

Abstract
Patients with Neurofibromatosis type 1 (NF1) and Neurofibromatosis type 2 (NF2) are predisposed to tumors of the nervous system. NF1 patients predominantly develop neurofibromas, and Malignant Peripheral Nerve Sheath Tumors (MPNST) while NF2 patients develop schwannomas and meningiomas. Here we quantified the drug sensitivities of NF1 and NF2 tumor cell lines in a high throughput platform. The platform contained a comprehensive collection of inhibitors of MEK, RAF, RAS, farnesyl transferase, PAK and ERK, representative drugs against many other cancer pathways including Wnt, Hedgehog, p53, EGF, HDAC, as well as classical cytotoxic agents recommended for treating MPNST, such as doxorubicin and etoposide. We profiled seven NF1-associated MPNST cell lines (ST88-14, ST88-3, 90-8, sNF02.2, T265, S462TY, SNF96.2), one sporadic MPNST cell line (STS26), one schwannoma from a NF2 patient (HEI193), one NF2-deficient malignant meningioma (KT21-MG-Luc5D), one mouse NF2 schwannoma (SC4) and one sporadic rat schwannoma (RT4-67 or RT4). NF1 cells were primarily distinguished from NF2 cells and the sporadic MPNST cell line by their sensitivity to MEK and ERK inhibitors, and to a smaller extent their sensitivity to BH3 mimetics and farnesyl transferase inhibitors. The platform was highly successful in predicting the effects of clinical trials for Neurofibromas.
Keywords: HTS, high throughput screen, signal transduction, von Recklinghausen disease, heat map, Spearman’s rank correlation coefficient, paradoxical activation
Introduction
Neurofibromatosis type 1 and neurofibromatosis type 2 are genetic disorders characterized by tumors of the nervous system. NF1 patients predominantly develop neurofibromas and Malignant Peripheral Nerve Sheath Tumors (MPNST) while NF2 patients develop schwannomas and meningiomas. While most of these tumors are benign, they cannot always be surgically resected and can sometimes become malignant.
From 30 to 50% of individuals with NF1 develop benign peripheral nerve sheath tumors, called plexiform neurofibromas (PNFs), which may transform to MPNST [1]. MPNST are a relatively rare sarcoma that can occur sporadically, but about half occur in patients with Neurofibromatosis type 1 (NF1). The overall incidence of MPNST in the general population is 1/100,000 but the lifetime risk of MPNST for NF1 patients is 8-13% [2,3]. MPNST are aggressive high grade sarcomas, with a high probability of local recurrence and distant metastasis [3]. MPNST patients have a 5-year survival rate of just 35%-50%, even with aggressive surgery and chemotherapy. In general, standard sarcoma treatment regimens are adapted for chemotherapy for MPNST, including surgical excision with radiation and chemotherapy with agents such as doxorubicin and ifosfamide, although MPNST do not respond well to cytotoxic chemotherapy [4]. To date, there have been no controlled trials for MPNST chemotherapy, so the effectiveness of chemotherapeutic agents for MPNST have been difficult to evaluate [5].
The predominant risk factor for MPNST is a diagnosis of NF1. NF1 is a dominantly inherited, tumor prone autosomal disorder with a worldwide incidence of 1 in 2500 [6]. Patients with NF1 have a life expectancy that is reduced by 10-15 years [2]. NF1 is caused by a mutation in the tumor suppressor NF1, which is located at 17q11.2. NF1 patients are born with a loss-of-function mutation in NF1. When Schwann cells acquire sporadic mutations in the other chromosomal copy, they initiate a benign tumor called a neurofibroma. If neurofibromas accumulate additional mutations in other genes including p53, CDKN2A, EED or SUZ12 the tumor can transform to a MPNST [7,8]. The NF1 gene product is a Ras-GAP called neurofibromin that functions as a negative regulator of Ras. As a consequence of neurofibromin loss, NF1-tumors have elevated levels of GTP-bound Ras and most research efforts to identify drugs have focused on Ras and its signaling pathways.
The predominant risk for schwannomas is neurofibromatosis type 2, a genetic disorder caused by mutations in NF2. Multiple schwannomas are a diagnostic criterion for NF2 and most sporadic schwannomas also have mutations in NF2. NF2 is located on a different chromosome than NF1, chromosome 22, and encodes a cytoskeletal protein that is a member of the ERM family of cytoskeletal proteins, called merlin. Merlin inactivates Pak kinases through direct interaction [9-11]. Merlin also interacts with Ex and KIBRA to activate Hippo signaling [12,13].
There have been several large scale projects to characterize the drug sensitivities of cancer cell lines. The first was the National Cancer Institute 60 cell line panel [14], while more recently surveys were initiated at GlaxoSmithKline [15], the Broad Institute and the Sanger Institute. Three of these groups periodically release data about compounds and cells. The Broad Institute created The Cancer Cell Line Encyclopedia (CCLE), and through The Cancer Therapeutics Response Portal currently lists 860 cell lines tested with 481 anticancer drugs [16] while the Sanger Institute’s Genomics of Drug Sensitivity in Cancer project (GDSC) lists 1074 cancer cell lines tested with 265 drugs [17], and GlaxoSmithKline project tested 311 cell lines. However, none of these profiling projects systematically profiled the cell lines commonly used in MPNST studies and many drugs under investigation for MPNST are not in their drug panels. One recent study analyzed two MPNST cell lines, only one of which was from and NF1 patient and none were from any NF2 patients [18].
Here, we surveyed the most commonly studied MPNST cell lines against a panel of 130 highly relevant drugs to NF1 and NF2. Most drugs in clinical use or in clinical trials for NF1 and NF2 were on the panel. Our drug panel included a comprehensive set of small molecule inhibitors of RAS signaling, PAK kinase inhibitors, representative probes against many other cancer pathways and classical cytotoxic agents such as doxorubicin and ifosfamide. We tested the drugs in a 384 well high throughput format at eight concentrations against seven NF1-associated MPNST cell lines (ST88-14, ST88-3, 90-8, sNF02.2, T265, S462TY, SNF96.2), one sporadic MPNST cell line (STS26), one schwannoma from a NF2 patient (HEI193), one NF2-deficient malignant meningioma (KT21-MG-Luc5D), one mouse NF2 schwannoma (SC4) and one sporadic rat schwannoma (RT4-67 or RT4). We found that some drugs clearly distinguished NF1 cells from NF2 cells and the sporadic tumor MPNST cell line derived from a patient that did not have NF1.
Materials and methods
Cell culture
Human NF1-derived MPNST cell lines ST88-14, T265, 90-8, and the sporadic human MPNST cell line STS26T were kindly provided by Dr. Nancy Ratner (University of Cincinnati, Cincinnati, OH, USA). ST88-3, S462TY, sNF02.2, sNF96.2 and KT21-MG1-Luc5D were provided by Dr. Jonathan Chernoff (Fox Chase Cancer Center, Philadelphia, PA, USA). The human schwannoma cell line, HEI193, which was immortalized from a NF2 patient [19], and SC4 cells, from NF2-/- mice, were generously provided by Dr. Marco Giovannini (University of California, Los Angeles, CA). The rat schwannoma, RT4-67 (RT4), was from Dr. Joseph Kissil (The Scripps Research Insitute, Jupiter Florida). ST88-14, T265, STS26T, ST88-3, S462TY, sNF96.2, sNF94.3, HEI193, KT21-MG1-Luc5D, SC4 and RT4 cell lines were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Life Technologies, NY, USA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, MO, USA) and 1% Penicillin Streptomycin (P/S; Gibco, Life Technologies, NY, USA). The 90-8 cell line was grown in RPMI 1640 (Corning, VA, USA) containing 10% FBS and 1% P/S. Cell lines were confirmed Mycoplasma negative and cryobanked.
Drugs and compound management
Drugs were purchased from Selleckchem (Houston, TX, USA) as stock solutions at a concentration of 10 mM in DMSO. A complete list of the drugs can be found in Table S1. Forty-four drugs were arrayed on 384 well plates and an 8-pt serial dilution of each drug was prepared in 100% DMSO using the Janus Automated workstation and serial dilution tool (SDT). Column one of each compound plate contained 100% DMSO as a solvent control. Column two contained 5 mM doxorubicin as a positive control for cytotoxicity. Multiple daughter plates for each source plate were prepared containing 5 µl of compound and stored at -40°C. Each daughter plate was thawed a maximum of 10 times.
High throughput profiling
Figure 1 shows an overview of the screening workflow. 2000 cells were plated in a volume of 25 μl per well of 384-well microplates (Corning 3707) using a MultidropTM Combi Reagent Dispenser (Thermo Scientific) and allowed to attach overnight at 37°C, 5% CO2 in a humidified chamber (Figure 1A). Drugs (50 nL) were transferred to assay plates using a 384 W, 50 nL slotted pin tool (V&P Scientific) and a JANUS Automated Workstation (Perkin Elmer) (Figure 1B), resulting in final concentrations of 4.6 nM, 14 nM, 41 nM, 123 nM, 370 nM, 1.11 μM, 3.33 μM, and 10 µM of each test compound. Plates were incubated for 72 hours at 37°C, 5% CO2. Cell viability was measured using the ATPlite Luminescence Assay (PerkinElmer). Assay plates were removed from the incubator for 1 hour to equilibrate to room temperature prior to adding 25 μL of ATPlite. Luminescence was measured on an EnVision Xcite Multilable Plate Reader (PerkinElmer), using the ultrasensitive luminescence measurement technology (Figure 1C).
Figure 1.
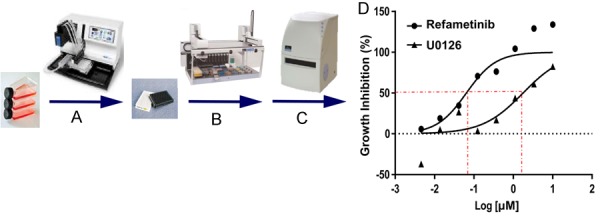
High Throughput Screening (HTS) workflow. A. Cells were plated in 384 well assay; B. 50 nL of compounds were transferred into each well via the JANUS Automated Workstation (Perkin Elmer) and then incubated for 72 hrs; C. Luminescence was measured by ATPlite Luminescence Assay; D. Raw data values were normalized and log-transformed to produce a dose-response curve. IC50s were determined to be the concentration of drug tested that resulted in 50% response.
Data analysis
The raw values of the DMSO and doxorubicin treated wells on each assay plate were aggregated and used to calculate z’-factors, as a measure of assay performance and data quality, with a z’-factor > 0.5 representing acceptable data. Raw data values of test wells were normalized to aggregate DMSO and doxorubicin plate control wells and expressed as normalized percent inhibition (NPI = ((DMSOavg-Test well)/(DMSOavg-Doxorubicinavg) × 100)). A non-linear fit with variable slope (GraphPad Prism 7) of Normalized Percent Inhibition and log10 concentration values was used to define IC50 values for each drug cell line combination (Figure 1D). Heat maps were also generated in GraphPad Prism.
Results and discussion
A database of drug sensitivity of MPNST cells
We profiled seven NF1-associated MPNST cell lines (ST88-14, ST88-3, 90-8, sNF02.2, T265, S462TY, SNF96.2), one sporadic MPNST cell line (STS26), one schwannoma from a NF2 patient (HEI193), one NF2-deficient malignant meningioma (KT21-MG-Luc5D), one mouse NF2 schwannoma (SC4) and one sporadic rat schwannoma (RT4). These are among the most common cell lines used in NF research. Several other cell lines were also tested, including sNF94.3, and human primary Schwann cells, but they did not grow well enough to yield adequate z-factors, as a result, the data were excluded.
Since about half of MPNST occur in patients with NF1, the drugs in our panel were biased towards the Ras pathway (Table S1). The two most druggable Ras pathways are the RAF/MEK/ERK pathway and the PI3K/AKT/mTOR pathway. Our panel included a comprehensive set of inhibitors against RAF (13 drugs), MEK1/MEK2 (16 drugs), ERK (5 drugs), and multiple drugs against PI3K, AKT and mTOR. Ras itself was targeted with farnesyl transferase inhibitors, deltarasin and salirasib, which prevent Ras processing and erastin, which induces ferroptosis and shows some preference for Ras tumors. We also tested representative drugs against many other cancer targets including EGFR, Wnt, HDAC, bromodomains and classical chemotherapy agents such as taxols. Many drugs in our panel are FDA approved for other cancers, while two, doxorubicin and ifosfamide, are recommended for MPNST.
Experiments were repeated at least twice for all cells except S462TY. We used non-linear curve fitting to calculate IC50s as a quantitative measure of cell line responses. Since the IC50 values of some compounds were extrapolated outside the concentration range we tested, we assigned IC50 values to them to indicate the limitations. In assigning values, inactive or weakly inhibitory compounds, i.e. failed to reach 100% inhibition, were assigned an IC50 of 25 μM (eg. IC50 ≥ 25 μM). In contrast, highly active compounds where the concentration series failed to titrate away the drugs activity were assigned an IC50 of 0.001 μM (eg. IC50 ≤ 0.001 μM) (Figure 2). Several potent compounds did not always cause 100% loss of viability at the highest dose, likely due to cytostatic effects rather than cytotoxic effects under our assay conditions. This was most common with rapamycin, Docetaxel and Paclitaxel. In the heat maps, the most active compounds are blue, while compounds in red are less active. Drugs were sorted into groups with similar targets.
Figure 2.

Drug sensitivity of NF1 and NF2 cell lines. Heatmap of drug IC50s in seven NF1-associated MPNST cell lines (ST88-14, ST88-3, 90-8, sNF02.2, T265, S462TY, SNF96.2), one sporadic MPNST cell line (STS26), one schwannoma from a NF2 patient (HEI193), one NF2-deficient malignant meningioma (KT21-MG-Luc5D), one mouse NF2 schwannoma (SC4) and one sporadic rat schwannoma (RT4). The IC50s were determined from eight drug concentrations and expressed as a heatmap. Drugs are segregated by drug target. Experiments were repeated at least twice for all cells except S462TY. The most active compounds are blue, while compounds in red are less active. Inactive compounds were assigned an IC50 of 25 μM (eg. ≥ 25 μM), and highly active compounds were assigned an IC50 of 0.001 μM (eg. IC50 ≤ 0.001 μM).
We used Spearman’s rank correlation coefficients (Rs) of the IC50s to compare how responses differed between cell lines (Figure 3). A heat map of the Spearman’s is shown in Figure 3A. The mean for replicates of a cell line was 0.85. This was independent of the assay system used as comparing ATPlite, resazurin and cell counting (after DAPI staining) yielded Spearman’s rank correlations of 0.83 to 0.88 (data not shown). The NF1 cell lines compared to each other yielded a mean Rs of 0.82. The NF1 cell lines compared to NF2 cell lines yielded a Rs of 0.73 and the NF2 vs. NF2 is 0.69, while the NF1 vs. sporadic is 0.67 and the NF2 vs. sporadic is 0.73 (Figure 3B). Thus, NF1 cell lines compared to each other are almost as similar as biological replicates, but differ significantly from the NF2 and sporadic cell lines. NF2 cell lines may be more heterogeneous than NF1 cell lines as a group.
Figure 3.
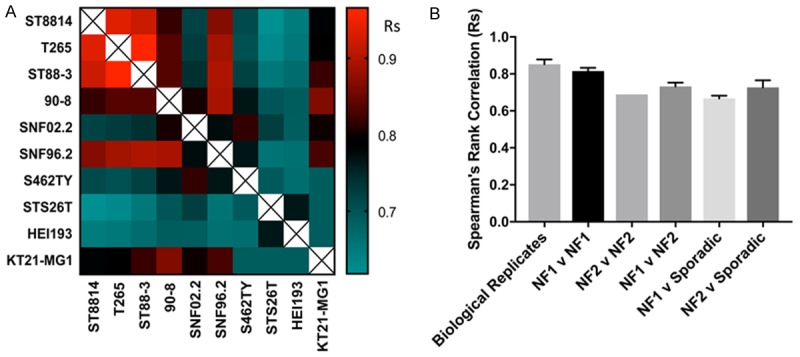
Concordance of Drug Sensitivity amongst cell lines and replicates. A. Heat map of Spearman’s rank correlation coefficients between IC50 values for indicated conditions. Warmer colors (red) represent higher correlation coefficients, while cooler colors (blue) represent lower correlation coefficients. B. Selected comparisons of Spearman’s rank correlation coefficients shown in part A. In this analysis, all Spearman’s rank correlation coefficients were included when multiple runs of the same human cell line were performed. Error bars represent standard error of the mean. Note that NF1 cell lines compared to each other are almost the same as biological replicates.
Several classes of drugs were potent inhibitors of all cell lines, with IC50s in the nanomolar range. These include proteasome inhibitors, HDAC inhibitors, topoisomerase inhibitors, microtubule destabilizers and, to a smaller extent, PAK inhibitors. All NF1 cell lines were sensitive to MEK inhibitors, but these drugs had little effect on the sporadic or NF2 cell lines. The bromodomain inhibitor JQ1 was the only drug in our panel that inhibited both NF1 and NF2 cells, but not the sporadic cell line. The activity of JQ1 in NF1 MPNST is likely occuring because most MPNST have deletions in SUZ12 or EED, two Polycomb repressive complex 2 components, which sensitizes cells to bromodomain inhibitors [7,8,20]. However, there is no known genetic mechanism that explains the sensitivity of the NF2 cell line to JQ1.
RAS processing inhibitors were modestly active in our drug screen. Deltarasin, a PDEδ inhibitor [21], and Tipifarnib, a farnesyl transferase inhibitor [22] both showed some activity. Cells were also sensitive to erastin, a ferroptosis activator. Though this was originally found to be specific to Ras mutant cells, erastin causes ferroptosis, a specific kind of oxidative cell death, which may not be that Ras selective [23]. We noted that these Ras inhibitors were not selective for the NF1 cells since they were cytotoxic to the NF2 cells.
RAF inhibitors had more effect on NF1 cell lines than NF2 cell lines, but even the NF1 cells were generally resistant to RAF inhibitors as a class. However, this is not an indication that RAF is a poor target, but instead a limitation of most of the RAF inhibitors. In cells with activated Ras, most RAF inhibitors cause a phenomenon called paradoxical hyperactivation of MAPK [24-26]. Paradoxical activation can be sufficiently high that BRAF inhibitors often cause secondary tumors, which often have Ras mutations [27-29]. In paradoxical activation, which occurs in the presence of activated Ras, RAF inhibitors cause the formation of BRAF/cRAF heterodimers, which activates cRAF. Paradoxical activation has been observed in patients treated with Sorafenib, a RAF inhibitor with other kinase targets, as well as in patients treated with more selective inhibitors of BRAF. The resistance of our NF1 MPNST to Sorafenib, PLX4720, GW5074, ZM336372, Vemurafenib and dabrafenib, which cause paradoxical activation, suggests our cells are prone to paradoxical activation [30,31]. However, several pan-RAF inhibitors were active in our cells including AZ628, TAK-632 and LY3009120. All of the active drugs are classified as “paradox breakers” and were particularly active in the NF1 lines [30,32]. These results indicate that dual inhibition of B-RAF and C-RAF is required for targeting RAF, and that the most effective drugs will be paradox breakers. New generations of RAF inhibitors specifically designed to be paradox breakers are being developed, which should be more effective [33].
Most MEK1/MEK2 inhibitors were active in NF1-associated MPNST, while they had little activity in the NF2 cell lines and the sporadic MPNST (Figure 4A). In fact, there was a linear correlation between the in vitro IC50s to MEK and the IC50s against NF1 MPNST [34] (Figure 4B). The primary difference in the cell lines is the extreme sensitivity of the NF1 cells to MEK inhibitors. Although one human NF2 cell line, KT21-MG was MEK sensitive, it was only comparable to the most resistant NF1 cell line. Omitting MEK from the Spearman’s correlations yielded no differences between NF1 and NF2 cells, while there was a substantial difference between NF1 and NF2 cells if only the MEK inhibitors were analyzed (Figures 4C, 4D, S1). Of note, Selumetinib is one of the weakest MEK inhibitors, both in our assays and in vitro, although it has shown promising results for unresectable plexiform neurofibromas in children and young adults [35,36]. All NF1 and NF2 cells except the sporadic line responded to BI-847325, a dual MEK/Aurora kinase inhibitor. As aurora kinase is overexpressed in MPNSTs but not neurofibromas, the dual action of this inhibitor may be especially potent [37]. The activity of BI-847325 against the NF2 cell line may indicate a role for aurora kinase in targeting NF2, since the cells are insensitive to MEK inhibitors.
Figure 4.
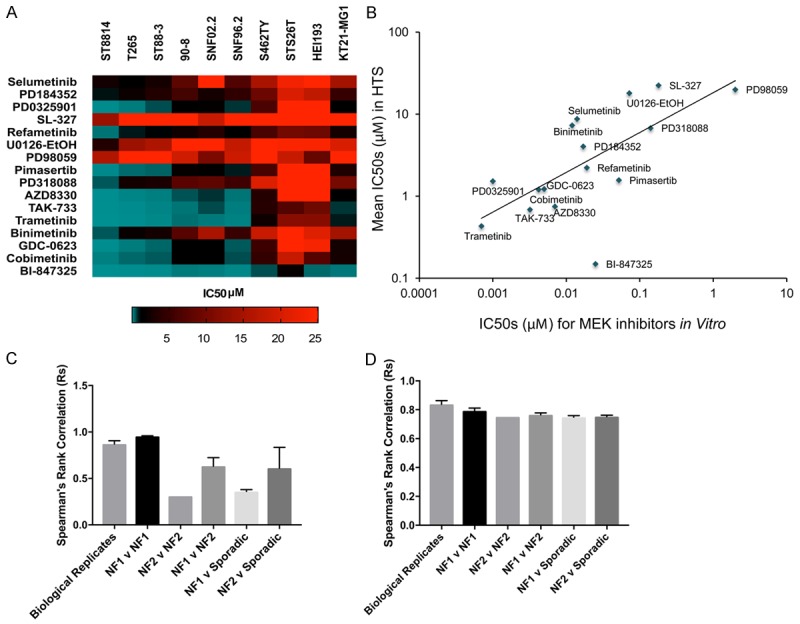
Distinguishing features between MPNST of different genetic origins. A. Heatmap of MEK inhibitors against NF1 and NF2 cell lines. NF1 cells are uniformly sensitive to MEK inhibitors whereas MPNST from NF2 do not respond to them. B. Relationship between IC50s of MEK inhibitors in vitro and the IC50s determined by HTS. C. Spearman’s rank correlation coefficients between IC50 values for MEK inhibitors alone. D. Spearman’s rank correlation coefficients between IC50 values for inhibitors excluding MEK inhibitors.
ERK inhibitor sensitivity followed a pattern similar to MEK sensitivity. The NF1 cells were the most sensitive, while the sporadic and NF2 cell lines were the least sensitive. Cells responded poorly to MEK5 inhibitors. There was only one JNK inhibitor, JNK inhibitor IX that was effective, and the sporadic cell line was among the cells most effectively inhibited by it.
Most cell lines were sensitive to three PI3K/mTOR inhibitors, PF04691502, Torkinib and especially AZD8055. These three mTOR inhibitors are dual inhibitors of PI3K and MTOR, or mTORC1 and mTORC2. Only one MPNST cell line, sNF02.2 is sensitive to rapamycin, although we found several others partially sensitive, likely due to the cytostatic effects of rapamycin. Rapamycin is selective for mTORC1 and causes feedback activation of Akt, limiting its effectiveness. Other studies have shown some activity of PI3K/mTOR inhibitors against MPNST, generally when combined with MEK inhibitors [38]. While most other studies tested only Rapamycin, our screens suggest that multi-targeted mTOR inhibitors may be more effective than rapamycin [39].
Pak kinases have been extensively studied in NF1 and NF2 [9,11,40-42]. Most cell lines responded to Pak1-3 inhibitors. One PAK inhibitor, PF-3758309, was especially potent to most MPNST cells and the NF2 Schwannoma cells. PF-3758309 inhibits Paks 1-6, indicating that most isoforms may need to be blocked for effective treatment or that alternative targets of PF-3758309 contribute to its potent activity in cells.
Other kinase inhibitors, including, EGF-R inhibitors, Akt inhibitors, GSK3 inhibitors and Jak inhibitors showed almost no activity. There was modest activity with the multi-receptor kinase inhibitor crizotinib, predominantly in NF1 cells. Additionally, although we did not observe activity with Jak inhibitors, inhibition of STAT with SH-4-54 or Niclosamide showed some activity.
Although the primary difference common to all NF1 cell lines were their sensitivity to MEK inhibitors, there are some exceptions. The NF2 cell line KT21-MG was partially sensitive to MEK inhibitors; this may be because it is from a malignant tumor, unlike our other NF2 cell line, HEI-193, which is derived from a benign tumor [19,43]. One NF1 cell line, sNF02.2, is more sensitive to rapamycin and other mTOR/PI3K/AKT inhibitors. The rodent cell lines behaved differently than human cell lines. Unexpectedly, the mouse NF2 cell line, SC4 was very sensitive to MEK inhibitors; since this cell line forms tumors in orthotopic xenografts, it may indicate a high level of transformation [42]. RT4, a rat schwannoma cell line, was sensitive to MEK and EGF receptor inhibitors, likely because it expresses a mutant erbB-2 [44].
Predictive value of HTS screening
Clinical trials of MPNST are rare, but several drugs in our panel, mostly RAS RAF and MEK inhibitors were investigated in neurofibroma trials and showed an effectiveness (or lack of infectiveness) as predicted by our panel. Tipifarnib, a farnesyl transferase inhibitor was not effective in a phase II trial [45,46], probably because K-RAS is the primary Ras isoform in NF1 that is activated by NF1 loss [47]. Sorafenib was not effective either, likely because of paradoxical activation [45]. However, the MEK inhibitor, Selumetinib was effective in a clinical trial for neurofibromas. Our data suggest that other MEK inhibitors may be more effective. Phase II clinical trials evaluated the effects of sorafenib [48] and a rapamycin analog in patients with distinct types of sarcoma only showed slight efficacy in treating MPNSTs [49]. Finally, doxorubicin a drug recommended for treating MPNST, was highly effective in all cells.
Other drugs that were tested in clinical trials or recommended for NF1 including Imatinib and ifosfamide were not effective. However, Imatinib acts on the tumor microenvironment and ifosfamide requires metabolic activation in the liver. Cyclophosphamide which, like ifosfamide is a highly cytotoxic nitrogen mustard also requires metabolic activation.
Acknowledgements
We thank Drs. Marco Giovannini, Nancy Ratner, Joseph Kissil and Jonathan Chernoff for generously providing cell lines. This work is supported by award number 2015-05-006 from the Children’s Tumor Foundation and R01HL134923 from the NIH.
Disclosure of conflict of interest
None.
Abbreviations
- HTS
High Throughput Screen
- NF1 or NF2
Neurofibromatosis types 1 or 2
- MPNST
Malignant Peripheral Nerve Sheath Tumors
- Rs
Spearman’s rank correlation coefficients
Figure S1
Table S1
References
- 1.Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65:205–211. doi: 10.1212/01.wnl.0000168830.79997.13. [DOI] [PubMed] [Google Scholar]
- 2.Evans DG, O’Hara C, Wilding A, Ingham SL, Howard E, Dawson J, Moran A, Scott-Kitching V, Holt F, Huson SM. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet. 2011;19:1187–1191. doi: 10.1038/ejhg.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, Maki RG. Malignant peripheral nerve sheath tumors. Oncologist. 2014;19:193–201. doi: 10.1634/theoncologist.2013-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zehou O, Fabre E, Zelek L, Sbidian E, Ortonne N, Banu E, Wolkenstein P, Valeyrie-Allanore L. Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1: a 10-year institutional review. Orphanet J Rare Dis. 2013;8:127. doi: 10.1186/1750-1172-8-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moretti VM, Crawford EA, Staddon AP, Lackman RD, Ogilvie CM. Early outcomes for malignant peripheral nerve sheath tumor treated with chemotherapy. Am J Clin Oncol. 2011;34:417–421. doi: 10.1097/COC.0b013e3181e9c08a. [DOI] [PubMed] [Google Scholar]
- 6.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15:290–301. doi: 10.1038/nrc3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, Qin LX, Viale A, Singer S, Zheng D, Berger MF, Chen Y, Antonescu CR, Chi P. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, Gokaslan ZL, Riggins GJ, Wolinksy JP, Wood LD, Montgomery EA, Hruban RH, Kinzler KW, Papadopoulos N, Vogelstein B, Bettegowda C. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1170–1172. doi: 10.1038/ng.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, O’Bryan JP, Gupta V, Ratner N, Der CJ, Jacks T, McClatchey AI. The Nf2 tumor suppressor, Merlin, functions in Rac-dependent signalling. Developmental Cell. 2001;1:63–72. doi: 10.1016/s1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 10.Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to Merlin. J Biol Chem. 2002;277:883–886. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- 11.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–849. doi: 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D. Kibra functions as a tumor suppressor protein that regulates hippo signaling in conjunction with merlin and expanded. Developmental Cell. 2010;18:288–299. doi: 10.1016/j.devcel.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, Snuderl M, Ladanyi M, Hanemann CO, Zhou P, Karajannis Matthias A, Giancotti FG. Merlin/NF2 loss-driven tumorigenesis linked to CRL4DCAF1-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell. 2014;26:48–60. doi: 10.1016/j.ccr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinstein JN, Myers TG, O’Connor PM, Friend SH, Fornace AJ Jr, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD. An information-intensive approach to the molecular pharmacology of cancer. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 15.Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, McNeil E, Moy C, Wegrzyn R, Auger K, Hardwicke MA, Wooster R. Molecular target class is predictive of in vitro response profile. Cancer Research. 2010;70:3677–86. doi: 10.1158/0008-5472.CAN-09-3788. [DOI] [PubMed] [Google Scholar]
- 16.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The cancer cell Line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–307. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teicher BA, Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, Ogle C, Reinhart R, Selby M, Connelly J, Harris E, Monks A, Morris J. Sarcoma cell line screen of oncology drugs and investigational agents identifies patterns associated with gene and microRNA expression. Mol Cancer Ther. 2015;14:2452–2462. doi: 10.1158/1535-7163.MCT-15-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hung G, Li X, Faudoa R, Xeu Z, Kluwe L, Rhim JS, Slattery W, Lim D. Establishment and characterization of a schwannoma cell line from a patient with neurofibromatosis 2. Int J Oncol. 2002;20:475–482. [PubMed] [Google Scholar]
- 20.De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, Clapp W, Bradner J, Vidaud M, Upadhyaya M, Legius E, Cichowski K. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. doi: 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 21.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, Waldmann H. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–42. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 22.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wildtype BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 27.Karajannis MA, Legault Gv, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, Harter DH, Goldberg JD, Hochman T, Merkelson A, Bloom MC, Sievert AJ, Resnick AC, Dhall G, Jones DTW, Korshunov A, Pfister SM, Eberhart CG, Zagzag D, Allen JC. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol. 2014;16:1408–1416. doi: 10.1093/neuonc/nou059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnault JP, Wechsler J, Escudier B, Spatz A, Tomasic G, Sibaud V, Aractingi S, Grange JD, Poirier-Colame V, Malka D, Soria JC, Mateus C, Robert C. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J. Clin. Oncol. 2009;27:e59–61. doi: 10.1200/JCO.2009.23.4823. [DOI] [PubMed] [Google Scholar]
- 29.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, Chapman PB, Kim MJ, Hayward R, Martin M, Yang H, Wang Q, Hilton H, Hang JS, Noe J, Lambros M, Geyer F, Dhomen N, Niculescu-Duvaz I, Zambon A, Niculescu-Duvaz D, Preece N, Robert L, Otte NJ, Mok S, Kee D, Ma Y, Zhang C, Habets G, Burton EA, Wong B, Nguyen H, Kockx M, Andries L, Lestini B, Nolop KB, Lee RJ, Joe AK, Troy JL, Gonzalez R, Hutson TE, Puzanov I, Chmielowski B, Springer CJ, McArthur GA, Sosman JA, Lo RS, Ribas A, Marais R. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arora R, Di Michele M, Stes E, Vandermarliere E, Martens L, Gevaert K, Van Heerde E, Linders JT, Brehmer D, Jacoby E, Bonnet P. Structural investigation of B-Raf paradox breaker and inducer inhibitors. J Med Chem. 2015;58:1818–1831. doi: 10.1021/jm501667n. [DOI] [PubMed] [Google Scholar]
- 31.Holderfield M, Nagel TE, Stuart DD. Mechanism and consequences of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640–645. doi: 10.1038/bjc.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, Van Horn RD, Yin T, Zhang X, Yadav V, Chen SH, Gong X, Ma X, Webster Y, Buchanan S, Mochalkin I, Huber L, Kays L, Donoho GP, Walgren J, McCann D, Patel P, Conti I, Plowman GD, Starling JJ, Flynn DL. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell. 2015;28:384–398. doi: 10.1016/j.ccell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, Zhang J, Lin J, Ewing T, Matusow B, Tsang G, Marimuthu A, Cho H, Wu G, Wang W, Fong D, Nguyen H, Shi S, Womack P, Nespi M, Shellooe R, Carias H, Powell B, Light E, Sanftner L, Walters J, Tsai J, West BL, Visor G, Rezaei H, Lin PS, Nolop K, Ibrahim PN, Hirth P, Bollag G. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 34.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 35.Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE, Rizvi TA, Wu J, Ershler R, Wolters P, Therrien J, Glod J, Belasco JB, Schorry E, Brofferio A, Starosta AJ, Gillespie A, Doyle AL, Ratner N, Widemann BC. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med. 2016;375:2550–2560. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves D, Widemann B, Kim MO, Dombi E, Sabo J, Hardiman Dudley A, Niwa-Kawakita M, Page GP, Giovannini M, Aronow BJ, Cripe TP, Ratner N. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340–347. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel AV, Eaves D, Jessen WJ, Rizvi TA, Ecsedy JA, Qian MG, Aronow BJ, Perentesis JP, Serra E, Cripe TP, Miller SJ, Ratner N. Ras-driven transcriptome analysis identifies aurora kinase a as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin Cancer Res. 2012;18:5020–5030. doi: 10.1158/1078-0432.CCR-12-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varin J, Poulain L, Hivelin M, Nusbaum P, Hubas A, Laurendeau I, Lantieri L, Wolkenstein P, Vidaud M, Pasmant E, Chapuis N, Parfait B. Dual mTORC1/2 inhibition induces anti-proliferative effect in NF1-associated plexiform neurofibroma and malignant peripheral nerve sheath tumor cells. Oncotarget. 2016;7:35753–35767. doi: 10.18632/oncotarget.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson AL, Anderson LK, Greeley AD, Keng VW, Rahrmann EP, Halfond AL, Powell NM, Collins MH, Rizvi T, Moertel CL, Ratner N, Largaespada DA. Co-targeting the MAPK and PI3K/AKT/mTOR pathways in two genetically engineered mouse models of schwann cell tumors reduces tumor grade and multiplicity. Oncotarget. 2014;5:1502–14. doi: 10.18632/oncotarget.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang Y, Marwaha S, Rutkowski JL, Tennekoon GI, Phillips PC, Field J. A role for Pak protein kinases in Schwann cell transformation. Proc Natl Acad Sci U S A. 1998;95:5139–5144. doi: 10.1073/pnas.95.9.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chow HY, Stepanova D, Koch J, Chernoff J. p21-Activated kinases are required for transformation in a cell-based model of neurofibromatosis type 2. PLoS One. 2010;5:e13791. doi: 10.1371/journal.pone.0013791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Licciulli S, Maksimoska J, Zhou C, Troutman S, Kota S, Liu Q, Duron S, Campbell D, Chernoff J, Field J, Marmorstein R, Kissil JL. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of NF2-associated schwannomas. J Biol Chem. 2013;288:29105–14. doi: 10.1074/jbc.M113.510933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka K, Sato C, Maeda Y, Koike M, Matsutani M, Yamada K, Miyaki M. Establishment of a human malignant meningioma cell line with amplified c-myc oncogene. Cancer. 1989;64:2243–2249. doi: 10.1002/1097-0142(19891201)64:11<2243::aid-cncr2820641110>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 44.Nikitin AYu, Ballering LA, Lyons J, Rajewsky MF. Early mutation of the neu (erbB-2) gene during ethylnitrosourea-induced oncogenesis in the rat Schwann cell lineage. Proc Natl Acad Sci U S A. 1991;88:9939–9943. doi: 10.1073/pnas.88.22.9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim A, Dombi E, Tepas K, Fox E, Martin S, Wolters P, Balis FM, Jayaprakash N, Turkbey B, Muradyan N, Choyke PL, Reddy A, Korf B, Widemann BC. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2013;60:396–401. doi: 10.1002/pbc.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Widemann BC, Dombi E, Gillespie A, Wolters PL, Belasco J, Goldman S, Korf BR, Solomon J, Martin S, Salzer W, Fox E, Patronas N, Kieran MW, Perentesis JP, Reddy A, Wright JJ, Kim A, Steinberg SM, Balis FM. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 2014;16:707–718. doi: 10.1093/neuonc/nou004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005;65:236–245. [PubMed] [Google Scholar]
- 48.Maki RG, D’Adamo DR, Keohan ML, Saulle M, Schuetze SM, Undevia SD, Livingston MB, Cooney MM, Hensley ML, Mita MM, Takimoto CH, Kraft AS, Elias AD, Brockstein B, Blachère NE, Edgar MA, Schwartz LH, Qin LX, Antonescu CR, Schwartz GK. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin. Oncol. 2009;27:3133–3140. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okuno S, Bailey H, Mahoney MR, Adkins D, Maples W, Fitch T, Ettinger D, Erlichman C, Sarkaria JN. A phase 2 study of temsirolimus (CCI-779) in patients with soft tissue sarcomas. Cancer. 2011;117:3468–3475. doi: 10.1002/cncr.25928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.