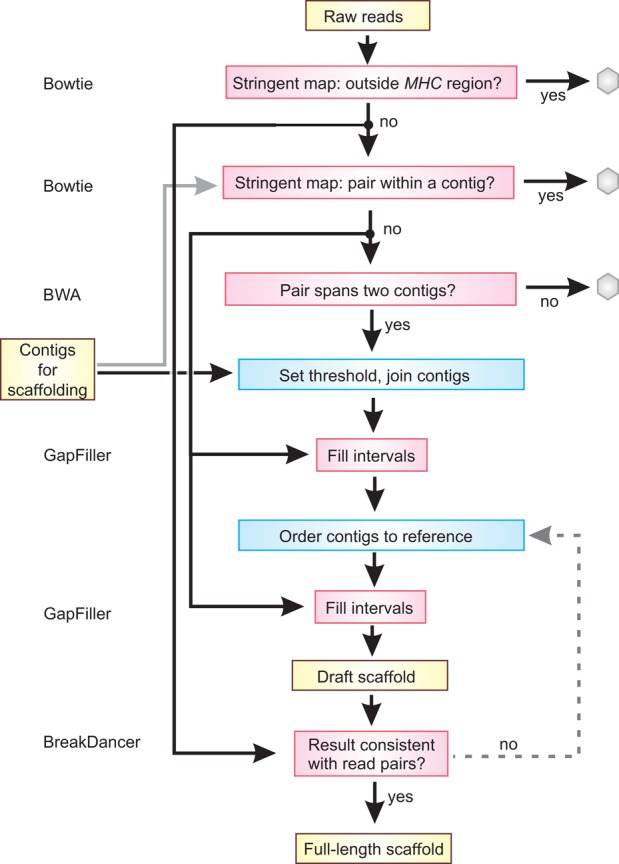
Pipeline for constructing scaffolds and the final sequence. Shown is an overview of the protocol for selecting reads as linkers, determining contig joins to make scaffolds, and ordering and making the final sequence. Yellow boxes denote data, red boxes denote existing computer programs that were used (these are named at the left), and blue boxes denote in-house scripts. Gray hexagons are used to show data that are not used further in the pipeline.
De novo contig assembly
To begin contig building (Fig. 5A), paired-end FASTQ files were first filtered using Bowtie 1.0.0 (Langmead et al. 2009) to retain just those read-pairs for which at least one read (100 bp) matched exactly to a nucleotide sequence in one of the eight MHC haplotype sequences (Horton et al. 2004). This value had been optimized to limit inclusion of repeats (specifically Alu) that did not derive from the MHC region. The filtered reads were then assembled using Velvet 1.1.04 (Zerbino and Birney 2008). The program was run with default parameters except for the k-mer value. This was determined empirically, in which Velvet was run with incremental values of k from 51 to 99. The value of k that returned the greatest length when all contigs >1 kbp were summed was used. The contigs from this assembly were ‘polished’ by removing 30 bp from each end, and contigs shorter than 101 bp were discarded. If a contig contained any poly(A) or poly(T) segments >10 bp or dinucleotide repeats >20 bp, it was split at those points into smaller contigs.
We next extended the contigs using paired-end information and a ‘greedy algorithm.’ If a contig was at least 1 kbp long, we found all read-pairs for which one read aligns perfectly to a sequence within the contig and the other read had a perfect match of 20 nt or more with one end of the contig. These overhanging reads were collected and used to extend the contig by 1 nt. Specifically, the immediate 3′ overhanging bases (only those with a q score greater than or equal to 30) were collected and the nucleotide distribution computed. If at least 3 nt were observed and the number of the most frequently observed base was greater than 10 times the number of observations for the next most frequent base, then the contig was extended with the most frequent base. This contig-extending process was repeated until one or both of the scoring criteria were violated. Velvet contigs of <1 kbp, were aligned to the eight known MHC haplotypes. Contigs with an exact alignment were selected, and 1 kbp of sequence from the known haplotype was appended to the 5′ end of the contig (relative to the extension site). These appended contigs then went through the same extension process outlined above. Following the extension, the 5′ sequence obtained from the known haplotype was removed from the contig. This process was performed for both ends of the contig. The resulting contigs were then input to Minimus2 (Sommer et al. 2007) to assemble overlapping contigs. N50 was calculated using QUAST 2.3 (Gurevich et al. 2013).
Filtering the de novo contigs
The contigs were mapped to the human genome reference (hg19 with the alternative MHC haplotypes removed) using BWA MEM (Li and Durbin 2009) with default parameters except with a k-mer value of 101 (instead of default 16) and sorted into the following bins:
Maps to the target MHC region with a mapping score 60;
No mapping to the genome, but BLASTs to MHC region (>99% contigs that did not map to hg19 were shown to be MHC region-specific);
Maps to the MHC region, but with low mapping score;
Maps equally to MHC region and elsewhere in hg19;
Does not map or BLAST to MHC region.
Bins 1–3 were used for de novo haplotype scaffold assembly (Fig. 5B). Bins 1–4 were used to estimate the region coverage. Bin 5 was not used. Because this process was used to sort the contigs, and the final scaffold orientations were determined using the haplotype sequence described below, our use of hg19 instead of GRCh38 will not affect the final haplotype sequences we obtained (the sequence of the MHC target is identical in hg19 and GRCh38/hg38).
De novo scaffold assembly
Scaffolds were constructed using information from read-pairs to determine which contigs should be adjacent and then join them together (Fig. 6). The following protocol was used: Sequence read-pairs that map stringently outside the target MHC region were removed using Bowtie 1.0.0. Contigs were filtered into five bins as described above, and bins 1–3 were pooled for each cell line for use as a mapping target. Read-pairs having both ends that map with high stringency within a single contig from this pool were identified and excluded using Bowtie 1.0.0. Those read-pairs with each end mapping to a different contig were then identified using BWA (Li and Durbin 2009), with k = 100. We termed these read-pairs as potential linkers. To be identified as a linker, each end of the read had to occur within 500 bp of the end of the respective contig. For each cell line, we set a threshold for a minimum number of linkers that would be required to make a join (shown in Supplemental Table S1). The threshold was based on empirical data derived from the COX and VAVY cell lines, which represent the same haplotype sequenced to two different read depths (Supplemental Fig. S5). For the VAVY cell line, we determined that a threshold of 80 linker reads was the optimum threshold because this produced the fewest scaffolds with minimal misassembly while retaining high coverage (Supplemental Fig. S5). This value corresponded to inclusion of 1260 potential contig joins in the scaffold assembly. For the remaining cell lines, the linkers were sorted in descending order by number of occurrences, and the threshold value was chosen to be the number of occurrences of the 1260th potential join. No threshold below 25 or above 80 was allowed (with the exception of COX and the other Sanger 8 cells that were sequenced to much higher depth; here, the threshold was set at 150). Potential joins were not allowed to become actual joins if more than one linker set was found that satisfied both the position and threshold criteria, or if a given contig was linked to more than one other contig in the same direction. Gaps were filled in the resulting scaffolds using GapFiller v1.10 (Boetzer and Pirovano 2012) with default parameters except for n = 18, d = 550, and using five iterations.
Ordering scaffolds and building the final haplotype
To produce the final haplotype sequence, the scaffolds were ordered with guidance from a reference haplotype and further joins made. The scaffolds were oriented by mapping them against the ‘superCOXmix’ reference haplotype, which is a mosaic of ‘superCOX_IIb,’ and the longest known segments of C4A/C4B and HLA class II. To construct this reference, the C4A/C4B segment of COX was supplemented with two copies of the same segment from PGF (thus, the mosaic reference has four copies of C4L and one of C4S, where the longest C4A/C4B we determined in the 95 cell lines has LLLL configuration—see section entitled ‘Complement C4 polymorphism and nomenclature’ in the Supplemental Material). The HLA class II region of COX (DRB1*03) was replaced with that from SSTO (DRB1*04). DRB1*04 is ∼300 kbp longer than DRB1*03 (Supplemental Fig. S3). To orient the scaffolds, their start positions were identified using BWA (k = 64). Scaffolds were excluded if they were <800 bp in length and the start and end positions fell within the coordinates of a previous contig unless either contig had been ‘soft-clipped’ in the mapping step. The scaffolds were then placed end-to-end in the order identified by the mapping step, separated by (20 × N) for downstream identification, and then subject to 15 cycles of GapFiller. The final scaffolds were trimmed to match the coordinates of the homozygous tracts defined previously using high-density SNP arrays. During optimization of the assembly process with the COX, VAVY, and PGF cell lines, to identify if any mistakes had been made we analyzed the final sequence using Bioedit, nucleotide BLAST (Altschul et al. 1990), and dottup (Rice et al. 2000). We also mapped sequence reads (from the non_hg19 set) back to the final scaffold using Bowtie 2 (Langmead and Salzberg 2012) and analyzed this result using BreakDancer (Chen et al. 2009) to identify any structural inconsistencies. None were found.
Genotyping complement component C4
To determine the number of C4A/C4B genes present in each cell, we randomly selected eight gene sequences from the MHC region data set (ABCF1, ATAT1, Complement C2, DHX16, KIFC1, TRIM27, VARS, and VPS52), trimmed them to the same length as the sequence of a C4BS gene (14,250 bp obtained from AL049547), and used them all as mapping targets. We then removed any reads that could map elsewhere in the MHC haplotype and calculated the copy number as the mean ratio of C4A/C4B reads to each of the eight targets, with guidance from previous results (Wu et al. 2007) to set the thresholds. We determined the ratio of C4A vs. C4B and L vs. S (described in the Supplemental Material) using virtual sequence probes and the method described for KIR (Norman et al. 2016). Again, the threshold values were set according to genotypes previously reported for 40 of the cell lines (Wu et al. 2007). The polymorphic sites and probes used to detect them are shown in Supplemental Table S5. Genotypes for the remaining SNP sites were obtained following alignment of sequence reads to a C4A reference sequence (NG_011638.1).
Data access
The raw sequence reads, contigs, and scaffolds from this study have been submitted to the EMBL-EBI European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena). All sequencing and assembled MHC haplotype data are linked to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) accession number PRJEB6763. ENA accession numbers are listed in Supplemental Table S1. All data are also available via ImmPort (https://immport.niaid.nih.gov/) with the project accession SDY691 (EXP13735). The CDS alleles have been deposited at GenBank (https://www.ncbi.nlm.nih.gov/nucleotide/) under accession numbers KP774801-KP774802, KY497260-KY497428, KY500 931-KY500951, and KY500134-KY500924 and are listed in Supplemental Tables S2 and S3. All scripts and filters are available at https://github.com/n0rmski/projectH and as Supplemental Scripts.
Supplementary Material
Acknowledgments
This study was supported by US National Institutes of Health (National Institute of Allergy and Infectious Diseases) grant U01 AI090905. We thank Steven G.E. Marsh for expert advice.
Author contributions: P.J.N., M.R., K.L.G., L.A.R., and P.P. designed the project. S.J.N., N.N.G., W.C., C.A., and P.J.N. generated data. P.J.N., T.R., L.A.G., T.D., T.M., and J.A.H. designed and implemented bioinformatics tools. P.J.N., L.A.G., T.R., E.E.W., S.J.N., M.S.W., and N.N.G. analyzed data. P.J.N. and P.P. wrote the paper, which was edited and approved by all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.213538.116.
References
- Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, Babrzadeh F, Gharizadeh B, Luo M, Plummer FA, et al. 2011. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334: 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad T, Neville M, Marshall SE, Armuzzi A, Mulcahy-Hawes K, Crawshaw J, Sato H, Ling KL, Barnardo M, Goldthorpe S, et al. 2003. Haplotype-specific linkage disequilibrium patterns define the genetic topography of the human MHC. Hum Mol Genet 12: 647–656. [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Ashouri E, Norman PJ, Guethlein LA, Han AS, Nemat-Gorgani N, Norberg SJ, Ghaderi A, Parham P. 2016. HLA class I variation in Iranian Lur and Kurd populations: high haplotype and allotype diversity with an abundance of KIR ligands. HLA 88: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, Quintana-Murci L. 2010. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 11: 17–30. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol 13: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DC, Sturrock RD. 1973. Ankylosing spondylitis and HL-A 27. Lancet 1: 904–907. [DOI] [PubMed] [Google Scholar]
- Chapman SJ, Hill AV. 2012. Human genetic susceptibility to infectious disease. Nat Rev Genet 13: 175–188. [DOI] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. 2009. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, Raychaudhuri S. 2012. Interrogating the major histocompatibility complex with high-throughput genomics. Hum Mol Genet 21: R29–R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rocío Baños-Lara M, Piao B, Guerrero-Plata A. 2015. Differential mucin expression by respiratory syncytial virus and human metapneumovirus infection in human epithelial cells. Mediators Inflamm 2015: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilthey A, Cox C, Iqbal Z, Nelson MR, McVean G. 2015. Improved genome inference in the MHC using a population reference graph. Nat Genet 47: 682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorak MT, Shao W, Machulla HK, Lobashevsky ES, Tang J, Park MH, Kaslow RA. 2006. Conserved extended haplotypes of the major histocompatibility complex: further characterization. Genes Immun 7: 450–467. [DOI] [PubMed] [Google Scholar]
- Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, Jostins L, Plant K, Andrews R, McGee C, et al. 2014. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343: 1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanter JM, Gignoux CR, Torgerson DG, Roth LA, Eng C, Oh SS, Nguyen EA, Drake KA, Huntsman S, Hu D, et al. 2014. Genome-wide association study and admixture mapping identify different asthma-associated loci in Latinos: the Genes-environments & Admixture in Latino Americans study. J Allergy Clin Immunol 134: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain RN. 1994. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 76: 287–299. [DOI] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata M, Matsushita I, Tanaka G, Tsuchiya T, Ito H, Tokunaga K, Ohashi J, Homma S, Kobashi Y, Taguchi Y, et al. 2011. Molecular cloning of two novel mucin-like genes in the disease-susceptibility locus for diffuse panbronchiolitis. Hum Genet 129: 117–128. [DOI] [PubMed] [Google Scholar]
- Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, Lush MJ, Povey S, Talbot CC Jr, Wright MW, et al. 2004. Gene map of the extended human MHC. Nat Rev Genet 5: 889–899. [DOI] [PubMed] [Google Scholar]
- Horton R, Gibson R, Coggill P, Miretti M, Allcock RJ, Almeida J, Forbes S, Gilbert JG, Halls K, Harrow JL, et al. 2008. Variation analysis and gene annotation of eight MHC haplotypes: the MHC Haplotype Project. Immunogenetics 60: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illing PT, Vivian JP, Purcell AW, Rossjohn J, McCluskey J. 2013. Human leukocyte antigen-associated drug hypersensitivity. Curr Opin Immunol 25: 81–89. [DOI] [PubMed] [Google Scholar]
- InanlooRahatloo K, Parsa AF, Huse K, Rasooli P, Davaran S, Platzer M, Kramer M, Fan JB, Turk C, Amini S, et al. 2014. Mutation in ST6GALNAC5 identified in family with coronary artery disease. Sci Rep 4: 3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen CE, Alford DR, Trautwein MR, Jalloh YK, Tarnacki JL, Kunnenkeri SK, Fici DA, Yunis EJ, Awdeh ZL, Alper CA. 2014. Dominant sequences of human major histocompatibility complex conserved extended haplotypes from HLA-DQA2 to DAXX. PLoS Genet 10: e1004637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P. 1988. HLA-A and B polymorphisms predate the divergence of humans and chimpanzees. Nature 335: 268–271. [DOI] [PubMed] [Google Scholar]
- Lechler R, Warrens A. 2000. HLA in health and disease, 2nd ed Academic Press, Cambridge, MA. [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, Zhao SD, Li J, Bradfield JP, Mohebnasab M, Steel L, Kobie J, Abrams DJ, Mentch FD, Glessner JT, et al. 2015. Genetic sharing and heritability of paediatric age of onset autoimmune diseases. Nat Commun 6: 8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SGE, Packer R, Heyes JM, Bolton B, Fauchet R, Charron D, Bodmer JG. 1996. The International Histocompatibility Workshop cell panel. In Genetic diversity of HLA functional and medical implications, Vol. 1 (ed. Charron D), pp. 26–28. EDK, Paris. [Google Scholar]
- Mayer WE, Jonker M, Klein D, Ivanyi P, van Seventer G, Klein J. 1988. Nucleotide sequences of chimpanzee MHC class I alleles: evidence for trans-species mode of evolution. EMBO J 7: 2765–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren PJ, Carrington M. 2015. The impact of host genetic variation on infection with HIV-1. Nat Immunol 16: 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelson E, Hurley CK, Ng J, Tilanus M, Carrington M, Marsh SGE, Rozemuller E, Pei J, Rosielle J, Voorter C, et al. 2006. 13th IHWS Shared Resources Joint Report. IHWG Cell and Gene Bank and reference cell panels. In Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatibilty Workshop and Conference, Vol. 1 (ed. Hansen JA), pp. 523–553. IHWG Press, Seattle. [Google Scholar]
- Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, Moretta L. 1996. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol 14: 619–648. [DOI] [PubMed] [Google Scholar]
- Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NV, Jenisch S, Weichenthal M, Abecasis GR, Lim HW, Christophers E, et al. 2006. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet 78: 827–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman PJ, Norberg SJ, Nemat-Gorgani N, Royce T, Hollenbach JA, Shults Won M, Guethlein LA, Gunderson KL, Ronaghi M, Parham P. 2015. Very long haplotype tracts characterized at high resolution from HLA homozygous cell lines. Immunogenetics 67: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman PJ, Hollenbach JA, Nemat-Gorgani N, Marin WM, Norberg SJ, Ashouri E, Jayaraman J, Wroblewski EE, Trowsdale J, Rajalingam R, et al. 2016. Defining KIR and HLA class I genotypes at highest resolution via high-throughput sequencing. Am J Hum Genet 99: 375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MC. 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham P, Moffett A. 2013. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol 13: 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersdorf EW, Malkki M, Horowitz MM, Spellman SR, Haagenson MD, Wang T. 2013. Mapping MHC haplotype effects in unrelated donor hematopoietic cell transplantation. Blood 121: 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, French M, Mallal S, Christiansen F. 1999. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev 167: 257–274. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. 2015. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 43: D423–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933. [DOI] [PubMed] [Google Scholar]
- Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. 1973. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med 288: 704–706. [DOI] [PubMed] [Google Scholar]
- Shayevitz C, Cohen OS, Faraone SV, Glatt SJ. 2012. A re-review of the association between the NOTCH4 locus and schizophrenia. Am J Med Genet B Neuropsychiatr Genet 159B: 477–483. [DOI] [PubMed] [Google Scholar]
- Sommer DD, Delcher AL, Salzberg SL, Pop M. 2007. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CA, Horton R, Allcock RJ, Ashurst JL, Atrazhev AM, Coggill P, Dunham I, Forbes S, Halls K, Howson JM, et al. 2004. Complete MHC haplotype sequencing for common disease gene mapping. Genome Res 14: 1176–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki PI. 1969. Selection of organ donors. N Engl J Med 280: 1304. [DOI] [PubMed] [Google Scholar]
- Trowsdale J, Knight JC. 2013. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet 14: 301–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Hemmings GP. 2000. The NOTCH4 locus is associated with susceptibility to schizophrenia. Nat Genet 25: 376–377. [DOI] [PubMed] [Google Scholar]
- Wu YL, Savelli SL, Yang Y, Zhou B, Rovin BH, Birmingham DJ, Nagaraja HN, Hebert LA, Yu CY. 2007. Sensitive and specific real-time polymerase chain reaction assays to accurately determine copy number variations (CNVs) of human complement C4A, C4B, C4-long, C4-short, and RCCX modules: elucidation of C4 CNVs in 50 consanguineous subjects with defined HLA genotypes. J Immunol 179: 3012–3025. [DOI] [PubMed] [Google Scholar]
- Yang SY, Milford E, Hammerling U, Dupont B. 1987. Description of the Reference Panel of B-Lymphoblastoid Cell Lines for factors of the HLA system: the B-Cell Line Panel designed for the Tenth International Histocompatibility Workshop. In Immunobiology of HLA: Histocompatibility Testing 1987, Vol. 1 (ed. Dupont B), pp. 11–19. Springer-Verlag, New York. [Google Scholar]
- Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu Y, Kitzmiller K, et al. 2007. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet 80: 1037–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.