

Abstract
Tumor cells require vascular supply for their growth, and they express proangiogenic growth factors that promote the formation of vascular networks. Many oncogenic mutations that may potentially lead to tumor angiogenesis have been identified. Somatic mutations in the small GTPase NRAS are the most common activating lesions found in human cancer and are generally associated with poor response to standard therapies. However, the mechanisms by which NRAS mutations affect tumor angiogenesis are largely unknown. Therefore, we investigated the role of NRASQ61K oncogene in tumor angiogenesis and analyzed tumors harboring NRASQ61K for potential sensitivity to a kinase inhibitor. Knock-in of the NRASQ61K allele in human normal epithelial cells triggered the angiogenic response in these cells. In cancer cells harboring oncogenic NRAS, a mitogen-activated protein kinase (MEK) inhibitor down-regulated the extracellular regulated protein kinase (ERK) pathway and inhibited the expression of proangiogenic molecules. In tumor xenografts harboring the NRASQ61K, the MEK inhibitor extensively modified tumor growth, causing abrogation of angiogenesis. Overall, our results provide a functional link between oncogenic NRAS and angiogenesis, and imply that tumor vasculature could be indirectly altered by targeting a genetic lesion on which cancer cells are dependent.
Keywords: NRAS, NSCLC, angiogenesis
Introduction
Tumor-induced angiogenesis is pivotal for growth, invasion, and metastasis [1]. At the clinical level, tumor angiogenesis is associated with more aggressive phenotypes and poor drug delivery to the tumor mass. One of the initial steps in tumor-induced angiogenesis is the secretion of multiple angiogenic factors from tumor cells, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and platelet-derived growth factor (PDGF) [2]. The roles of VEGF and vascular endothelial growth factor receptor 2 (VEGFR2) in tumor angiogenesis are well established. Accordingly, anti-angiogenic agents that target VEGFR2 prolong the survival of cancer patients, but other angiogenic factors become activated during cancer progression. To date, several mutations in genes encoding cellular signaling proteins have been identified, and these “driver mutations” have been associated with tumor angiogenesis and other neoplastic processes [3]. Understanding the mechanisms by which the genetic variations in oncogenes affect the angiogenic process remains essential in order to increase the efficacy of the current cancer therapies.
Oncogenic mutations in three RAS genes have been identified in virtually all types of human cancers, with each mutation exhibiting a characteristic incidence and RAS gene association [4]. In particular, mutations in KRAS and NRAS (but rarely in the HRAS) are frequently reported. Recently, it has been suggested that alterations in oncogenes involved in the intracellular NRAS signaling cascade play a central role in regulating tumor progression [5]. Directly targeting oncogenic NRAS is an extremely challenging problem for rational drug design, and no clinically available mechanism-based therapies currently exist for ~30% of human cancers with oncogenic NRAS mutations, including non-small cell lung cancer (NSCLC), hepatocellular carcinoma, myeloid leukemia, thyroid carcinoma, and melanoma [6]. The most common NRAS mutation is Q61K, an amino acid substitution from glutamine (Q) to lysine (K), which results in constitutive activation of NRAS kinase activity. NRASQ61K is involved in the onset and progression of several cancers, including melanoma, papillary thyroid, colorectal, and ovarian tumors, and is often associated with a particularly poor prognosis. NRASQ61K transduces signals from activated growth factor receptors and other extracellular stimuli to the nucleus, thereby regulating cellular processes that include proliferation, differentiation, and survival [7]. Cancer-associated NRAS mutations encode proteins that accumulate in guanosine triphosphate (GTP)-bound active conformation because of their defective intrinsic GTP hydrolysis and resistance to GTPase-activating proteins. Thus, there is an increasing interest in developing anti-cancer drugs targeting components of the NRAS-regulated phosphoinositide 3 kinase/protein kinase B (PI3K/AKT) cascade and the mitogen-activated protein kinase (MAPK) pathway, which includes Raf, MEK, and ERK.
The NRAS-mediated signaling pathways, particularly those involved in cell survival, have been well studied. However, the role of NRAS in angiogenesis, another process causing malignant transformation of tumors, has been rarely reported. Therefore, the current study aimed to investigate potential effects of NRAS alterations on tumor angiogenesis, and test whether clinically successful kinase inhibitors that target NRAS-mutant tumors could reverse tumor angiogenesis and growth.
Materials and methods
Cell lines and transfection
BEAS-2B, H1299, H2087, human umbilical vein endothelial cells (HUVECs), and H1975 cell lines were purchased from the American Type Culture Collection (ATCC). They were cultured in DMEM, RPMI 1640, or F12 medium supplemented with 10% fetal calf serum (FCS; Gibco, Invitrogen, USA), and 1% penicillin/streptomycin (Gibco). Cell were maintained at 37°C in a humidified atmosphere containing 5% CO2. A small (or short) interfering RNA (siRNA) specifically targeting NRASQ61K gene (5’-GAAGUGCAUACACCGAGAC-3’) was constructed by Nanjing GenScript Biotechnology Co., Ltd. Cells were transfected with control or NRAS-specific siRNA (0.5 μg/well in 96-well plates and 3 μg/well in 6-well plates). Plasmid harboring a constitutively active mutant NRASQ61K was a gift from Dr. Channing Der (Addgene plasmid # 12543) [8]. Cells were transfected with the NRASQ61K plasmid (0.15 μg/well in 96-well plates and 2 μg/well in 6-well plates). Transient transfection was performed by using the Lipofectamine® RNAiMAX reagent (Invitrogen), per manufacturer’s instructions.
Chorioallantoic membrane (CAM) assay
Fertilized white leghorn chicken embryos were incubated for 3 days at 37°C and 70% humidity. A small hole was made over the air sac at the end of the egg, and a second hole was made directly over the embryonic CAM. After 10 days, 1 × 106 BEAS-2B wild-type (WT) or NRAS Q61K knock-in cells were mixed with 50 μL of serum-free DMEM plus 50 μL of Matrigel and dropped onto the CAM to form a plug. After 48 hours, CAMs were fixed with PBS, containing 3.7% paraformaldehyde, for 10 minutes at room temperature, and images were acquired with a Nikon digital color camera [9].
In vivo matrigel plug assay
A Matrigel plug assay was performed in BALB/c mice, as described previously, with some modifications. Matrigel (500 µL) containing HUVECs, BEAS-2B WT, or NRAS Q61K knock-in cells was inoculated subcutaneously into the right flanks of BALB/c mice. Each treatment group contained six mice. After 10 days, the Matrigel plugs were removed and hemoglobin content was determined according to Drabkin’s method [10].
Cell viability assay
Briefly, cells (3 × 104 cells per well) were seeded in 96-well plates, and exposed to various concentrations of kinase inhibitors for 24 hours. Cell viability was measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Three independent experiments, each in triplicate, were performed [11].
Co-immunoprecipitation assay
Cells were lysed in a culture dish by adding 0.5 mL of ice-cold RIPA lysis buffer. The supernatants were collected by centrifugation at 15,000 × g for 10 minutes at 4°C, incubated with IgG or NRAS in the presence or absence of trametinib (10 nM) at 4°C, followed by incubation with anti-NRAS antibody for 4 hours. Then, the supernatants were incubated with protein G-Sepharose (Santa Cruz) for 4 hours. After the removal of supernatant by brief centrifugation (6,000 × g), the protein G-Sepharose beads were washed three times with lysis buffer and then boiled for 5 minutes in loading buffer. Immunoprecipitates were further analyzed by western blotting with anti-B-Raf and anti-C-Raf antibodies.
Wound healing assay
We examined the migration of HUVECs by a wound healing assay. Briefly, cells were grown on 3.5-cm plates with M199. Once the cells reached confluence, we inflicted a uniform wound in each plate using a pipette tip, and washed the wounded layers with PBS to remove cell debris. Then, cells were cultured with supernatant from BEAS-2B WT or NRAS Q61K knock-in cells, which were cultured under serum starvation conditions. We evaluated the wound closure at 24 hours using bright-field microscopy [12].
Invasion assay
To determine the effect of NRASQ61K on HUVEC invasion in vitro, we performed cell invasion assays by using Matrigel-coated Boyden chamber inserts (8 μm; BD Biosciences). Cells were seeded in the upper chamber and allowed to invade the lower chamber containing 500 μL supernatant from BEAS-2B WT or NRASQ61K knock-in cells, which were cultured under serum starvation conditions. After incubation for 7 hours, noninvasive cells were removed with cotton swabs, and invasive cells were fixed with cold 4% paraformaldehyde and stained with 1% crystal violet. Images were acquired with an inverted microscope, and migrated cells in five random fields were quantified by manual counting. Three independent experiments were performed, each done in triplicates [13].
Endothelial cell capillary-like tube formation assay
HUVECs were pretreated with the supernatant from BEAS-2B WT or NRASQ61K knock-in cells for 1 hour and then seeded onto the Matrigel layer in 48-well plates at a density of 4 × 104 cells per well. After incubation for 8 hours, angiogenesis was assessed on the basis of formation of capillary-like structures. Tubes in randomly chosen microscopic fields were photographed [14]. Three independent experiments were performed.
ELISA
H1299 and H2087 cells (7 × 105) were plated in 6-well plates and treated for 24 hours with trametinib or vehicle. Supernatants were collected and VEGFA ELISA was performed with a Quantikine Immunoassay Kit (DVE00; R&D Systems) following the manufacturer’s instructions.
Real-time PCR
Total RNA was isolated by using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). The total RNA concentration was detected spectrophotometrically by measuring the absorbance at 260 nm (OD260). Reverse transcription (RT) was performed using Superscript III Reverse Transcriptase (Invitrogen), per the manufacturer’s protocol. Real-time PCR was performed on an ABI Prism 7500 Sequence detection system (Applied Biosystems, CA) with the KAPA SYBR® qPCR Kit (Kapa Biosystems, USA), according to the manufacturer’s instructions. The primer sequences used are shown in Supplementary Table 1. The target gene mRNA levels in the control cells were normalized to the β-actin mRNA levels, which were defined as ‘1’. Results were obtained from three independent experiments. For gene expression assay, the total RNA was extracted from cells by using the phenol-chloroform method (Trizma; Sigma-Aldrich). RNA samples were treated with DNase-free kit (Ambion; Applied Biosystems), and RNA levels were quantified with an RNA 6000 Nano Assay Kit in an Agilent 2100 Bioanalyzer (Agilent Technologies). Single-strand cDNA synthesis from total RNA was performed by using TaqMan reverse transcription reagents (Applied Biosystems), as recommended by the manufacturer. All quantitative real-time PCR reactions were performed with ABI PRISM 7900HT Fast Real-Time PCR system (Applied Biosystems). The expression of the housekeeping gene, TATA-binding protein (TBP), was used to normalize for variations in input cDNA. All measurements were performed in technical duplicates. Raw data were analyzed using SDS v2.2 (Applied Biosystems) to define relative quantity (RQ), and the gene expression data analysis suite (GEDAS; http://sourceforge.net/projects/gedas) was used for hierarchical clustering. The following TaqMan Gene Expression Assays (Applied Biosystems) were used for the analysis: CXCL-3-Hs00171061_m1, CD44-Hs01075862_m1, MMP-2-Hs01548727_m1, cMET-Dm01843535_g1, CD31-Hs00169777_m1, CCL-2-Hs00234140_m1, IL11-Hs01055413_g1, VEGFA-Hs00900055_m1, bRAF-Hs00269944_m1, cRAF-Hs00234119_m1, IL-6-Hs00985639_m1, MMP-14-Hs01037003_g1, CXCL-12-Hs03676656_mH, VEGF-C-Hs00153458_m1, DLL4-Hs01117333_m1, IL8-Hs00174103_m1, COX-2-Mm03294838_g1, PI3K-Hs01046353_m1, bFGF-Cf03460065_g1, ERK1/2-Hs00946872_m1, FGFR1-Hs00915142_m1, AKT-Hs01086102_m1, MMP-9-Hs00234579_m1, aFGF-Ss03374827_m1, PTEN-Hs02621230_s1, Src-Hs01082246_m1, TGFBR3-Hs00234257_m1, FGFR2-Hs01552926_m1, BCAR1-Hs01547079_m1, E-cadherin-Hs01023894_m1, Rap1-Sc04159738_s1, STAT3-Hs00374280_m1, Paxillin-Hs01104424_m1, TP53-Hs01034249_m1, FAK-Hs00164611_m1, Hif1a-Hs00153153_m1, GSK-3β-Ce02424746_m1, MAP3K7-Hs00177373_m1, EGFR-Hs01076078_m1, PDGFB-Hs00966522_m1, mTOR-Hs00234508_m1, PDGFRB-Hs01019589_m1, Gsk3a-Hs00997938_m1, TGFA-Hs00608187_m1, HDAC9-Hs00206843_m1, and CXCL2-Hs00601975_m1.
Western blot analysis
Whole-cell extracts were prepared by using lysis buffer supplemented with various protein inhibitors. Equal protein aliquots from each lysate were subjected to SDS-PAGE (8%) and blotted onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad). The blots were probed with antibodies specific for VEGFA (Santa Cruz, sc-507), phospho-ERKTyr204 (Santa Cruz, sc-7976), total-ERK (Santa Cruz, sc-514302), phospho-AKTSer473 (Santa Cruz, sc-24500), total-AKT (Santa Cruz, sc-377457), B-Raf (Santa Cruz, sc-55522), C-Raf (Santa Cruz, sc-52827), NRAS (Santa Cruz, sc-519), caspase-3 (Santa Cruz, sc-65497), CD31 (Abcam, ab28364), NRASQ61K (Wuhan New Biotechnology, 26193), and GAPDH (Santa Cruz, sc-36-5062). The protein levels were detected by chemiluminescence [15]. Protein concentration was determined by using Micro BCA Protein Assay Kit (Pierce Biotechnology).
Mouse xenografts
H1299 (5 × 106 cells per mouse) or H2087 (8 × 106 cells per mouse) were injected subcutaneously into the right posterior flanks of 7-week-old immunodeficient NOD/SCID female mice (six mice per group). On the seventh day, mice with the appropriate tumor size (100-200 mm3) were divided randomly into six treatment groups; the mice in each group were treated with vehicle or trametinib. The mice were administered trametinib or vehicle (30% PEG400, 0.5% Tween-80, and 5% propylene glycol) daily via intragastric injections. Tumor volume and mice body weight were measured every 3 days. Tumor volume was calculated as: volume (mm3) = 0.5 × length (mm) × width (mm)2. After sacrificing the mice on day 25, deparaffinized tumor sections were stained with specific antibodies, including anti-CD31 (Abbiotec) and anti-Ki-67 antibodies. Antigens were detected by using avidin-biotin-HRP complex (Thermo scientific) and 3,3’-diaminobenzidine as a chromogen [16]. Nuclei were counterstained with hematoxylin. All animal experiments were carried out in compliance with the guidelines of the Cangzhou Central Hospital of Hebei Province.
Statistical analysis
Data are expressed as mean ± standard error (SE). Statistical analysis was performed by using the t-test for independent means and Bonferroni’s post hoc test. Differences were considered statistically significant at P < 0.05.
Results
NRASQ61K mutant gene drives angiogenesis
To study the specific influence of NRASQ61K mutant on angiogenesis, we performed knock-in of the NRASQ61K construct in normal (non-neoplastic) human bronchial epithelial cells (BEAS-2B). We used the chicken CAM model and Matrigel plug assay to assess whether NRASQ61K could modulate angiogenesis. Compared to wild type (WT) cells, NRASQ61K overexpressing cells growing on CAM were characterized by a disorganized vasculature with several hemorrhagic areas. Quantification of chicken endothelium within the epithelial plugs indicated that NRASQ61K expression increased the recruitment of endothelial cells (Figure 1A). The effects of KRAS G12C mutation (KRASG12C), the most frequent KRAS mutation, on angiogenesis were also evaluated by using the CAM assay. When compared to WT cells, the KRASG12C-transfected cells growing on CAM exhibited negligible angiogenesis (Supplementary Figure 1). To confirm the proangiogenic effects of NRASQ61K in vivo, a Matrigel plug assay was performed in athymic nude mice. As shown in Figure 1B, NRASQ61K-expressing cells demonstrated a marked increase in angiogenesis in the plugs, as indicated by a bright red color. However, angiogenesis was negligible and hemoglobin levels were lower in the WT cells. Cells spontaneously form capillary-like structures on Matrigel, which is a critical step in tumor angiogenesis. Therefore, we investigated the effects of NRASQ61K expression on tubulogenesis. As shown in Figure 1C, NRASQ61K expression significantly increased the capillary-like network in vitro.
Figure 1.
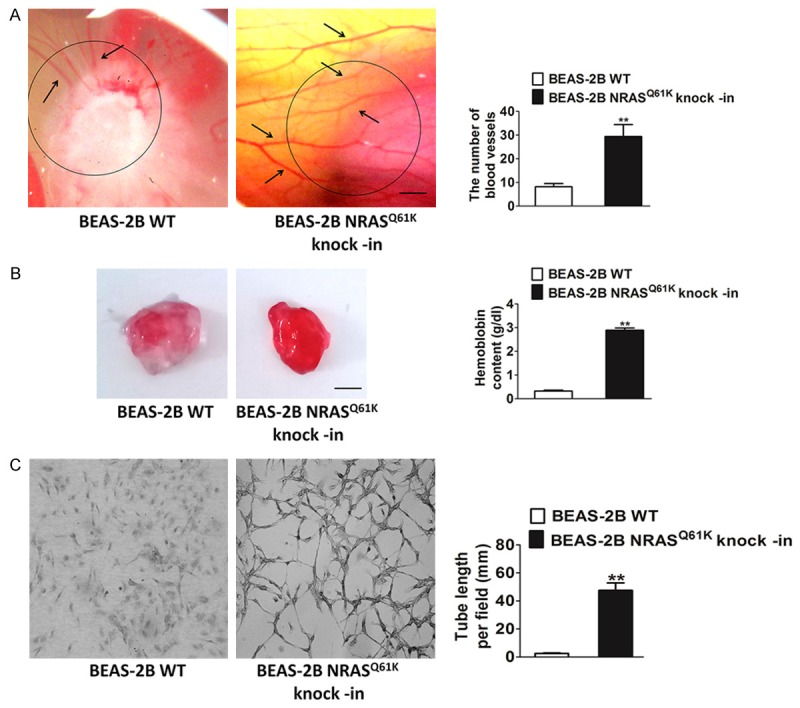
Knock-in of the NRASQ61K into BEAS-2B cells enhances angiogenesis. A. Representative images of BEAS-2B cells plated on the CAM. Scale bar: 1 cm. Qualitative assessment of angiogenesis in the CAM assay. Data are from three independent experiments and are mean ± SD. n = 6, **P < 0.01 compared with wild type cells. B. Representative images of BEAS-2B cells growth in the plugs. Quantitative analysis of hemoglobin levels in BEAS-2B plugs. Data are from three independent experiments and are mean ± SD. n = 6, **P < 0.01 compared with wild type cells. Scale bar: 0.5 cm. C. Photopictographs of a typical experiment showing the tube formation in wild type cells and NRASQ61K knock-in cells. NRASQ61K increased the capillary lengths of BEAS-2B cells (Scale bar represents 50 μm). Data are from three independent experiments and are mean ± SD. n = 3, **P < 0.01 compared with wild type cells.
NRASQ61K knock-in triggers up-regulation of angiogenic factors
We next compared the transcriptional profiles of genes involved in angiogenesis between NRASQ61K knock-in and parental BEAS-2B cells. This comprehensive approach revealed that oncogenic NRASQ61K enhanced the expression of several proangiogenic molecules, including VEGFA and acidic fibroblast growth factor (aFGF), bFGF, PDGF, transforming growth factor-β (TGF-β), and chemokines such as interleukin (IL)-6 and IL-8 (Figure 2A). The finding that oncogenic NRASQ61K triggered the up-regulation of factors known to modulate angiogenesis led us to hypothesize that the expression of NRASQ61K in epithelial cells might affect the endothelium. Among all the angiogenesis factors identified, we focused on VEGFA because it is a well-known regulator and an important prognostic marker of tumor angiogenesis. NRASQ61K knock-in cells released and expressed higher amounts of VEGFA (Figure 2B-D). Therefore, the supernatant from NRASQ61K knock-in cells enhanced HUVEC migration (Figure 2E) and invasion (Figure 2F) compared with the supernatant from control cells. The chemotactic effect of oncogenic NRASQ61K on the mobility of HUVECs was abrogated by an anti-VEGFA blocking antibody (Figure 2G, 2H). These observations support the pivotal role of VEGFA in NRASQ61K-driven angiogenesis.
Figure 2.

NRASQ61K knock-in increases VEGFA-mediated migration of HUVECs. A. NRASQ61K up-regulates the expression of pro-angiogenic factors in NRASQ61K knock-in BEAS-2B cells. Gene expression analysis was performed by real-time PCR comparing parental BEAS-2B with NRASQ61K clone. Differentially modulated genes were selected and analyzed by hierarchical clustering. B. Quantification of secreted VEGFA in BEAS-2B cells by ELISA. Data are from three independent experiments and are mean ± SD. n = 3, **P < 0.01 compared with wild type cells. C. Effect of NRASQ61K on VEGFA gene mRNA level. BEAS-2B cells were treated with NRASQ61K plasmid or vector, and VEGFA mRNA level were detected by real-time PCR. Data are from three independent experiments and are mean ± SD. n = 3, **P < 0.01 compared with wild type cells. D. Western blot shown that the expression of VEGFA was increased in cells transfected with NRASQ61K plasmid. Data were from three independent experiments. E. Migration of HUVECs was enhanced by the supernatant of BEAS-2B NRASQ61K knock-in cells compared with the wild type counterpart. Scale bar represents 50 μm. F. Invasion of HUVECs was enhanced by the supernatant of BEAS-2B NRASQ61K knock-in cells compared with the wild type BEAS-2B cells. Scale bar represents 50 μm. G and H. VEGFA blocking antibody (VEGFA-Ab) abolished NRASQ61K-induced cell migration and invasion. Data are from three independent experiments and are mean ± SD. n = 3, **P < 0.01 compared with wild type cells. ##P < 0.01 compared with NRASQ61K knock-in cells.
MEK inhibitor exerts cytostaticity in tumor cells harboring NRASQ61K
To identify potential therapies for patients with NRAS-mutant tumors, we tested the sensitivity of NRAS-mutant NSCLC cell lines to various kinase inhibitors by using in vitro cell growth inhibition assays (Figure 3A and Table 1). The lung cancer cell lines H1299 and H2087 were selected because these cells frequently harbor the NRASQ61K mutation. None of these cells were sensitive to the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), gefitinib; C-Raf inhibitor, regorafenib; c-MET inhibitor, crizotinib; B-Raf inhibitor, dabrafenib; or anaplastic lymphoma kinase (ALK) inhibitor, ceritinib. Interestingly, both lines were sensitive to the MEK inhibitor, trametinib. NSCLC cell line H1975 carrying WT NRAS was used as control and was found to be insensitive to MEK inhibition (Supplementary Figure 2). To further verify the dependency of MEK inhibition on NRASQ61K, we performed siRNA-mediated knockdown experiments (Supplementary Figure 3A). As expected, NRASQ61K knockdown inhibited the growth of the NRAS-mutant cell lines H1299 and H2087 (Supplementary Figure 3B); however, the inhibition of cell proliferation by the MEK inhibitor was abolished by NRASQ61K knockdown (Figure 3B). Consistent with these data, the MEK inhibitor trametinib inactivated ERK and AKT phosphorylation in the NRASQ61K mutated cells (Figure 3C). We then measured RAS-Raf association in the two NRASQ61K-mutant lung cancer cell lines, H1299 and H2087, by using co-immunoprecipitation. As predicted, treatment with trametinib decreased the association of B-Raf and C-Raf with NRAS (Figure 3D).
Figure 3.
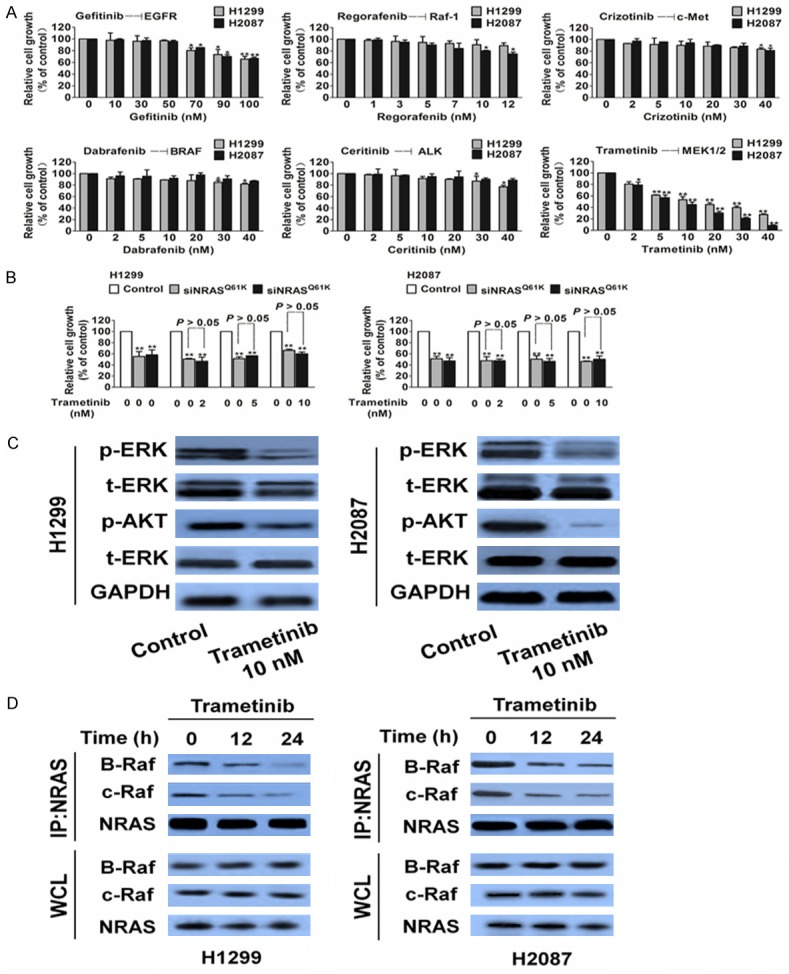
MEK inhibits proliferation and turns off ERK signaling in tumor cells carrying NRASQ61K. A. Proliferation of H1299 and H2087 cells were assessed with kinase inhibitors. Data are from three independent experiments and are mean ± SD. n = 3, **P < 0.01, compared to control. B. The proliferation inhibitory effects of trametinib on H1299 and H2087 cells were abolished by NRASQ61K siRNA. Data are from three independent experiments and are mean ± SD. n = 3, **P < 0.01 versus control. C. Biochemical analysis of phospho-ERK and phospho-AKT (Ser473 and Thr308) in H1299 and H2087. GAPDH was used as loading control. D. Co-immunoprecipitation (IP) of B-Raf and C-Raf with RAS from NRASQ61K cell lines after treatment with trametinib. Data were from three independent experiments. WCL, whole cell lysate.
Table 1.
In vitro profile of kinases inhibitors against H1299 and H2087 cells. The assays were performed in three independent experiments
| Kinases inhibitor | Cell lines | IC50 (nM) |
|---|---|---|
| Gefitinib | H1299 | > 100 ± 9.7 |
| H2087 | > 100 ± 3.2 | |
| Regorafenib | H1299 | 31 ± 0.2 |
| H2087 | 24 ± 4.0 | |
| Crizotinib | H1299 | 59 ± 1.7 |
| H2087 | 53 ± 2.2 | |
| Dabrafenib | H1299 | 67 ± 3.2 |
| H2087 | 59 ± 4.5 | |
| Ceritinib | H1299 | 52 ± 3.1 |
| H2087 | 60 ± 6.0 | |
| Trametinib | H1299 | 12 ± 1.4 |
| H2087 | 8 ± 2.6 |
Trametinib exerts growth inhibition and anti-angiogenic activities in tumors harboring NRASQ61K
We next examined the effect of the MEK inhibitor trametinib in immunocompromised mice that were subcutaneously injected with H1299 and H2087 cells. Trametinib induced a prolonged cytostatic effect and the tumors volumes showed evident shrinkage at the end of the experiments in both xenograft models (Figure 4A). There was no significant difference in body weight between the trametinib- and vehicle-treated groups (Figure 4B). This was further supported by our findings from cell proliferation and apoptosis assays by using Ki-67 staining (Figure 4C) and caspase-3 inactivation (Figure 4D), respectively. We found that trametinib markedly inhibited tumor cell proliferation; whereas it was ineffective in inducing apoptosis (Figure 4D). Similar to the in vitro observations, trametinib treatment of H1299 and H2087 tumors decreased the phosphorylation of ERK and AKT (Figure 4D). To further examine whether trametinib suppressed angiogenesis, tumor tissues were stained with a CD31-specific antibody. CD31 is a widely used endothelial marker for quantifying angiogenesis by calculating microvessel density (MVD). Tumor sections stained with anti-CD31 antibody revealed that trametinib inhibited MVD (Figure 4E).
Figure 4.
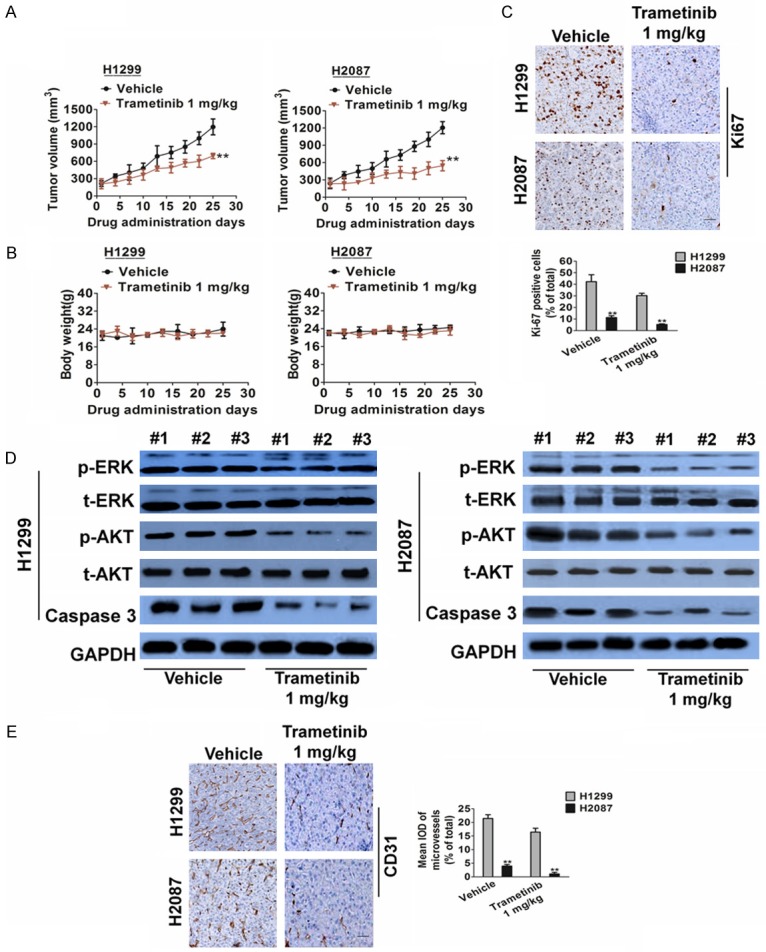
Trametinib treatment inhibits angiogenesis in H1299 and H2087 xenograft models. A. Representative images of H1299 and H2087 tumors. Tumor growth curve of H1299 and H2087 xenografts. H1299 and H2087 xenografts were treated with trametinib 1 mg/kg or vehicle for 25 days after tumor volume reached an average of 100 to 200 mm3. Data are presented as means ± SD, n = 6, **P < 0.01 versus vehicle group. B. Body weight changes in trametinib and vehicle treated mice. Data are presented as means ± SD, n = 6. C. Representative images of Ki-67 staining in H1299 and H2087 xenografts. Scale bar represents 50 μm. Quantification of proliferating cells by Ki-67 staining in H1299 and H2087 xenografts. Data are presented as means ± SD, n = 6, **P < 0.01 versus vehicle group. D. Biochemical analysis of ERK, AKT and Caspase in protein extract of H1299 and H2087 xenografts. Protein loading was normalized by GAPDH. Three independent samples were evaluated. E. Tumor tissues were prepared for immunohistochemistry detection with antibody against CD31 (Scale bar represents 50 μm). Data are presented as means ± SD, n = 6, **P < 0.01 versus vehicle group.
Western blot analysis further verified our findings. The trametinib treatment group had lower levels of CD31 and VEGFA expression than the control group (Figure 5A). We also found that trametinib decreased C-Raf and B-Raf levels by targeting the NRAS signaling pathway in NSCLC cells. Therefore, we sought to identify any changes in NRAS signaling upon treatment with the drug. A similar trend was also seen in vitro. In vivo, the levels of C-Raf, B-Raf, and NRAS were significantly lower in the trametinib group than in the control (Figure 5B).
Figure 5.
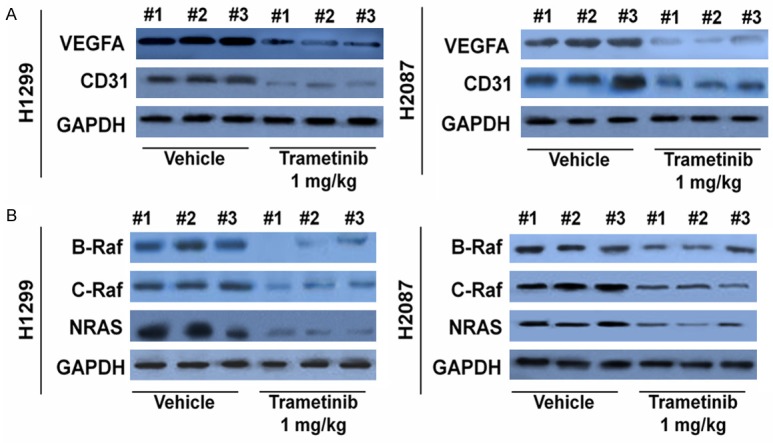
Trametinib treatment led to decreased VEGFA levels and NRAS signaling in vivo. A. VEGFA and CD31 levels from tumor samples were analyzed through Western blot analysis. Lower CD31 and VEGFA levels were measured in the trametinib treatment group. B. Similarly, through Western blot, trametinib treatment had the most profound decrease in CRAF, BRAF and NRAS protein levels in tumor samples.
Secretion of proangiogenic factors in cancer cells harboring NRASQ61K
We further assessed whether the anti-angiogenic effect of trametinib was direct (on the tumor vasculature) or indirect (via epithelial cells) by in vitro experiments with HUVECs. We found that HUVECs proliferation (Figure 6A), migration (Figure 6B), and invasion (Figure 6C) were unaffected by drug treatment, ruling out a possible direct effect of trametinib inhibition on the endothelial compartment. Next, in vivo Matrigel plug angiogenesis assays were performed to test the effects of trametinib treatment on angiogenesis under normal conditions. The hemoglobin levels were comparable in Matrigels that contained trametinib or vehicle (Figure 6D). Western blot analysis of CD31 and VEGFA levels in plugs tissues further verified that the anti-angiogenic effects of trametinib were less pronounced under normal conditions (Figure 6E).
Figure 6.
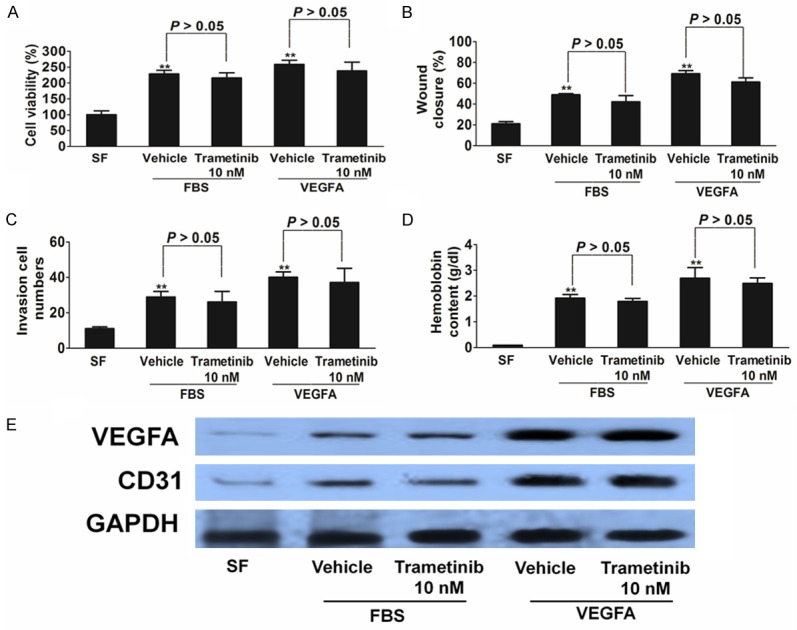
Trametinib does not affect HUVECs proliferation and mobility. A-C. HUVECs were used to evaluate proliferation, migration, and invasion. HUVECs were stimulated by FBS or 50 ng/mL VEGFA and were treated with trametinib or vehicle. Cells proliferation is shown as mean percentage of cells viability compared with serum-Free untreated samples (SF) ± SD in quadruplicate. HUVECs migration is represented as mean number of migrated cells ± SD in triplicate. In both cases, representative results of three independent experiments are shown. D. Quantitative analysis of hemoglobin levels in HUVECs plugs. Data are from three independent experiments and are mean ± SD. n = 6, **P < 0.01 compared with wild type cells. E. VEGFA and CD31 levels from plugs tissues were analyzed through Western blot analysis.
We next examined whether trametinib modulated the production of angiogenic factors by cancer cells, which in turn might have influenced tumor angiogenesis. For this, we analyzed the effects of trametinib on the expression of angiogenic factors in tumor cells harboring mutant NRASQ61K. We found that several angiogenic mediators (e.g., IL-6, IL-8, and VEGFA) were down-regulated in both H1299 and H2087 cells upon trametinib treatment (Figure 7A-C). On the contrary, the expression of these genes did not change significantly in H1975 cells carrying WT NRAS (Supplementary Figure 4). Taken together, these results indicate that trametinib modifies the proangiogenic effects of NRASQ61K. To further verify the dependency of trametinib inhibition on the presence of NRASQ61K, we performed siRNA-mediated knockdown experiments (Supplementary Figure 3). As expected, NRASQ61K knockdown led to the inhibition of VEGFA expression in tumor cells (Figure 7D). Moreover, trametinib suppressed VEGFA in knockdown cells, which was consistent with the NRASQ61K knock-down group, thus suggesting that trametinib-mediated inhibition of tumor angiogenesis is partly dependent on NRASQ61K (Figure 7D).
Figure 7.
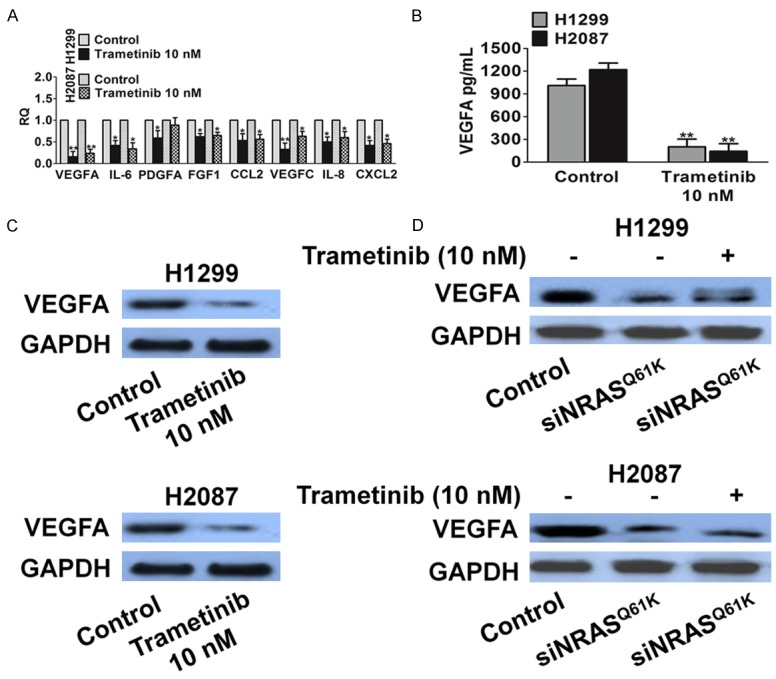
Trametinib treatment down-regulates the expression of proangiogenic factors. A. Expression of proangiogenic factors was evaluated by real-time PCR. H1299 and H2087 cells were treated for 24 h with trametinib. Data are expressed as relative quantity (RQ) of trametinib compared with vehicle-treated samples. Bars show mean ± SD of triplicate measurements. B. Quantification of secreted VEGFA in cells supernatant by ELISA after 24 h of the indicated treatments. Data are expressed in pg/mL and bars represent mean ± SD of duplicate observations. C. Biochemical analysis of VEGFA in H1299 and H2087 cells. Protein loading was normalized by GAPDH. D. VEGFA was measured in H1299 and H2087 cell after trametinib treated in the presence of siNRASQ61K. GAPDH expression was used as a loading control.
Discussion
NSCLC is one of the most lethal types of cancer and is associated with significant mortality and morbidity worldwide. Despite improvements in conventional treatments including surgery and chemoradiotherapy for NSCLC, overall survival remains poor [17]. These findings suggest that improvements in NSCLC treatment and patient outcome are urgently needed. Angiogenesis, the formation of blood vessels within a solid tumor, is not only essential for primary tumor growth but also vital for tumor invasion and metastasis. Recent advances have been made in targeting molecularly defined subsets of NSCLC that depend on specific molecular alterations for tumor angiogenesis [18]. These findings are helping clinicians to match patients with appropriate drugs, a process often referred to as personalized medicine [19]. Tumor angiogenesis is pivotal for NSCLC growth, invasion, and metastasis, as it ensures the supply of oxygen and nutrients to cancer cells and other cells constituting the tumor. Therapies aimed at inhibiting angiogenesis are promising treatments for NSCLC.
VEGFs play central roles in tumor angiogenesis via the activation of VEGF receptor tyrosine kinases (RTKs), including VEGFR1 and VEGFR2, on the surface of endothelial cells. Most clinical and basic research on angiogenesis and anti-angiogenic treatment has focused on RTKs [20]. Unfortunately, the clinical benefit conferred by these therapies is variable and other angiogenic factors switch on during cancer progression, which facilitates tumor initiation and induces resistance to RTK inhibitors. Among these, the EGFR tyrosine kinase family and related downstream pathways play a critical role in cancer development, and over recent years, they have been validated as targets for NSCLC treatment [21]. The development of first-generation TKIs targeting EGFR has had a considerable impact on patient outcomes. However, despite dramatic and sustained responses and the discovery of specific patient subgroups that may derive clinical benefit, resistance to EGFR TKIs inevitably develops. For example, EGFR kinase inhibitors are mainly active in cancer cells carrying EGFR mutations; likewise, the occurrence of other potential driver mutations such as NRAS mutations impairs the efficacy of EGFR TKIs in colorectal cancer. RAS family proteins (including KRAS and NRAS) play important roles in the EGFR signaling pathway. Downstream signaling of EGFR activates the RAS and Raf genes that are members of this pathway and can harbor oncogenic mutations in 30-60% (KRAS) and 1-3% (NRAS) of cases, respectively [22]. NRAS is a GTPase related to KRAS, originally identified in neuroblastoma cell lines as the third RAS family member, following KRAS and HRAS. It has been shown that activating mutations in KRAS or NRAS lead to consecutive activation of the RAS-Raf pathway downstream of EGFR, and consequently result in resistance to anti-EGFR therapy. The observation that subtle molecular changes in KRAS oncogene in tumor cells can have major impact on the tumor vasculature provides new insights into the role of KRAS mutations in angiogenesis. However, the mechanism by which the activation of NRAS oncogene modulates the angiogenic progression is largely unknown.
In the present work, we explored the possibility that the mutations in NRAS oncogene can affect the angiogenic potential of cancer cells. We utilized a normal epithelial cell model, in which the NRAS mutation was introduced, in order to recapitulate the situation in neoplastic lesions. We report that NRASQ61K mutant epithelial cells exhibited upregulation of proangiogenic factors, and that NRASQ61K knock-in contributed to endothelial cell migration and invasion. In tumor cells, blockage of NRAS with a specific kinase inhibitor, trametinib, not only exerted a cytostatic activity, but also influenced angiogenesis in vivo. Treatment with the MEK inhibitor trametinib strongly affected angiogenesis in H1299 and H2087 cell lines. Specifically, histological analysis of CD31-stained tumor sections revealed that the trametinib-treated group exhibited significantly lower MVD than the vehicle-treated group. In addition to exerting angiogenesis inhibitory activities on NRASQ61K-mutant cancer cells, trametinib affected tumor growth by attenuating the ERK signaling in tumor tissues. Notably, trametinib treatment did not affect endothelial cells directly, but downregulated the expression of angiogenic factors in tumor cells by switching off the ERK pathway. Therefore, our findings suggest that pharmacological inhibition of a mutant oncogenic protein could downregulate proangiogenic mediators in targeted tumor cells, leading to abrogation of tumor angiogenesis.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kubota Y. Tumor angiogenesis and anti-angiogenic therapy. Keio J Med. 2012;61:47–56. doi: 10.2302/kjm.61.47. [DOI] [PubMed] [Google Scholar]
- 2.Pircher A, Hilbe W, Heidegger I, Drevs J, Tichelli A, Medinger M. Biomarkers in tumor angiogenesis and anti-angiogenic therapy. Int J Mol Sci. 2011;12:7077–7099. doi: 10.3390/ijms12107077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sato Y. Molecular diagnosis of tumor angiogenesis and anti-angiogenic cancer therapy. Int J Clin Oncol. 2003;8:200–206. doi: 10.1007/s10147-003-0342-8. [DOI] [PubMed] [Google Scholar]
- 4.Sawada J, Li F, Komatsu M. R-Ras inhibits VEGF-induced p38MAPK activation and HSP27 phosphorylation in endothelial cells. J Vasc Res. 2015;52:347–359. doi: 10.1159/000444526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawada J, Li F, Komatsu M. R-Ras protein inhibits autophosphorylation of vascular endothelial growth factor receptor 2 in endothelial cells and suppresses receptor activation in tumor vasculature. J Biol Chem. 2015;290:8133–8145. doi: 10.1074/jbc.M114.591511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat Rev. 2017;53:61–69. doi: 10.1016/j.ctrv.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Kinsler VA, O’Hare P, Jacques T, Hargrave D, Slater O. MEK inhibition appears to improve symptom control in primary NRAS-driven CNS melanoma in children. Br J Cancer. 2017;116:990–993. doi: 10.1038/bjc.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khosravi-Far R, White MA, Westwick JK, Solski PA, Chrzanowska-Wodnicka M, Van Aelst L, Wigler MH, Der CJ. Oncogenic Ras activation of Raf/mitogen-activated protein kinaseindependent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923–3933. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandracchia D, Tripodo G, Trapani A, Ruggieri S, Annese T, Chlapanidas T, Trapani G, Ribatti D. Inulin based micelles loaded with curcumin or celecoxib with effective anti-angiogenic activity. Eur J Pharm Sci. 2016;93:141–146. doi: 10.1016/j.ejps.2016.08.027. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Hou M, Lu G, Ciccone N, Wang X, Zhang H. The prognosis of anti-angiogenesis treatments combined with standard therapy for newly diagnosed glioblastoma: a metaanalysis of randomized controlled trials. PLoS One. 2016;11:e0168264. doi: 10.1371/journal.pone.0168264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Wang L, Liu Z, Zu C, Xing F, Yang P, Yang Y, Dang X, Wang K. MicroRNA-494 inhibits cell proliferation and invasion of chondrosarcoma cells in vivo and in vitro by directly targeting SOX9. Oncotarget. 2015;6:26216–26229. doi: 10.18632/oncotarget.4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian J, Hachim MY, Hachim IY, Dai M, Lo C, Raffa FA, Ali S, Lebrun JJ. Cyclooxygenase-2 regulates TGFbeta-induced cancer stemness in triple-negative breast cancer. Sci Rep. 2017;7:40258. doi: 10.1038/srep40258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang HL, Jiang Y, Wang YH, Chen T, He HJ, Liu T, Yang T, Yang LW, Chen J, Song ZQ, Yao W, Wu B, Liu G. FBXO31 promotes cell proliferation, metastasis and invasion in lung cancer. Am J Cancer Res. 2015;5:1814–1822. [PMC free article] [PubMed] [Google Scholar]
- 14.Cantarella G, Risuglia N, Dell’eva R, Lempereur L, Albini A, Pennisi G, Scoto GM, Noonan DN, Bernardini R. TRAIL inhibits angiogenesis stimulated by VEGF expression in human glioblastoma cells. Br J Cancer. 2006;94:1428–1435. doi: 10.1038/sj.bjc.6603092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Zhu L, Fang J, Ge Z, Li X. LRG1 modulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via HIF-1alpha activation. J Exp Clin Cancer Res. 2016;35:29. doi: 10.1186/s13046-016-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Yao J, Sun H, He K, Tong D, Song T, Huang C. MicroRNA-101 suppresses progression of lung cancer through the PTEN/AKT signaling pathway by targeting DNA methyltransferase 3A. Oncol Lett. 2017;13:329–338. doi: 10.3892/ol.2016.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, Grosveld F, Philipsen S. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K, Zhang M, Zhu J, Hong W. Screening of gene mutations associated with bone metastasis in nonsmall cell lung cancer. J Cancer Res Ther. 2016;12:C186–C190. doi: 10.4103/0973-1482.200597. [DOI] [PubMed] [Google Scholar]
- 19.Zang J, Hu Y, Xu X, Ni J, Yan D, Liu S, He J, Xue J, Wu J, Feng J. Elevated serum levels of vascular endothelial growth factor predict a poor prognosis of platinum-based chemotherapy in non-small cell lung cancer. Onco Targets Ther. 2017;10:409–415. doi: 10.2147/OTT.S124124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao F, Vasquez SX, Su F, Roberts S, Shah N, Grijalva V, Imaizumi S, Chattopadhyay A, Ganapathy E, Meriwether D, Johnston B, Anantharamaiah GM, Navab M, Fogelman AM, Reddy ST, Farias-Eisner R. L-5F, an apolipoprotein A-I mimetic, inhibits tumor angiogenesis by suppressing VEGF/basic FGF signaling pathways. Integr Biol (Camb) 2011;3:479–489. doi: 10.1039/c0ib00147c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tate CM, Mc Entire J, Pallini R, Vakana E, Wyss L, Blosser W, Ricci-Vitiani L, D’Alessandris QG, Morgante L, Giannetti S, Larocca LM, Todaro M, Benfante A, Colorito ML, Stassi G, De Maria R, Rowlinson S, Stancato L. A BMP7 variant inhibits tumor angiogenesis in vitro and in vivo through direct modulation of endothelial cell biology. PLoS One. 2015;10:e0125697. doi: 10.1371/journal.pone.0125697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramani A, Alsidawi S, Jagannathan S, Sumita K, Sasaki AT, Aronow B, Warnick RE, Lawler S, Driscoll JJ. The brain microenvironment negatively regulates miRNA-768-3p to promote K-ras expression and lung cancer metastasis. Sci Rep. 2013;3:2392. doi: 10.1038/srep02392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.