

Abstract
While metagenomics has emerged as a technology of choice for analyzing bacterial populations, the assembly of metagenomic data remains challenging, thus stifling biological discoveries. Moreover, recent studies revealed that complex bacterial populations may be composed from dozens of related strains, thus further amplifying the challenge of metagenomic assembly. metaSPAdes addresses various challenges of metagenomic assembly by capitalizing on computational ideas that proved to be useful in assemblies of single cells and highly polymorphic diploid genomes. We benchmark metaSPAdes against other state-of-the-art metagenome assemblers and demonstrate that it results in high-quality assemblies across diverse data sets.
Metagenome sequencing has emerged as a technology of choice for analyzing bacterial populations and the discovery of novel organisms and genes (Tyson et al. 2004; Venter et al. 2004; Yooseph et al. 2007; Arumugam et al. 2011). In one of the early metagenomics studies, Venter et al. (2004) attempted to assemble the complex Sargasso Sea microbial community but, as the study stated, failed. On the other side of the spectrum of metagenomics studies, Tyson et al. (2004) succeeded in assembling a simple microbial community consisting of a few species.
These landmark studies (Tyson et al. 2004; Venter et al. 2004) used conventional assembly tools—namely, Celera (Myers et al. 2000) and JAZZ (Aparicio et al. 2002)—with minor modifications. Since they were published, many specialized metagenomic assemblers have been developed (Koren et al. 2011; Laserson et al. 2011; Peng et al. 2011, 2012; Boisvert et al. 2012; Namiki et al. 2012; Haider et al. 2014; Li et al. 2016). However, bioinformaticians are still struggling to bridge the gap between assembling simple and complex microbial communities (for a review, see Gevers et al. 2012). Meanwhile, many researchers succeeded in isolating abundant population genomes out of complex metagenomes (Hess et al. 2011; Dupont et al. 2012; Iverson et al. 2012; CL Dupont, D Kaul, A Bankevich, DB Rusch, RA Richter, J Zhang, J Stuzka, V Montel, A Young, AE Allen, in prep.) by complementing de novo assembly with a partition of contigs into bins based on coverage depth, sequence composition, mate-pair information, and other criteria (Dick et al. 2009; Wu and Ye 2011; Wu et al. 2014). However, this approach often faces difficulties because high fragmentation of metagenomic assemblies negatively affects both the accuracy of binning and the contiguity of genomes attributed to specific bins. Thus, development of better assemblers remains an important goal in metagenomics.
Recent applications of single-cell (Kashtan et al. 2014) and TruSeq Synthetic Long Reads (TSLRs) (Sharon et al. 2015) technologies revealed an enormous microdiversity of related strains within various microbial communities. While strains share most of the genomic sequence, they often have significant variation arising from mutations, insertions of mobile elements, genome rearrangements, or horizontal gene transfer. For example, single-cell sequencing revealed that the wild Prochlorococcus (the most abundant photosynthetic bacteria on earth) population can be viewed as a “federation” of hundreds of distinct subpopulations (some differing in <5% of positions) (Kashtan et al. 2014; Biller et al. 2015). Moreover, nearly all analyzed single cells carried at least one gene cassette not found in other cells from the same subpopulation. By using TSLRs, Sharon et al. (2015) showed that the most abundant species in their sediment samples are represented by dozens of related strains. Moreover, investigators argued that this microdiversity was responsible for the poor reconstruction of the corresponding genomes from short-read libraries. But microdiversity is just one of many metagenomic assembly challenges that we discuss below.
First, widely different abundance levels of various species in a microbial sample result in a highly nonuniform read coverage across different genomes. Moreover, coverage of most species in a typical metagenomic data set is much lower than in a typical sequencing project of a cultivated sample. As a result, standard assembly techniques aimed at isolate genomes with high and rather uniform coverage generate fragmented and error-prone metagenomic assemblies.
Second, various species within a microbial community often share highly conserved genomic regions. Besides complicating the assembly and fragmenting contigs, such “interspecies repeats,” together with low coverage of most species, may trigger intergenomic assembly errors.
Third, many bacterial species in a microbial sample are represented by strain mixtures, that is, multiple related strains with varying abundances (Biller et al. 2014; Kashtan et al. 2014; Rosen et al. 2015; Sharon et al. 2015). Although various studies outside the field of metagenomics extensively addressed a similar challenge of assembling two haplomes within a highly polymorphic eukaryotic genome (Dehal et al. 2002; Vinson et al. 2005; Donmez and Brudno 2011; Kajitani et al. 2014; Safonova et al. 2015), assembly of many closely related bacterial strains is a somewhat different problem with unique computational challenges. While some studies described the initial steps toward identification of complex strain variants (Koren et al. 2011; Peng et al. 2011; Nijkamp et al. 2013), popular metagenomic assembly tools (Boisvert et al. 2012; Peng et al. 2012; Li et al. 2016) still include only rudimentary procedures for assembling strain mixtures with high level of microdiversity.
We note that each of the challenges described above has already been addressed in the course of development of the SPAdes assembly toolkit, albeit in applications outside the field of metagenomics. SPAdes was initially developed to assemble data sets with nonuniform coverage, one of the key challenges of single-cell assembly (Bankevich et al. 2012; Nurk et al. 2013). exSPAnder repeat resolution module of SPAdes (Prjibelski et al. 2014; Vasilinetc et al. 2015; Antipov et al. 2016) was developed to accurately resolve genomic repeats by combining multiple libraries sequenced with various technologies. Lastly, dipSPAdes (Safonova et al. 2015) was developed to address the challenge of assembling a two-haplome mixture within a highly polymorphic diploid genome.
While these recently developed SPAdes tools address challenging assembly problems, metagenomic assembly is arguably an even more difficult problem with data set sizes that dwarf most other DNA sequencing projects. Nevertheless, despite the fact that SPAdes was not designed for metagenomics applications, various groups successfully applied it to their metagenomics studies (McLean et al. 2013; Nurk et al. 2013; Coates et al. 2014; Cotten et al. 2014; Bertin et al. 2015; García-López et al. 2015; Kleigrewe et al. 2015; Kleiner et al. 2015; Miller et al. 2016; Tsai et al. 2016; Xie et al. 2016). However, while SPAdes indeed works well for assembling low-complexity metagenomes like cyanobacterial filaments (Coates et al. 2014), or MDA-amplified mixtures of a small number of randomly selected bacterial cells (Nurk et al. 2013), its performance deteriorates in the case of complex bacterial communities.
Our novel metaSPAdes software combines new algorithmic ideas with proven solutions from the SPAdes toolkit to address various challenges of metagenomic assembly. Below we describe algorithmic approaches used in metaSPAdes and benchmark it against the state-of-the-art metagenomic assemblers IDBA-UD (Peng et al. 2012), Ray-Meta (Boisvert et al. 2012), and MEGAHIT (Li et al. 2015).
Results
Outline of metaSPAdes pipeline
metaSPAdes first constructs the de Bruijn graph of all reads using SPAdes, transforms it into the assembly graph using various graph simplification procedures, and reconstructs paths in the assembly graph that correspond to long genomic fragments within a metagenome (Bankevich et al. 2012; Nurk et al. 2013). metaSPAdes works across a wide range of coverage depths and attempts to maintain a trade-off between the accuracy and the contiguity of assemblies. Responding to the microdiversity challenge, metaSPAdes focuses on reconstructing a consensus backbone of a strain mixture, thus ignoring some strain-specific features corresponding to rare strains.
Benchmarking challenge
Genome assemblers are usually benchmarked on isolates with known reference genomes using various metrics (Salzberg et al. 2012; Gurevich et al. 2013). Benchmarking of metagenomic assemblers is a more difficult task because no reference metagenomes are available for microbial communities of even moderate complexity.
One approach to address this issue relies on identifying reference genomes closely related to some genomes in a metagenome (Koren et al. 2011; Treangen et al. 2013). However, this approach is limited since (1) closely related reference genomes are available only for a fraction of species in a metagenome, and (2) differences between identified references and their counterparts in a metagenome are often misinterpreted as assembly errors (see “Analysis of the HMP Dataset” in the Supplemental Material). Another approach to benchmarking metagenomic assemblers uses synthetic data sets with known community members. Such data sets can be obtained by sequencing the mixtures of bacteria with known genomes (Turnbaugh et al. 2007; Shakya et al. 2013), mixed from isolate sequencing data (Mavromatis et al. 2007), or simulated from reference sequences (Richter et al. 2008; Mende et al. 2012). However, while synthetic data sets proved to be useful in various benchmarking efforts, they are typically less complex than real metagenomes (Koren et al. 2011; Peng et al. 2012).
We benchmarked metaSPAdes against three popular metagenomic assemblers—IDBA-UD (Peng et al. 2012), Ray-Meta (Boisvert et al. 2012), and MEGAHIT (Li et al. 2015)—across diverse synthetic and real data sets using metaQUAST (Mikheenko et al. 2016). The data sets were preprocessed as described in “Data Preprocessing” in the Supplemental Material.
Data sets
We analyzed the following data sets.
Synthetic community data set
Synthetic community data set (SYNTH) is a set of reads from the genomic DNA mixture of 64 diverse bacterial and archaeal species (SRA acc. no. SRX200676) (Shakya et al. 2013) that was used for benchmarking the Omega assembler (Haider et al. 2014). It contains 109 million Illumina HiSeq 100-bp paired-end reads with mean insert size of 206 bp. Since the reference genomes for all 64 species forming the SYNTH data set are known, we used them to assess the quality of various SYNTH assemblies.
Human Microbiome Project data set
Human Microbiome Project data set (HMP) is a female tongue dorsum data set (SRA acc. no. SRX024329) generated by the Human Microbiome Project (The Human Microbiome Project Consortium 2012) that was used for benchmarking by Peng et al. (2011), Treangen et al. (2013), and Mikheenko et al. (2016). It contains 75 million Illumina HiSeq 95-bp paired-end reads with mean insert size of 213 bp. Although the genomes comprising the HMP sample are unknown, we cautiously selected three reference genomes that are similar to the genomes within the sample for benchmarking.
Marine metagenome data set
Marine metagenome data set (MARINE) is a 300-m-depth marine metagenome data set (SRA acc. no. SRX1991080) originating from the functional genomics study of an oxygen minimum zone in the equatorial Pacific (http://genome.jgi.doe.gov/FungenequPacific/FungenequPacific.info.html). It contains 48 million Illumina HiSeq 150-bp paired-end reads with mean insert size of 245 bp.
Aquifer sediment data set
Aquifer sediment data set (SOIL; SRA acc. no. SRX2021633) is one of the Illumina data sets from the study of the sediment samples collected in an aquifer adjacent to the Colorado River (Castelle et al. 2013; Hug et al. 2013), containing 32 million Illumina HiSeq 150-bp paired-end reads with mean insert size of 460 bp (even though aquifer sediments represent a different environment than soil, we took a liberty to refer to this data set as SOIL). Sharon et al. (2015) improved on the original analysis using TSLR sequencing (Kuleshov et al. 2014; McCoy et al. 2014). Since the TSLR technology results in greatly improved metagenomics assemblies (Kuleshov et al. 2015; Bankevich and Pevzner 2016; CL Dupont, D Kaul, A Bankevich, DB Rusch, RA Richter, J Zhang, J Stuzka, V Montel, A Young, AE Allen, in prep.), this data set provides a unique opportunity to benchmark various metagenomic assemblers based on how well they reconstruct genomic regions captured by TSLRs.
Supplemental Materials “CAMI Datasets” and “Analysis of CAMI Datasets” also present results of benchmarking on two synthetic data sets, simulated from reference genomes within the “Critical Assessment of Metagenome Interpretation” (CAMI) initiative (http://www.cami-challenge.org/).
Assembly parameters
metaSPAdes from SPAdes v3.10 prerelease package (see “Software Availability” section), MEGAHIT v1.0.6.1, IDBA-UD v1.1.1, and Ray-Meta v2.3.1 have been launched in 16 threads with (mostly) default parameters. IDBA-UD was launched with read error-correction enabled as recommended in the manual for metagenomic data. Ray-Meta was launched with a k-mer size equal to 31. Supplemental Table S1 provides information about the running time and memory footprints of various assemblers. All assemblers were benchmarked using metaQUAST from QUAST v4.5 package (Gurevich et al. 2013) (with “-m 1000 --scaffolds” options). metaQUAST classifies a position in a scaffold as an intragenomic misassembly if its flanking regions align to nonconsecutive regions of the same reference genome and as an intergenomic misassembly if they align to different reference genomes.
Benchmarking
We benchmarked four assemblers (metaSPAdes, MEGAHIT, UDBA-UD, and Ray-Meta) on four data sets (SYNTH, HMP, MARINE, and SOIL) across a wide array of metrics described below.
Scaffold length statistics
Table 1 provides the scaffold length statistics and demonstrates that, with respect to the total length of scaffolds (>1 kb), Ray-Meta generated inferior results compared with other assemblers on all data sets. metaSPAdes significantly improved the total scaffold length in the case of the most diverse SOIL data set (21% and 40% increase compared with IDBA-UD and MEGAHIT, respectively). Although all tools generated assemblies with similar total scaffold length for the HMP data set, metaSPAdes significantly improved on the length of the 1000 longest scaffolds (36.5 Mb) compared with MEGAHIT (26.6 Mb) and IDBA-UD (29.4 Mb). Similarly, while metaSPAdes and IDBA-UD resulted in assemblies of similar total length on MARINE data set (269.5 and 273.7 Mb, respectively), metaSPAdes significantly improved on the length of the 1000 longest scaffolds compared with IDBA-UD (31.6 Mb vs. 19.3 Mb). MEGAHIT resulted in an inferior assembly compared with metaSPAdes and IDBA-UD on this data set.
Table 1.
The total length of scaffolds (in megabases) for all data sets and all assemblers
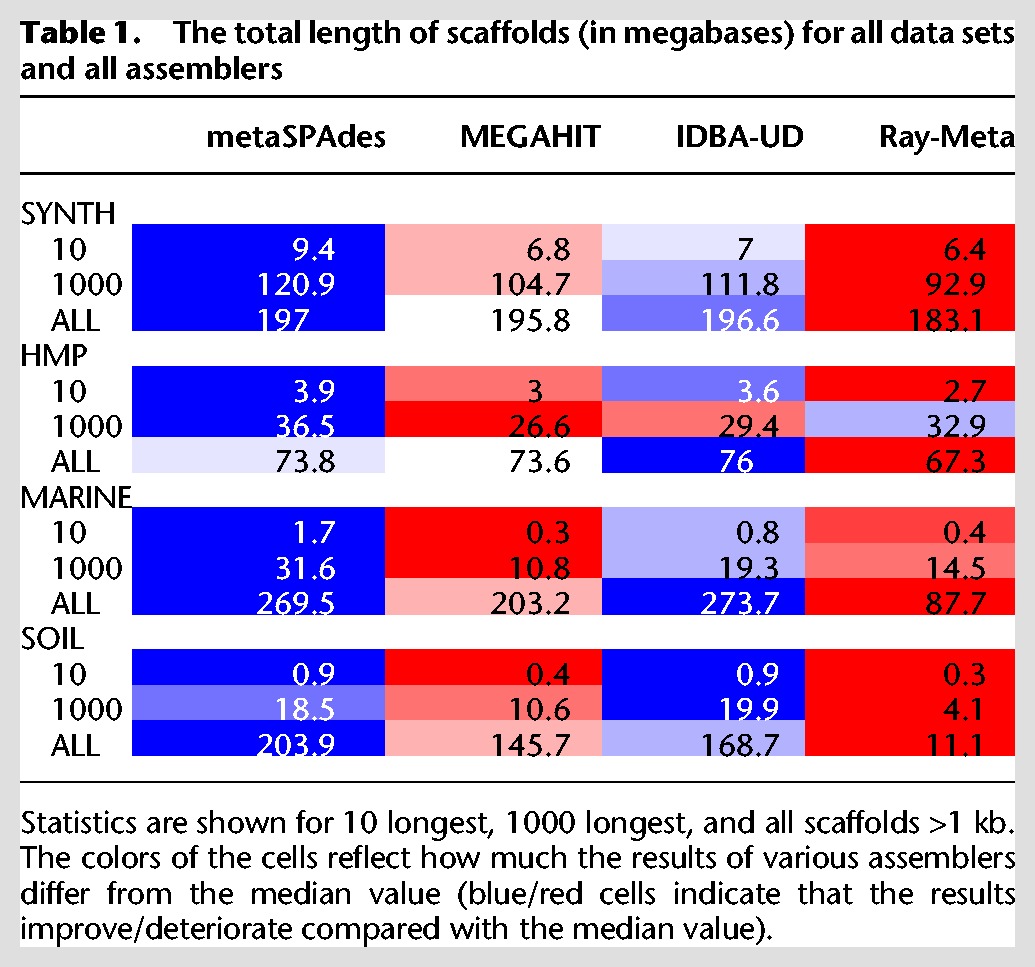
Figure 1 provides cumulative scaffold length plots illustrating that metaSPAdes improves the contiguity of assemblies over all other assemblers for the HMP, MARINE, and SOIL data sets. A surprising conclusion of our benchmarking is that IDBA-UD (often viewed as a slower predecessor of MEGAHIT) improved on the contiguity of MEGAHIT assemblies on all data sets. “The summary of Nx statistics” in the Supplemental Material presents Nx plots across all data sets (for details on the Nx statistics, see metaQUAST manual).
Figure 1.

The cumulative scaffold lengths plots. On the x-axis, scaffolds are ordered from the longest to the shortest. The y-axis shows the total length of x longest scaffolds in the assembly.
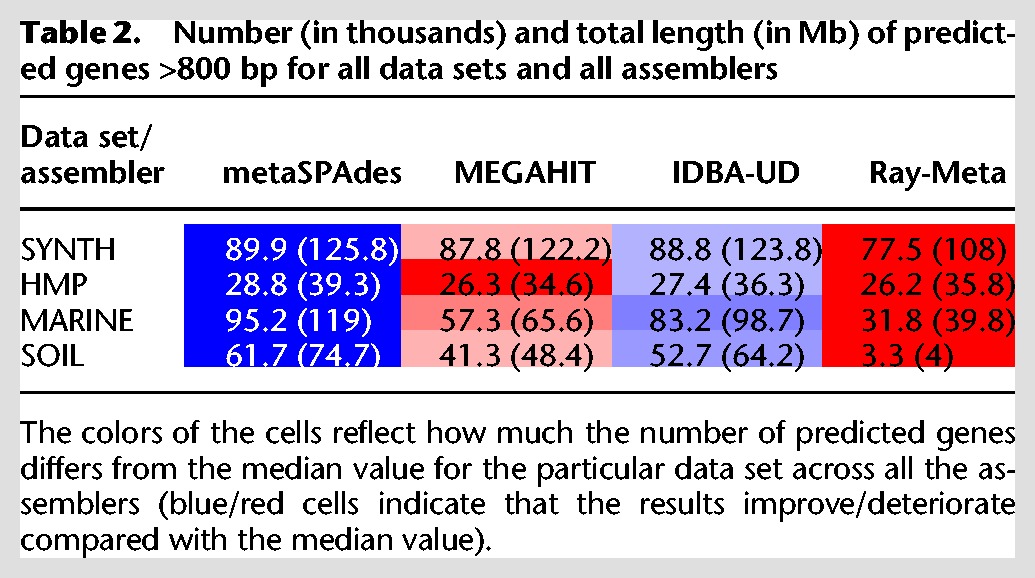
Gene prediction statistics
To further evaluate how fragmented the resulting assemblies are, we used MetaProdigal v2.6.2 (Hyatt et al. 2012) to predict the complete genes (option -c) in each assembly. Predicted genes were then passed through CD-HIT v4.6 (Li and Godzik 2006; Fu et al. 2012) clustering software (with 99% similarity) to correct for potential advantage of more redundant assemblies and to retain only the longest predicted gene in a cluster. Table 2 reports the number and the total length of predicted genes >800 bp (length threshold was set to filter less reliable short gene predictions). In the case of the most complex MARINE and SOIL data sets, the number of predicted long genes in metaSPAdes assemblies is significantly larger compared with other assemblers (14% and 66% increase for the MARINE data set and 17% and 49% increase for the SOIL data set compared with IDBA-UD and MEGAHIT, respectively). Although we cannot rule out many false-positive gene predictions, there is no reason to believe that their rate significantly varies across various assemblers.
Table 2.
Number (in thousands) and total length (in Mb) of predicted genes >800 bp for all data sets and all assemblers
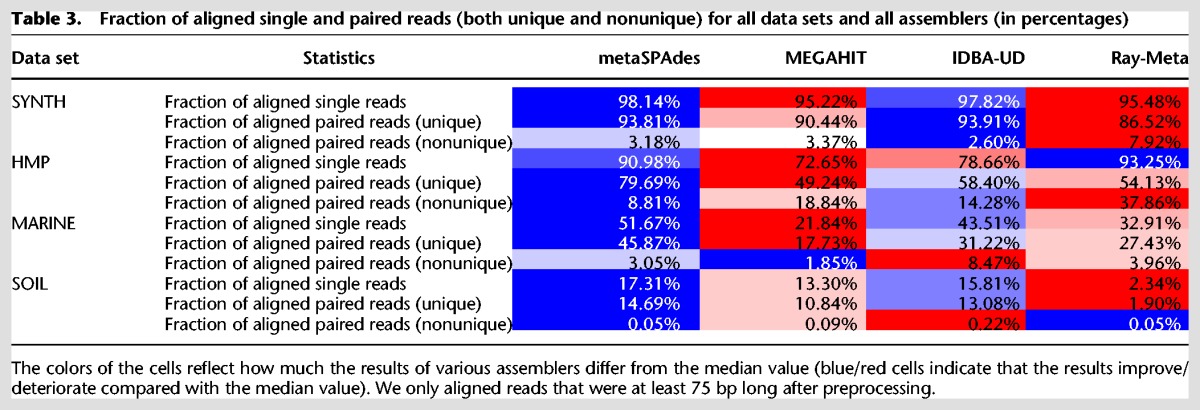
Read alignment statistics
For each data set and assembler, we further aligned read-pairs to scaffolds (>1 kb) with Bowtie 2 v2.2.4 (Langmead and Salzberg 2012). A read-pair is classified as aligned if both reads align to the same scaffold within 1 kb from each other with proper orientation. We further distinguished between uniquely and nonuniquely aligned read-pairs and reported the fractions of aligned single reads and read-pairs for all data sets and assemblers. Better assemblies are characterized by higher fractions of uniquely aligned read-pairs and smaller fractions of nonuniquely aligned read-pairs.
Table 3 illustrates that metaSPAdes resulted in a substantially higher fraction of uniquely aligned read-pairs for the HMP and MARINE data sets compared with all other assemblers. It also shows that only 15%, 11%, 13%, and 2% of read-pairs in the SOIL data set align to assemblies generated by metaSPAdes, MEGAHIT, IDBA-UD, and Ray-Meta, respectively, confirming high diversity of this community. The small fraction of the uniquely aligned read-pairs for this complex data set suggests that the vast majority of genomes in this metagenome have low depth of read coverage, thus preventing their assemblies. Table 3 also provides the fraction of nonuniquely aligned read-pairs (that align to multiple regions in an assembly), thus revealing the potential redundancies (duplicated fragments) in the assemblies. metaSPAdes assemblies have lower rates of nonunique paired-read alignments, indicating that they are less redundant. For example, in the case of the HMP data set, metaSPAdes had <9% of nonuniquely aligned read-pairs compared with 14%, 19%, and 38% for IDBA-UD, MEGAHIT, and Ray-Meta, respectively.
Table 3.
Fraction of aligned single and paired reads (both unique and nonunique) for all data sets and all assemblers (in percentages)
Below we discuss benchmarking results for each data set in more details. metaQUAST reports the NGA50 statistics (NG50 statistics corrected for assembly errors) to evaluate the quality of assembly of individual genomes within a metagenome. To compute NGA50, the contigs are first broken into smaller segments at the identified misassembly breakpoints. NGA50 for a given reference genome is the maximal value such that the broken segments (that align to this reference) of at least that length cover at least half of the bases of the reference.
SYNTH data set
Details about the references comprising the SYNTH data set are given in Supplemental Table S2. Assembly statistics for all assemblers and references is summarized in Supplemental Table S3. “Analysis of the SYNTH Dataset” in the Supplemental Material discusses significant differences in the performance of various assemblers even for this rather simple data set. Assembly statistics for 20 most abundant references is summarized on Figure 2.
Figure 2.

metaQUAST statistics for 20 most abundant species comprising the SYNTH data set. The NGA50 statistics (top left), the fraction of the reconstructed genome compared with the total genome length (top right), the number of intragenomic misassemblies (bottom left), and the number of intergenomic misassemblies (bottom right) for 20 most abundant species comprising the SYNTH data set. References are denoted by their RefSeq IDs (see Supplemental Table S2) and arranged in the decreasing order of the coverage depths.
HMP data set
Since the genomes comprising bacterial communities are typically unknown, the HMP consortium identified a number of reference genomes (listed at HMP Shotgun Community profiling SRS077736) similar to the genomes present in the HMP data set (Treangen et al. 2013). However, our attempt to use this resource for reliable quality assessment faced difficulties: Only three genomes in this list (Streptococcus salivarius SK126, Neisseria subflava NJ9703, and Prevotella melaninogenica ATCC 25845) were at least 70% covered by contigs generated by assemblers included in this study. Moreover, we revealed substantial differences between these references and the genomes in the sample, making metaQUAST analysis unreliable (see “Analysis of the HMP Dataset” in the Supplemental Material).
MARINE data set
Based on the fraction of aligned reads (Table 3), we conclude that MARINE data set represents a more diverse community than the HMP data set but is less diverse that the SOIL data set. Tables 1 through 3 illustrate that metaSPAdes results in more contiguous and complete assembly of the MARINE data set than all other assemblers.
SOIL data set
We compared assemblies of the SOIL data set against the set of contigs obtained by Bankevich and Pevzner (2016) from TSLRs for all three samples described by Sharon et al. (2015) (which improves on the TSLR assemblies from the original study). Contigs >20 kb (total length 103 Mb) were selected as a “reference genome” for computing metaQUAST statistics (with additional “--fragmented” option). Results are summarized in Table 4. Only 27.6 Mb (≈13.6%) of the total length of the metaSPAdes scaffolds >1 kb (196 Mb) overlapped with TSLR contigs, covering just ≈26% of the total length of the TSLR assembly (best result across all assemblers).
Table 4.
Comparison of long scaffolds (>1 kb) generated by various metagenomic assemblers for the SOIL data set against TSLR contigs generated by Bankevich and Pevzner (2016)

Additional benchmarks
In addition to the benchmarking results presented above, the Supplemental Materials also include benchmarking of all assemblers on two CAMI data sets (“CAMI Datasets” and “Analysis of the CAMI Datasets”), comparison of metaSPAdes and SPAdes assemblies on the SYNTH data set (“Benchmarking SPAdes against metaSPAdes”), and discussion of how the novel algorithms proposed in this work affect the quality of assemblies (“Effect of Novel Algorithmic Approaches in metaSPAdes on Assembly Quality”).
Discussion
metaSPAdes addresses a number of challenges in metagenomic assembly and implemented several novel features, such as efficient assembly graph processing to address the microdiversity challenge, a new repeat resolution approach that utilizes rare strain variants to improve the consensus assembly of strain mixtures, and fast algorithms for constructing assembly graphs and error-correcting reads.
These features contributed to improvements in metaSPAdes assemblies (compared with the state-of-the-art assemblers MEGAHIT, IDBA-UD, and Ray-Meta) and enabled us to scale metaSPAdes for analyzing large metagenomes.
In addition to the intrinsic biological challenges discussed in this article, the field of metagenomic assembly also faces technological challenges caused by innovations in sequencing and library preparation techniques. For example, recently introduced high-quality jumping (mate-pair) libraries (such as Nextera Mate Pair Libraries) have a potential to significantly improve assembly quality (Vasilinetc et al. 2015). However, metagenomic assemblers have not caught up with this technological innovation yet. Another example is the TSLR technology (Kuleshov et al. 2014; McCoy et al. 2014), whose first metagenomic applications highlighted the need for developing methods to reliably combine it with the paired-end libraries (Kuleshov et al. 2015; Sharon et al. 2015; Bankevich and Pevzner 2016). metaSPAdes now faces the challenge of incorporating these emerging technologies into its assembly pipeline.
Methods
Detecting and masking strain variation
Genomic differences between related strains often result in “bulges” and “tips” in the de Bruijn graphs that are not unlike artifacts caused by sequencing errors in genome assembly (Pevzner et al. 2004; Zerbino and Birney 2008). For example, a sequencing error often results in a bulge formed by two short alternative paths between the same vertices in the de Bruijn graph, a “correct” path with high coverage and an “erroneous” path with low coverage. Similarly, a substitution or a small indel in a rare strain (compared with an abundant strain) often results in a bulge formed by a high-coverage path corresponding to the abundant strain and an alternative low-coverage path corresponding to the rare strain.
Aiming at the consensus assembly of a strain mixture, metaSPAdes masks the majority of strain differences using a modification of the SPAdes procedures for masking sequencing errors (the algorithms for removal of tips, “simple” bulges [Bankevich et al. 2012], and “complex” bulges [Nurk et al. 2013]). metaSPAdes uses more aggressive settings than the ones used in assemblies of isolates; for example, it collapses larger bulges and removes longer tips than SPAdes. We note that the bulge projection approach in SPAdes improves on the originally proposed bulge removal approach (Pevzner et al. 2004; Zerbino and Birney 2008) used in most existing assemblers since it stores valuable information about the processed bulges (see “Bulge Projection Approach” in the Supplemental Material). This feature is important for the repeat resolution approach in metaSPAdes described below.
Analyzing filigree edges in the assembly graph
In addition to single-nucleotide variants and small indels, strain variation is often manifested as highly diverged regions, insertions of mobile elements, rearrangements, large deletions, parallel gene transfer, etc. The green edges in the assembly graph shown in Figure 3 result from an additional copy of a mobile element in a rare strain2 (compared with the abundant strain1), while the blue edge corresponds to a horizontally transferred gene (or a highly diverged genomic region) in a rare strain3 (compared to the abundant strain1). Such edges fragment contigs corresponding to the abundant strain1; for example, the green edges in Figure 3 (bottom right) break the edge c into three shorter edges. We note that the edges in the assembly graph are condensed; that is, they represent nonbranching paths formed by k-mers.
Figure 3.

The de Bruijn graphs of three strains and their strain mixture. The figure shows only a small subgraph of the de Bruijn graph. The abundant strain (strain1) is shown by thick lines, and the rare strains (strain2 and strain3) are shown by thin lines. The genomic repeat R is shown in red. (Top left) The de Bruijn graph of the abundant strain1. (Top right) The rare strain2 differs from the abundant strain1 by an insertion of an additional copy or repeat R. The two breakpoint edges resulting from this insertion are shown in green. These filigree edges are not removed by the graph simplification procedures in the standard assembly tools aimed at isolates. (Bottom left) The rare strain3 differs from the abundant strain1 by an insertion of a horizontally transferred gene (or a highly diverged genomic region). (Bottom right) The de Bruijn graph of the mixture of three strains.
We refer to edges originating from rare strain variants within the assembly graph of a strain mixture as filigree edges. Traditional genome assemblers use a global threshold on read coverage to remove the low-coverage edges (that typically result from sequencing errors) from the assembly graph during the graph simplification step. However, this approach does not work well for metagenomic assemblies, since there is no global threshold that (1) removes edges corresponding to rare strains and (2) preserves edges corresponding to rare species. Similarly to IDBA-UD and MEGAHIT, metaSPAdes analyzes the coverage ratios between adjacent edges in the assembly graph, classifying edges with low-coverage ratios as potential filigree edges.
We denote the coverage of an edge e in the assembly graph as cov(e) and define the coverage cov(v) of a vertex v as the maximum of cov(e) over all edges e incident to v. Given an edge e incident to a vertex v and a threshold ratio (the default value is 10), a vertex v predominates an edge e if its coverage is significantly higher than the coverage of the edge e; that is, if ratio · cov(e) < cov(v). An edge (v,w) is weak if it is predominated by either v or w. Note that filigree edges are often classified as weak since their coverage is much lower than the coverage of adjacent edges resulting from abundant strains.
metaSPAdes disconnects all weak edges from their predominating vertices in the assembly graph. Disconnection of a weak edge (v,w) in the assembly graph from its starting vertex v (ending vertex w) is simply a removal of its first (last) k-mer rather than removal of the entire condensed edge. We emphasize that, in contrast to IDBA-UD and MEGAHIT, we disconnect rather than remove weak edges in the assembly graph since our goal is to preserve the information about rare strains whenever possible, that is, when it does not lead to a deterioration of the consensus backbone.
Repeat resolution with exSPAnder
exSPAnder (Prjibelski et al. 2014; Vasilinetc et al. 2015; Antipov et al. 2016) is a module of SPAdes that combines various sources of information (e.g., paired reads or long error-prone reads) for resolving repeats and scaffolding in the assembly graph. Starting from a path consisting of a single condensed edge in the assembly graph, exSPAnder iteratively attempts to extend it to a longer path that represents a contiguous segment of the genome (genomic path). To extend a path, exSPAnder selects one of its extension edges (edges that start at the terminal vertex of this path). Choice of the extension edge is controlled by the decision rule that evaluates whether a particular extension edge is sufficiently supported by the data, while other extension edges are not (given the existing path). exSPAnder further removes overlaps between generated genomic paths (overlap reduction step) and outputs the strings spelled by the resulting paths as a set of contigs.
metaSPAdes modifies the decision rule of exSPAnder to account for the local read coverage, denoted localCov, of the specific genomic region that is being reconstructed during the path extension process. For details, see “Modifying the Decision Rule in exSPAnder for Metagenomic Data” in the Supplemental Material. The value localCov is estimated as the minimum across the average coverages of the edges in the path that is being extended. Taking minimum (rather than the average) coverage excludes the repetitive edges in the path from consideration and typically underestimates the real coverage of the region, making the decision rule more conservative.
A new metagenomic decision rule in metaSPAdes
Some intergenomic repeats between species of different abundances can be resolved based on the differences in the depth of read coverage (Haider et al. 2014; Namiki et al. 2012). metaSPAdes introduces an additional metagenomics-specific decision rule that filters out unlikely path extensions using the coverage estimate of the region that is being reconstructed (Fig. 4). It often allows metaSPAdes to pass through long inter-species repeats during reconstruction of abundant species. metaSPAdes applies a new decision rule described below only if the paired reads failed to provide sufficient evidence to discriminate between extension edges.
Figure 4.

Applying the metagenomics-specific decision rule for repeat resolution. The figure shows only a small subgraph of the assembly graph. (A) The path that is currently being extended (formed by green edges) along with its blue extension edges e and e′. (B) The short-edge traversal from the end of the extension edge e. The dotted curve shows the boundary frontier(e) of the traversal. The edges in the set next(e) are shown in red with low-coverage edges represented as dashed arrows (other edges in next(e) are represented as solid arrows). Since all edges in next(e) have low coverage, the edge e is ruled out as an unlikely extension candidate. (C) The short-edge traversal from the end of the extension edge e′. (D) Since e′ is a single extension edge that was not ruled out (there is a solid edge in next(e′)), it is added to the growing path and the extension process continues.
An edge in the assembly graph is called long if its length exceeds a certain threshold (1500 bp by default) and short otherwise. We say that a long edge e2 follows a long edge e1 in a genomic path if all edges between the end of e1 and the start of e2 in this path are short.
While considering an extension edge e, metaSPAdes performs a directed traversal of the graph (Fig. 4B), starting from the end of e and walking along the short edges. We define the set of all vertices that are reached by this traversal as frontier(e) and consider the set next(e) of all long edges starting in frontier(e). This procedure is aimed at finding nonrepetitive long edges that can follow e in the (unknown) genomic path. We classify an edge in the set next(e) as a low-coverage edge if the coverage estimate of the region that is being reconstructed, localCov, exceeds its coverage at least by a factor β (the default value β = 2). If all edges in next(e) are low-coverage edges, then e is considered an unlikely candidate for an extension of the current path. If all but a single edge e′ represent unlikely extensions, the path is extended by e′ (Fig. 4C).
Utilizing strain differences for repeat resolution in metaSPAdes
Safonova et al. (2015) showed that differences between haplomes can be used to improve the quality of consensus assembly of a highly polymorphic diploid genome. metaSPAdes capitalizes on the similar observation that the differences between strains can be, somewhat counter-intuitively, used to improve the quality of consensus assembly of a strain mixture. In particular, contigs generated prior to masking strain differences in assembly graph and thus representing genomic fragments of individual strains (strain-contigs) often provide additional long-range information for reconstruction of a strain-mixture backbone.
Inspired by dipSPAdes (Safonova et al. 2015), metaSPAdes uses the following pipeline that includes two launches of exSPAnder (Fig. 5).
Generating strain-contigs. After constructing the assembly graph (that encodes both abundant and rare strains), we launch exSPAnder to generate a set of strain-contigs representing both rare and abundant strains (Fig. 5C). Strain-contigs are not subjected to the default overlap reduction step in exSPAnder.
Transforming assembly graph into consensus assembly graph. metaSPAdes identifies and masks rare strain variants, resulting in the consensus assembly graph (Fig. 5D).
Generating strain-paths in the consensus assembly graph. Capitalizing on the bulge projection approach (see “Bulge Projection Approach” in the Supplemental Material), metaSPAdes reconstructs paths in the consensus assembly graph corresponding to strain-contigs, referred to as strain-paths (Fig. 5E).
Repeat resolution using strain-paths. This step utilizes the hybrid mode of exSPAnder originally developed to incorporate long error-prone Pacific Biosciences and Oxford Nanopore reads in the repeat resolution process (Ashton et al. 2014; Labonté et al. 2015; Antipov et al. 2016). Instead of working with long error-prone reads, we modified exSPAnder to work with virtual reads spelled by the strain-paths to facilitate resolution of repeats in the consensus assembly graph (Fig. 5F).
Figure 5.

Repeat resolution in metagenomic assembly. (A) One of two identical copies of a long (longer than the insert size) repeat R (red) in the abundant strain has mutated into a unique genomic “green” region R′ in the rare strain. (B) The assembly graph resulting from a mixture of reads from the abundant and rare strains. Two alternative paths between the start and the end of the green edge (one formed by a single green edge and another formed by two black and one red edge) form a bulge. (C) The strain-contig spanning R′ (shown by green dashed line) constructed by exSPAnder at the “generating strain-contigs” step. (D) Masking of the strain variation at the “transforming assembly graph into consensus assembly graph” step leads to a projection of a bulge (formed by red and green edges) and results in the consensus assembly graph shown in E. The blue arrows emphasize that SPAdes projects rather than deletes bulges, facilitating the subsequent reconstruction of strain-paths in the consensus assembly graph. (E) Reconstruction of the strain-path (green dotted line), corresponding to a strain-contig (green dashed line) at the “generating strain-paths in the consensus assembly graph” step. (F) At the “repeat resolution using strain-paths” step, metaSPAdes utilizes both strain-paths and paired reads to resolve repeats in the consensus graph. The green dotted strain-path from E is used as additional information to reconstruct the consensus contig cRd spanning the long repeat.
Note that in the example in Figure 5, the long red repeat with multiplicity 2 in the abundant strain is resolved because of the variants (diverged green copy of the repeat) in the rare strain.
Scaling metaSPAdes
Since some metagenomic data sets contain billions of reads, metagenomic assemblers have to be optimized with respect to both speed and memory footprint (Nagarajan and Pop 2013). “Reducing Running Time and Memory Footprint of metaSPAdes” in the Supplemental Material describes efforts to scale metaSPAdes for assembling large metagenomic data sets.
Software availability
The latest version of the SPAdes toolkit that includes metaSPAdes is available from http://cab.spbu.ru/software/spades. The source code for the v3.10 prerelease package used for benchmarking in this article is available as a Supplemental Material or alternatively can be accessed as git revision 3d6df0c62ca31a187cb7c2209c892e6e5711229e at https://github.com/ablab/spades. The QUAST v4.5 package is available from https://github.com/ablab/quast/tree/release_4.5.
Supplementary Material
Acknowledgments
We thank Chris Dupont, Rob Knight, Mihai Pop, and Bahar Behsaz for useful comments. We also thank Alla Lapidus, who brought our attention to the field of metagenomics. This work was supported by the Russian Science Foundation (grant 14-50-00069).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.213959.116.
References
- Antipov D, Korobeynikov A, McLean JS, Pevzner PA. 2016. hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics 32: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia J-M, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. 2002. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297: 1301–1310. [DOI] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J, et al. 2011. Enterotypes of the human gut microbiome. Nature 473: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, Wain J, O'Grady J. 2014. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol 33: 296–300. [DOI] [PubMed] [Google Scholar]
- Bankevich A, Pevzner PA. 2016. TruSPAdes: barcode assembly of TruSeq synthetic long reads. Nat Methods 13: 248–250. [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin MJ, Schwartz SL, Lee J, Korobeynikov A, Dorrestein PC, Gerwick L, Gerwick WH. 2015. Spongosine production by a Vibrio harveyi strain associated with the sponge Tectitethya crypta. J Nat Prod 78: 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biller SJ, Berube PM, Lindell D, Chisholm SW. 2015. Prochlorococcus: the structure and function of collective diversity. Nat Rev Microbiol 13: 13–27. [DOI] [PubMed] [Google Scholar]
- Boisvert S, Raymond F, Godzaridis É, Laviolette F, Corbeil J. 2012. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol 13: R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Wu D, Tringe SG, Singer SW, Eisen JA, Banfield JF. 2013. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun 4: 2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates RC, Podell S, Korobeynikov A, Lapidus A, Pevzner P, Sherman DH, Allen EE, Gerwick L, Gerwick WH. 2014. Characterization of cyanobacterial hydrocarbon composition and distribution of biosynthetic pathways. PLoS One 9: e851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten M, Oude Munnink B, Canuti M, Deijs M, Watson SJ, Kellam P, van der Hoek L. 2014. Full genome virus detection in fecal samples using sensitive nucleic acid preparation, deep sequencing, and a novel iterative sequence classification algorithm. PLoS One 9: e93269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, et al. 2002. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298: 2157–2167. [DOI] [PubMed] [Google Scholar]
- Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF. 2009. Community-wide analysis of microbial genome sequence signatures. Genome Biol 10: R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez N, Brudno M. 2011. Hapsembler: an assembler for highly polymorphic genomes. In Research in computational molecular biology (ed. Bafna V, Sahinalp SC), Vol. 6577, pp. 38–52. Springer, Berlin, Heidelberg. [Google Scholar]
- Dupont CL, Rusch DB, Yooseph S, Lombardo M-J, Alexander Richter R, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, et al. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28: 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-López R, Vázquez-Castellanos JF, Moya A. 2015. Fragmentation and coverage variation in viral metagenome assemblies, and their effect in diversity calculations. Front Bioeng Biotechnol 3: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Pop M, Schloss PD, Huttenhower C. 2012. Bioinformatics for the human microbiome project. PLoS Comput Biol 8: e1002779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, Ahn T-H, Bushnell B, Chai J, Copeland A, Pan C. 2014. Omega: an overlap-graph de novo assembler for metagenomics. Bioinformatics 30: 2717–2722. [DOI] [PubMed] [Google Scholar]
- Hess M, Sczyrba A, Egan R, Kim T-W, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, et al. 2011. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331: 463–467. [DOI] [PubMed] [Google Scholar]
- Hug LA, Castelle CJ, Wrighton KC, Thomas BC, Sharon I, Frischkorn KR, Williams KH, Tringe SG, Banfield JF. 2013. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Locascio PF, Hauser LJ, Uberbacher EC. 2012. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28: 2223–2230. [DOI] [PubMed] [Google Scholar]
- Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. 2012. Untangling genomes from metagenomes: revealing an uncultured class of marine euryarchaeota. Science 335: 587–590. [DOI] [PubMed] [Google Scholar]
- Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashtan N, Roggensack SE, Rodrigue S, Thompson JW, Biller SJ, Coe A, Ding H, Marttinen P, Malmstrom RR, Stocker R, et al. 2014. Single-cell genomics reveals hundreds of coexisting subpopulations in wild prochlorococcus. Science 344: 416–420. [DOI] [PubMed] [Google Scholar]
- Kleigrewe K, Almaliti J, Tian IY, Kinnel RB, Korobeynikov A, Monroe EA, Duggan BM, Di Marzo V, Sherman DH, Dorrestein PC, et al. 2015. Combining mass spectrometric metabolic profiling with genomic analysis: a powerful approach for discovering natural products from cyanobacteria. J Nat Prod 78: 1671–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner M, Hooper LV, Duerkop BA. 2015. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 16: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Treangen TJ, Pop M. 2011. Bambus 2: scaffolding metagenomes. Bioinformatics 27: 2964–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov V, Xie D, Chen R, Pushkarev D, Ma Z, Blauwkamp T, Kertesz M, Snyder M. 2014. Whole-genome haplotyping using long reads and statistical methods. Nat Biotechnol 32: 261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov V, Jiang C, Zhou W, Jahanbani F, Batzoglou S, Snyder M. 2015. Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome. Nat Biotechnol 34: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonté JM, Swan BK, Poulos B, Luo H, Koren S, Hallam SJ, Sullivan MB, Woyke T, Wommack KE, Stepanauskas R. 2015. Single-cell genomics-based analysis of virus-host interactions in marine surface bacterioplankton. ISME J 9: 2386–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laserson J, Jojic V, Koller D. 2011. Genovo: de novo assembly for metagenomes. J Comput Biol 18: 429–443. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31: 1674–1676. [DOI] [PubMed] [Google Scholar]
- Li D, Luo R, Liu C-M, Leung C-M, Ting H-F, Sadakane K, Yamashita H, Lam T-W. 2016. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102: 3–11. [DOI] [PubMed] [Google Scholar]
- Mavromatis K, Ivanova N, Barry KW, Shapiro HJ, Goltsman E, McHardy AC, Rigoutsos I, Salamov A, Korzeniewski F, Land M, et al. 2007. Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nat Methods 4: 495–500. [DOI] [PubMed] [Google Scholar]
- McCoy RC, Taylor RW, Blauwkamp TA, Kelley JL, Kertesz M, Pushkarev D, Petrov DA, Fiston-Lavier A-S. 2014. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS One 9: e106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JS, Lombardo M-J, Badger JH, Edlund A, Novotny M, Yee-Greenbaum J, Vyahhi N, Hall AP, Yang Y, Dupont CL, et al. 2013. Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc Natl Acad Sci 110: E2390–E2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende DR, Waller AS, Sunagawa S, Järvelin AI, Chan MM, Arumugam M, Raes J, Bork P. 2012. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS One 7: e31386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko A, Saveliev V, Gurevich A. 2016. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32: 1088–1090. [DOI] [PubMed] [Google Scholar]
- Miller IJ, Weyna TR, Fong SS, Lim-Fong GE, Kwan JC. 2016. Single sample resolution of rare microbial dark matter in a marine invertebrate metagenome. Sci Rep 6: 34362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196–2204. [DOI] [PubMed] [Google Scholar]
- Nagarajan N, Pop M. 2013. Sequence assembly demystified. Nat Rev Genet 14: 157–67. [DOI] [PubMed] [Google Scholar]
- Namiki T, Hachiya T, Tanaka H, Sakakibara Y. 2012. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res 40: e155–e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijkamp JF, Pop M, Reinders MJT, de Ridder D. 2013. Exploring variation-aware contig graphs for (comparative) metagenomics using MaryGold. Bioinformatics 29: 2826–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, et al. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20: 714–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HCM, Yiu SM, Chin FYL. 2011. Meta-IDBA: a de novo assembler for metagenomic data. Bioinformatics 27: 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HCM, Yiu SM, Chin FYL. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Pevzner PA, Tang H, Tesler G. 2004. De novo repeat classification and fragment assembly. Genome Res 14: 1786–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prjibelski AD, Vasilinetc I, Bankevich A, Gurevich A, Krivosheeva T, Nurk S, Pham S, Korobeynikov A, Lapidus A, Pevzner PA. 2014. ExSPAnder: a universal repeat resolver for DNA fragment assembly. Bioinformatics 30: 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter DC, Ott F, Auch AF, Schmid R, Huson DH. 2008. MetaSim: a sequencing simulator for genomics and metagenomics ed. D. Field. PLoS One 3: e3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MJ, Davison M, Bhaya D, Fisher DS. 2015. Fine-scale diversity and extensive recombination in a quasisexual bacterial population occupying a broad niche. Science 348: 1019–1023. [DOI] [PubMed] [Google Scholar]
- Safonova Y, Bankevich A, Pevzner P. 2015. dipSPAdes: assembler for highly polymorphic diploid genomes. J Comput Biol 22: 528–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, et al. 2012. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res 22: 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya M, Quince C, Campbell JH, Yang ZK, Schadt CW, Podar M. 2013. Comparative metagenomic and rRNA microbial diversity characterization using archaeal and bacterial synthetic communities. Environ Microbiol 15: 1882–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I, Kertesz M, Hug LA, Pushkarev D, Blauwkamp TA, Castelle CJ, Amirebrahimi M, Thomas BC, Burstein D, Tringe SG, et al. 2015. Accurate, multi-kb reads resolve complex populations and detect rare microorganisms. Genome Res 25: 534–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Koren S, Sommer DD, Liu B, Astrovskaya I, Ondov B, Darling AE, Phillippy AM, Pop M. 2013. MetAMOS: a modular and open source metagenomic assembly and analysis pipeline. Genome Biol 14: R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y-C, Conlan S, Deming C, Segre JA, Kong HH, Korlach J, Oh J. 2016. Resolving the complexity of human skin metagenomes using single-molecule sequencing. MBio 7: e01948–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. 2007. The human microbiome project. Nature 449: 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43. [DOI] [PubMed] [Google Scholar]
- Vasilinetc I, Prjibelski AD, Gurevich A, Korobeynikov A, Pevzner P. 2015. Assembling short reads from jumping libraries with large insert sizes. Bioinformatics 31: 3262–3268. [DOI] [PubMed] [Google Scholar]
- Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, et al. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304: 66–74. [DOI] [PubMed] [Google Scholar]
- Vinson JP, Jaffe DB, O'Neill K, Karlsson EK, Stange-Thomann N, Anderson S, Mesirov JP, Satoh N, Satou Y, Nusbaum C, et al. 2005. Assembly of polymorphic genomes: algorithms and application to Ciona savignyi. Genome Res 15: 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y-W, Ye Y. 2011. A novel abundance-based algorithm for binning metagenomic sequences using l -tuples. J Comput Biol 18: 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y-W, Tang Y-H, Tringe SG, Simmons BA, Singer SW. 2014. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Ren M, Yang C, Yi H, Li Z, Li T, Zhao J. 2016. Metagenomic analysis reveals symbiotic relationship among bacteria in microcystis-dominated community. Front Microbiol 7: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W, et al. 2007. The Sorcerer II global ocean sampling expedition: expanding the universe of protein families. PLoS Biol 5: 0432–0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.