

Abstract
Non-small cell lung cancer (NSCLC) sensitive to first-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) often acquires resistance through secondary EGFR mutations, including the T790M mutation, aberrant c-Met receptor activity, or both. We assessed the ability of the highly selective c-Met inhibitor tepotinib to overcome EGFR TKI resistance in various xenograft models of NSCLC. In models with EGFR-activating mutations and low c-Met expression (patient explant-derived LU342, cell line PC-9), EGFR TKIs caused tumors to shrink, but growth resumed upon cessation of treatment. Tepotinib combined with EGFR TKIs delayed tumor regrowth, while tepotinib alone was ineffective. In patient explant-derived LU858, which has an EGFR-activating mutation and expresses high levels of c-Met/HGF, EGFR TKIs had no effect on tumor growth. Tepotinib combined with EGFR TKIs caused complete tumor regression and tepotinib alone caused tumor stasis. In cell line DFCI081 (activating EGFR mutation, c-Met amplification), EGFR TKIs were ineffective, whereas tepotinib alone induced complete tumor regression. Finally, in a ‘double resistant’ EGFR T790M-positive, high c-Met model (cell line HCC827-GR-T790M), the EGFR TKIs erlotinib, afatinib, and rociletinib, as well as tepotinib as a single agent or in combination with erlotinib or afatinib, slowed tumor growth, but only tepotinib in combination with rociletinib induced complete tumor regression. We conclude that tepotinib can overcome acquired resistance to EGFR TKIs. Based on these data, clinical trials of tepotinib in combination with EGFR TKIs in patients with NSCLC with acquired resistance to first-generation EGFR TKIs are warranted.
Keywords: Tepotinib, EGFR, EGFR T790M, c-Met, hepatocyte growth factor, NSCLC, resistance, erlotinib, afatinib, rociletinib
Introduction
Lung cancer is the most common cause of cancer mortality worldwide, and non-small cell lung cancer (NSCLC) accounts for 85% of the total. Treatments have improved with increased understanding of the molecular drivers of the disease, but a high unmet need for better therapies remains, particularly for patients developing resistance to existing therapies.
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are widely used for the treatment of patients with NSCLC [1]. The first-generation EGFR TKIs gefitinib and erlotinib are effective against tumors that express active mutant forms of EGFR, such as EGFR del19 and EGFR L858R. These mutant forms are expressed in 10-35% of NSCLCs and are associated with aggressive tumor growth [2-4]. However, despite the initial efficacy of EGFR TKIs against NSCLC in a proportion of patients with tumors harboring these mutations, resistance to treatment ultimately develops [5].
In approximately 50% of cases, resistance is associated with further mutation of the EGFR, most commonly a point mutation leading to the substitution of methionine for the gatekeeper residue threonine at position 790 in the kinase domain (T790M). This substitution increases the affinity of the EGFR kinase domain for ATP, which outcompetes the binding of first-generation EGFR TKIs. Second-generation inhibitors such as afatinib are similarly outcompeted, but because they bind irreversibly to EGFR, they nevertheless exhibit some activity against EGFR T790M [6]. Third-generation EGFR TKIs such as rociletinib (CO-1686) and osimertinib bind to EFGR T790M with high affinity and have demonstrated good efficacy in patients with T790M-mediated resistance to first-generation EGFR TKIs [6].
In about 20% of cases, NSCLC resistance to first-generation EGFR TKIs is associated with aberrant c-Met activity [7]. c-Met is a receptor tyrosine kinase [8] that is normally activated by dimerization and transphosphorylation induced by its only ligand, hepatocyte growth factor (HGF). Normal c-Met activity is critical for embryogenesis, and in the adult plays a role in liver regeneration and in wound healing [9]. Aberrant c-Met activity in NSCLC can arise through mutation or amplification of the MET gene, through increased c-Met and/or HGF expression induced by other mechanisms [10], and is associated with poor prognosis [11-14]. Such aberrant c-Met activity mediates acquired resistance to EGFR TKIs by activating signaling pathways downstream from EGFR, most importantly PI3K/mTOR, rendering activation by EGFR redundant for tumor growth. The sensitivity to EGFR inhibition of cancer cells that are resistant to EGFR TKIs, including lung cancer models resistant to gefitinib due to aberrant c-Met activity, can be restored by inhibiting c-Met signaling [15,16].
Drugs with c-Met inhibitory activity include HGF-neutralizing antibodies (e.g., rilotumumab), HGF antagonists (e.g., onartuzumab), bivalent c-Met antibodies (e.g., emibetuxumab), and c-Met TKIs (e.g., non-selective: cabozantinib, glesatinib; selective: tepotinib, capmatinib). c-Met TKIs can inhibit both HGF-dependent and HGF-independent c-Met signaling [17].
Tepotinib (MSC2156119J) is a potent, highly selective c-Met TKI with >1,000-fold selectivity for c-Met over 236 of 241 kinases tested, and >50-fold selectivity over the other five [18]. This selectivity enables dosing at a level that is not expected to affect other tyrosine kinase receptors, reducing the risk of off-target toxicities. Tepotinib monotherapy has already been shown to effectively inhibit tumor growth in c-Met-overexpressing liver cancer models, in which c-Met is an oncogenic driver [19]. The properties of tepotinib provide a rationale for its use in combination with EGFR inhibitors to reverse the emergence of resistance mediated by aberrant c-Met activity.
To investigate the potential for tepotinib to overcome resistance to EGFR inhibitors, we assessed the effects of tepotinib and various EGFR inhibitors, alone and in combination, in NSCLC xenograft models with discrete aberrations of c-Met and/or EGFR.
Materials and methods
Xenograft models
Two xenograft models derived from patient carcinoma explants were studied: LU342, which expresses EGFR del19 and low levels of c-Met; and LU0858, which expresses EGFR L858R and high levels of c-Met. Patient-derived explants were maintained subcutaneously in nude mice and harvested for inoculation into the right flank of recipients by excising 3 mm3 (LU0858) or 2 mm3 (LU342) sections from well-developed regions of the tumor.
Three xenograft models derived from cell lines were used: PC-9 (provided by Dr Roman Thomas, Max Planck Institute, Cologne, Germany), which expresses EGFR del19 and low levels of c-Met; DFCI08 (patient-derived cell line), which expresses EGFR del19 and has amplification of the MET locus; and HCC827-GR-T790M (provided by Dr Pasi Janne, Dana-Farber Cancer Center, Boston, MA, USA), which expresses endogenous EGFR del19, exogenous EGFR T790M, and has amplification of the MET locus. Cell lines were maintained in growth medium at 37°C, 5% CO2 (PC-9: RPMI1640 medium with 10% FBS; DFCI08: RPMI1640 medium with 10% FBS; HCC-827-GR-T790M: RPMI1640 medium with 10% FBS). To generate xenografts, cultured cells were suspended in 100 µL PBS or PBS/Matrigel (BD Biosciences, San Jose, CA, USA) and injected subcutaneously into the right flanks of immunodeficient mice (PC-9: 5×106 cells/animal; DFCI08: 107 cells/30% Matrigel/animal; HCC827-GR-T790M; 2.5×106 cells/15% Matrigel/animal).
Treatment and administration
Tepotinib 100 mg/kg, afatinib 5 mg/mg, gefitinib 150 mg/kg, and rociletinib (CO-1686) 100 mg/kg were prepared in 20% Solutol/80% 100 mM sodium acetate buffer pH 5.5. Erlotinib 20 mg/kg was prepared in 0.5% hydroxypropylmethyl cellulose/0.25% Tween 20. Solutions were administered by oral gavage at the indicated doses. Cetuximab 50 mg/kg was prepared in 0.9% sterile saline. Pemetrexed 200 mg/kg was freshly prepared in 0.9% sterile saline. Cisplatin 5 mg/kg was supplied as a ready-to-use injectable solution. Solutions were administered once-weekly by intraperitoneal (IP) injection.
Mice were randomly assigned to treatment groups of 8-10 when tumors were 100-250 mm3. Dosing schedules are summarized in Table 1.
Table 1.
Dosing schedules
| Model | LU342 | PC-9 | LU858 | DCFI08 | HCC827-GR-T790M |
|---|---|---|---|---|---|
| Treatment (days) | 22 | 70 | 12 | 18-21 | 32 |
| Observation period (days) | 78 | 128 | 46 | 80 | 60 |
| Tepotinib 100 mg/kg | (5+2) w | (5+2) w | (5+2) w | Daily | Daily |
| Cetuximab 50 mg/kg | 3×1 w | - | - | - | - |
| Erlotinib 20 mg/kg | (5+2) w | - | - | Daily | Daily |
| Gefitinib 150 mg/kg | (5+2) w | - | (5+2) w | - | - |
| Afatinib 5 mg/kg | (5+2) w | (5+2) w | (5+2) w | Daily | Daily |
| Rociletinib 10 mg/kg | - | - | Daily | Daily | |
| Erlotinib + tepotinib 20/100 mg/kg | (5+2) w | - | - | Daily | Daily |
| Gefitinib + tepotinib 150/100 mg/kg | (5+2) w | - | (5+2) w | - | - |
| Afatinib + tepotinib 5/150 mg/kg | (5+2) w | (5+2) w | (5+2) w | Daily | Daily |
| Rociletinib + tepotinib 10/150 mg/kg | - | - | - | Daily | Daily |
(5+2) w, treated for 5 days, untreated for 2 days of each week. 3×1 w, three weekly doses.
Endpoints
Tumor length (L) and width (W) were measured at regular intervals using calipers. Tumor volume was calculated as L × W2/2. The percentage treatment/control (T/C) value was used to quantify the effect of treatment on tumors, calculated as follows: for treated tumors that grew, T/C = 100 × (end voltreatment - start voltreatment)/(end volcontrol - start volcontrol); and for treated tumors that regressed, T/C = 100 × (end voltreatment - start voltreatment)/start voltreatment. End volumes for mice in all groups were measured when tumors in control mice reached the maximum permitted volume. Tumor progression was defined as >73% increase in tumor volume and tumor regression as >63% decrease in tumor volume during the period of treatment. Tumors <20 mm3 or impalpable were defined as being in complete remission. Tumor growth delay was measured as the difference between treatment and control groups in time required to reach a mean tumor volume of 600 mm3. Tumor-free survival was defined as tumors remaining <20 mm3 for the period of observation following withdrawal of treatment.
Body weight was recorded twice weekly. The mean body weight change (%) for each group was determined relative to the mean body weight of the group at Day 0.
Analysis plan
The mean tumor volume and the standard error of the mean (SEM) were determined for each group at each time point. Statistical analysis of difference in tumor volume among the groups and the analysis of potential drug synergy were conducted on the data obtained when tumors in control mice reached the maximum permitted volume. The statistical significance of differences between groups at the end of treatment was determined by one-way analysis of variance (ANOVA) (Kruskal-Wallis, Dunn’s post hoc test). P<0.05 was considered to be statistically significant. All data were analyzed using SPSS 16.0.
Pairs of groups were compared using an independent-samples t-test. For comparisons among three or more groups, a one-way ANOVA was performed followed by multiple comparison procedures. The potential synergistic effect between treatments was analyzed using two-way ANOVA. P≤0.05 was considered to be statistically significant. All data were analyzed using SPSS 16.0.
The statistical significance of treatment on tumor volume at Day 15 was determined using repeated measures two-way ANOVA followed by Bonferroni multiple comparisons. P≤0.05 was considered to be statistically significant.
Results
Tumor models with activating EGFR mutations and low c-Met expression
The first model of a tumor with activating EGFR mutation and low c-Met expression tested was patient-derived LU432. Treatment started when tumors reached a volume of 150 mm3 (Day 1). Tumors in vehicle-treated mice reached a mean volume of 1,124 mm3 by Day 21 (Figure 1). Tumors in mice treated with tepotinib as a single agent grew less quickly, with a T/C value of 75% on Day 21 (P>0.05). Afatinib, gefitinib, and cetuximab as single agents or in combination with tepotinib all caused profound shrinkage of LU342 tumors, with T/C values of -96% to -100% (P<0.001) on Day 21. Tumors in mice that received these treatments did not regrow between withdrawal of treatment on Day 21 and the end of the study on Day 80.
Figure 1.
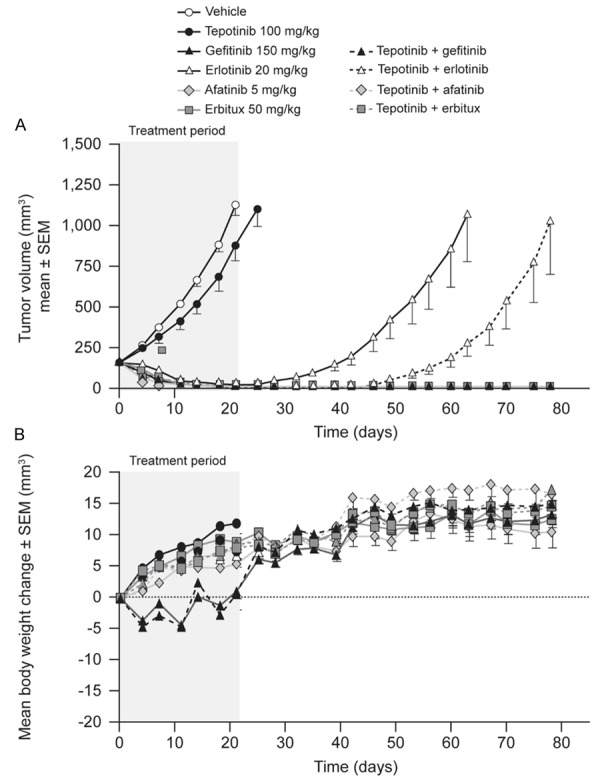
Effects of therapy on tumor growth (A) and body weight (B) in mice bearing patient-derived human lung carcinoma explant LU342 tumors (EGFR del19, c-Metlow).
Erlotinib as a single agent or in combination with tepotinib caused significant tumor regression, with T/C values of -91% (P<0.001) and -95% (P≤0.001), respectively. Tumor regrowth occurred in both groups, but tumors took 15 d longer to reach 600 mm3 in mice dosed with both agents compared to those treated with erlotinib alone.
Mean body weight gain was consistent in all groups and no gross clinical abnormalities were observed in any treatment group, suggesting no significant toxicity.
PC-9 xenografts were also tested. Mice with PC-9-derived tumors commenced treatment when the mean tumor volume was 214 mm3. Tumors in vehicle-treated mice achieved a mean volume of 1,265 mm3 over 28 d (Figure 2). Tepotinib as a single agent had no antitumor effect. Afatinib 5 mg/kg induced complete tumor regression (P≤0.0001), although there were signs of tumor growth after Day 56 when the dose was reduced to 1 mg/kg. Further tumor growth occurred after Day 70, when afatinib treatment was withdrawn. Tepotinib in combination with afatinib caused complete tumor regression with no regrowth during the period of observation. Consistent mean body weight gain in all groups and a lack of gross clinical abnormalities suggested that significant toxicity did not occur.
Figure 2.
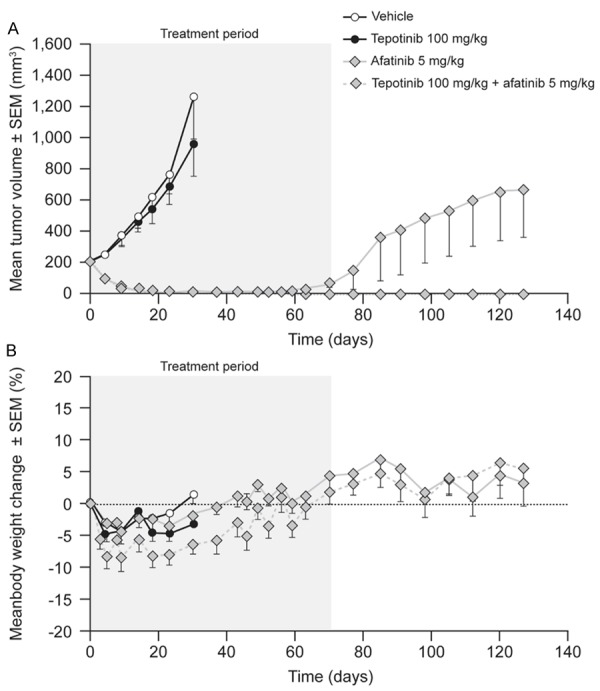
Effects of therapy on tumor growth (A) and body weight (B) in mice bearing human lung carcinoma PC-9 xenografts (EGFR del19, c-Metlow).
Tumor models with activating EGFR mutations and high HGF/c-Met expression
First, the patient-derived LU858 model, which carries an activating EGFR mutation and expresses high levels of HGF and c-Met, was tested. The mean LU858 tumor volume in vehicle-treated mice increased from 154 mm3 to 1,030 mm3 over 10 d of treatment (Figure 3). Dosing with tepotinib caused tumor stasis throughout the treatment period, but growth commenced after withdrawal of treatment. Gefitinib and afatinib as single agents had no effect on tumor growth, although gefitinib or afatinib in combination with tepotinib induced complete tumor regression over the treatment period. Tumors in mice receiving combination therapy started to grow when treatment was withdrawn; regrowth was delayed by approximately 10 d compared to that of tumors in mice dosed with tepotinib alone.
Figure 3.
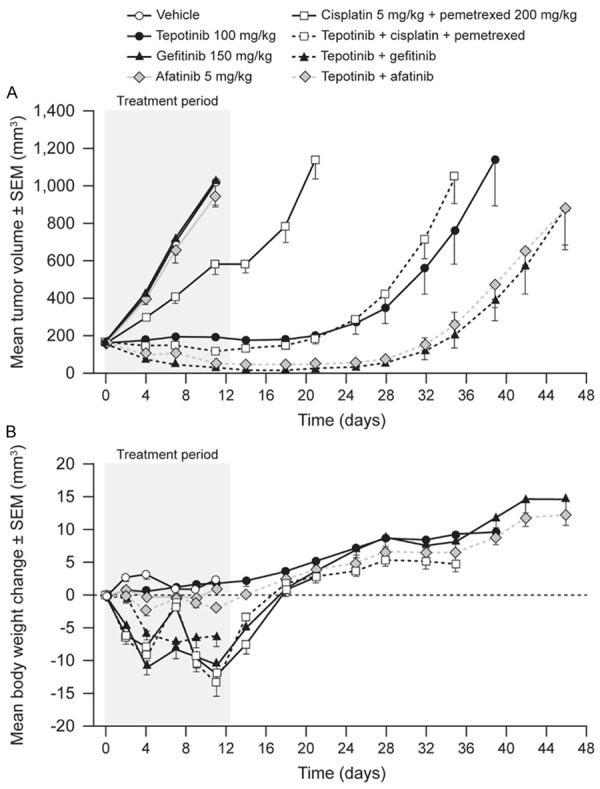
Effects of therapy on tumor growth (A) and body weight (B) in mice bearing patient-derived human lung carcinoma explant LU858 tumors (EGFR L858R, c-Methigh).
Groups of mice receiving cisplatin/pemetrexed or gefitinib showed a loss in mean body weight during the dosing period, indicating that cisplatin/pemetrexed and gefitinib had toxicity in this study. Loss of body weight was not increased with the addition of tepotinib. Due to the observed toxicity of cisplatin/pemetrexed, the efficacy of tepotinib in combination with cisplatin/pemetrexed was not assessed.
Then, DFCI081 cells, which express an activating EGFR mutation and have amplification of the MET locus, were analyzed. Treatment of mice implanted with DFCI081 cells was started when the mean tumor volume was 109 mm3. Tumors in mice dosed with vehicle grew to a mean volume of 1,390 mm3 in 18 d (Figure 4). Tepotinib treatment caused complete tumor regression in 10/10 treated mice, with a T/C of -97% (P≤0.0001). Conversely, treatment with the EGFR inhibitors erlotinib, afatinib, and rociletinib had no significant effect (P>0.05) on tumor growth. Treatment with tepotinib combined with any of the EGFR inhibitors showed no additional effect beyond that seen with tepotinib alone, although the profound tumor shrinkage caused by tepotinib precluded detection of any additional antitumor effects. No tumor regrowth was detected during the 80-day observation period in any of the groups treated with combination therapy except for the tepotinib/erlotinib combination group, in which five of 10 tumors relapsed approximately 30 d following withdrawal of treatment.
Figure 4.
Effects of therapy on tumor growth (A) and body weight (B) in mice bearing DFCI081 xenografts (EGFR del19, c-Methigh).
No treatment-related effects on mean body weight were seen in any treatment group.
Model of EGFR TKI-resistant tumors with high HGF/c-Met expression
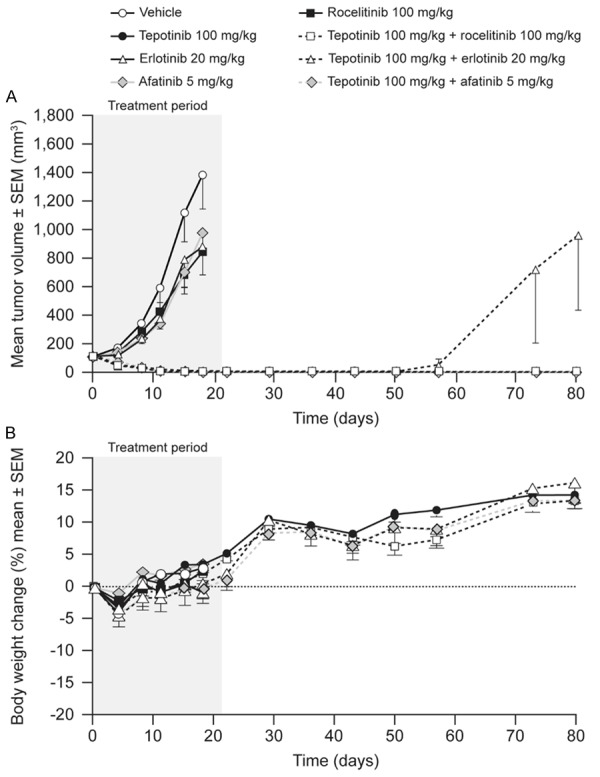
HCC827-GR-T790M cells express endogenous EGFR del19, exogenous EGFR T790M, and have amplification of the MET locus. The mean volume of HCC827-GR-T790M tumors in vehicle-treated mice increased from 112 mm3 to 633 mm3 over 14 d of dosing (Figure 5). Tepotinib as a single agent slowed tumor growth, with a T/C of 45% (P = 0.01). The EGFR inhibitors erlotinib and afatinib showed no significant antitumor activity, while the third-generation EGFR inhibitor rociletinib was the most active single agent, slowing tumor growth with a T/C value of 18% (P≤0.0001). Tumor growth was reduced further when tepotinib was combined with erlotinib (T/C = 22%, P = NS) or afatinib (T/C = 12%, P<0.01). The combination of tepotinib with rociletinib was the most effective, causing complete tumor regression over the treatment period of 21 d (P≤0.0001), with no regrowth during the observation period following withdrawal of treatment.
Figure 5.
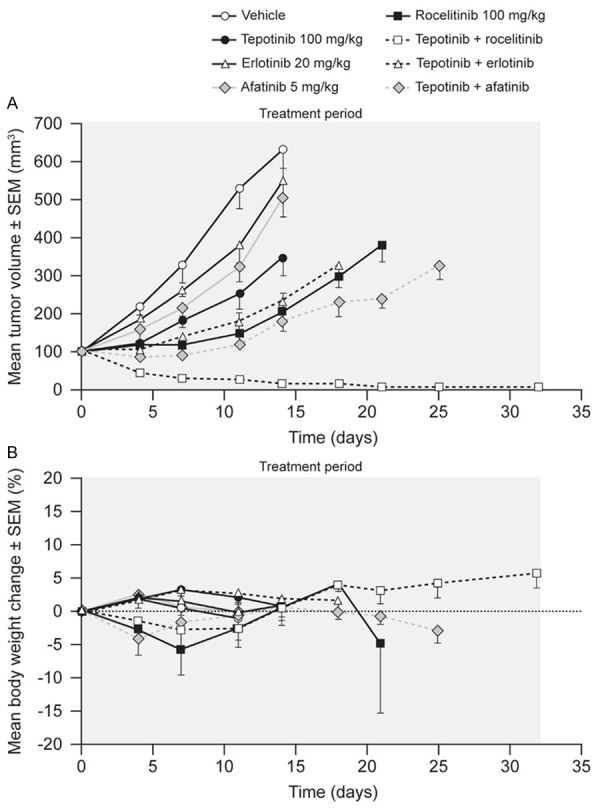
Effects of therapy on tumor growth (A) and body weight (B) in mice bearing HCC827-GR-T790M xenografts (EGFRdel19, Transgenic EGFRT790M, c-Methigh).
Single-agent therapies had no effect on body weight. Combination therapies caused initial reductions in mean body weight of less than 10%, followed by return to baseline within 10 d of treatment start. A summary of the treatment effects in each model is provided in Table 2.
Table 2.
Summary of models and the effects of therapies on tumor growth
| First-generation EGFR TKI resistance type | EGFR TKI-sensitive | c-Met-mediated EGFR TKI-resistant | c-Met- and T790M- mediated EGFR TKI-resistant | |||||||
|
|
|
|
||||||||
| Xenograft model | LU342 | PC-9 | LU858 | DFCI081 | HCC827-GR-T790M | |||||
|
|
|
|
||||||||
| Model type | Explant | Cell line | Explant | Cell line | Cell line | |||||
|
|
|
|
||||||||
| EGFR status | Del19 | Del19 | L858R mutation | Del19 | Del19+T790M transgene | |||||
|
|
|
|
||||||||
| c-Met status | Low | Low | High | High | High | |||||
|
|
|
|
||||||||
| Experimental period | Treatment | Observation | Treatment | Observation | Treatment | Observation | Treatment | Observation | Treatment | Observation |
|
|
|
|
||||||||
| Duration from first dose (d) | 22 | 78 | 70 | 128 | 12 | 46 | 18-21 | 80 | 32 | 60 |
|
| ||||||||||
| Single-agent treatment | ||||||||||
| Tepotinib | No effect | NT | No effect | NT | Stasis | Regrowth | Complete regression | No regrowth | No effect | NT |
| Cetuximab | Complete regression | No regrowth | NT | NT | NT | NT | NT | NT | NT | NT |
| Erlotinib | Complete regression | Regrowth | NT | NT | NT | NT | No effect | NT | No effect | NT |
| Gefitinib | Complete regression | No regrowth | NT | NT | No effect | NT | NT | NT | NT | NT |
| Afatinib | Complete regression | No regrowth | Complete regression | Regrowth | No effect | NT | No effect | NT | No effect | NT |
| Rociletinib | NT | NT | NT | NT | NT | NT | No effect | NT | Slowed growth | NT |
| Combination treatment | ||||||||||
| Erlotinib + tepotinib | Complete regression | Delayed regrowtha | NT | NT | NT | NT | Complete regression | No regrowth | Slowed growth | NT |
| Gefitinib + tepotinib | Complete regression | NT | NT | NT | Complete regression | Delayed regrowthb | NT | NT | NT | NT |
| Afatinib + tepotinib | Complete regression | No regrowth | Complete regression | No regrowth | Complete regression | Delayed regrowthb | Complete regression | No regrowth | Slowed growth | NT |
| Rociletinib + tepotinib | NT | NT | NT | NT | NT | NT | Complete regression | No regrowth | Complete regression | No regrowth |
d, days; NT, not tested.
Compared to EGFR inhibitor monotherapy.
Compared to tepotinib monotherapy.
Discussion
We tested whether tepotinib, a potent and highly selective c-Met inhibitor, could inhibit the growth of NSCLC xenograft models with activating EGFR mutations with or without aberrant c-Met activation, either as a single agent or in combination with different types of EGFR TKI. Our data demonstrate that tumors expressing low levels of c-Met were insensitive to tepotinib, whereas models with aberrant c-Met showed varying degrees of sensitivity to c-Met despite being resistant to EGFR inhibition. This included a model expressing EGFR T790M and aberrant c-Met, which is resistant to first- and second-generation EGFR TKIs and the growth of which is completely inhibited only by tepotinib combined with a third-generation EGFR TKI.
The data also suggest that when EGFR signaling is partially inhibited, low-level c-Met activation can be critical to the survival of tumors, compensating for the loss of EGFR signaling in EGFR-dependent tumors. In contrast, tumors expressing low levels of c-Met were, as expected, insensitive to single-agent tepotinib treatment, while therapeutic benefit could be increased by combination with EGFR TKIs. In several instances, EGFR TKIs caused effective tumor shrinkage, but tumors regrew when treatment was stopped. In some instances, tumor regrowth was delayed or prevented when tepotinib was combined with an EGFR TKI. This suggests a potential benefit in continuing c-Met inhibitor therapy if EGFR TKI therapy is withdrawn due to toxicity or perceived lack of benefit. Combination therapy may also be expected to delay the emergence of c-Met-mediated resistance to EGFR TKI.
Tumors with aberrant c-Met exhibited different degrees of sensitivity to single-agent tepotinib, ranging from entirely insensitive to fully sensitive (complete tumor regression). All three models tested were, as expected, resistant to EGFR TKI monotherapy, but were fully sensitive to treatment with an EGFR TKI in combination with tepotinib. This is consistent with aberrant c-Met activity being sufficient to induce resistance to EGFR TKI. Insensitivity to tepotinib and EGFR TKI monotherapies, but sensitivity to combined tepotinib/EGFR TKI therapy in one model, implies redundant oncogenic signaling by EGFR and c-Met, such that signaling by either receptor is sufficient to maintain tumors and both must be inhibited to prevent tumor growth. Conversely, sensitivity to tepotinib monotherapy indicates dependence on c-Met signaling for tumor growth and survival, and that this cannot be maintained by EGFR signaling alone. This supports the conclusion that as initially sensitive tumors become resistant to inhibition of EGFR signaling, they become increasingly dependent on c-Met activity. Although this implies that EGFR TKI-resistant tumors with aberrant c-Met activity might be expected to respond to c-Met inhibitor monotherapy, unchecked EGFR signaling could provide a sustained growth signal. Effective treatment of EGFR TKI-resistant NSCLC due to aberrant c-Met expression is therefore anticipated to require therapy with an EGFR TKI in combination with a c-Met inhibitor to provide simultaneous inhibition of both pathways.
One xenograft model expressed a transgenic EGFR T790M mutant form in addition to an endogenous activating EGFR del19 mutant form and high levels of c-Met. Signaling by either EGFR T790M or c-Met therefore had the potential to confer resistance to first-generation EGFR TKI inhibitors only effective against the EGFR del19 form. This ‘double resistant’ model was fully sensitive only to tepotinib in combination with the third-generation EGFR TKI inhibitor rociletinib, which is effective against EGFR del19 and EGFR T790M mutant forms [6]. This suggests that this combination therapy approach might be necessary in clinical practice to optimize benefit for patients with ‘double resistant’ tumors.
Our findings support the results of previous preclinical research using NSCLC models showing that c-Met inhibitors can effectively overcome resistance to EGFR TKI treatment [20-27]. These studies also highlighted differences between agents that have c-Met inhibitory activity. For example, the growth of a modified PC-9 cell line engineered to express HGF could be inhibited by a combination of erlotinib and the c-Met monoclonal antibody onartuzumab [26]. However, it is unclear whether this antibody would be active in tumors that highly overexpress c-Met, resulting in HGF-independent activation of the c-Met pathway. In another example, the relatively non-specific c-Met inhibitor crizotinib was shown to be able to overcome the resistance of NSCLC xenografts to EGFR TKI when combined with an EGFR TKI, but triggered a number of adverse events. Strong efficacy in the absence of toxicity in our study highlights the potential benefit of selective agents, which can help to avoid toxicity when combination therapies are considered [25].
It is hypothesized that c-Met inhibitors restore tumor sensitivity to EGFR TKIs by preventing the activation of redundant bypass signaling. It has been consistently shown that the phosphorylation of ERK and AKT signaling molecules in EGFR TKI-resistant NSCLC cell lines can be effectively inhibited by a combination of c-Met and EGFR TKIs, but not by either agent alone [20,23]. Activated ERK and AKT are strongly implicated in oncogenesis [28,29]. These observations provide a mechanistic rationale for treating EGFR TKI-resistant NSCLC with a combination of EGFR TKI and c-Met inhibitors.
Interestingly, HGF can induce the development of EGFR TKI-resistant clones in preclinical models, and c-Met inhibitors can delay this process [23,24]. This provides a rationale for considering treating patients with tumors with activating EGFR mutations with an EGFR TKI in combination with a c-Met inhibitor to delay the emergence of resistance.
Our preclinical data indicate that clinical studies of tepotinib with EGFR TKIs in NSCLC are warranted. An ongoing clinical trial of tepotinib in NSCLC is examining its ability to overcome resistance to EGFR TKIs in both c-Met-positive, T790M-negative and c-Met-positive, T790M-positive NSCLC (NCT01982955) [30]. The phase Ib part confirmed that the recommended dose of tepotinib for use in combination with gefitinib 250 mg/d q3w is 500 mg/d and that the combination is well tolerated and has antitumor activity. The phase II part of the study is ongoing and will randomize approximately 156 patients with c-Met-positive, T790M-negative tumors who have failed first-line EGFR TKI therapy to tepotinib 500 mg/d combined with gefitinib or platinim/pemetrexed, while also examining the role of the combination in a cohort of patients with c-Met-positive, T790M-positive NSCLC. This trial will determine whether the promise of combining the c-Met inhibitor with EGFR inhibitors shown in preclinical models translates into clinical benefit for patients with NSCLC. The efficacy and safety of tepotinib 500 mg/d as monotherapy are also being assessed in an ongoing phase II trial in patients with advanced lung adenocarcinoma harboring MET exon 14-skipping mutations and wild-type EGFR (NCT02864992) [31]. In contrast to wild-type c-Met, which has a role in acquired resistance to EGFR TKI, c-Metdel14 produced by MET with exon 14-skipping mutations is a primary oncogenic driver in lung adenocarcinoma. Tumor biomarkers will be used to select patients for these phase II trials of tepotinib to avoid the inclusion of patients with tumors unlikely to respond to c-Met inhibition.
Our preclinical studies provide new evidence that the selective c-Met inhibitor tepotinib in combination with an EGFR TKI is effective in NSCLC xenografts harboring c-Met abnormalities that are resistant to EGFR TKI treatment. Efficacy is observed whether or not the NSCLC xenografts harbor the EGFR T790M mutation and whether tepotinib is combined with first/second-generation or third-generation EGFR TKIs.
Acknowledgements
Friedhelm Bladt’s current affiliation is Experimental Medicine Oncology, Bayer Pharmaceuticals, Berlin, Germany. Uz Stammberger’s current affiliation is Novartis Institutes for BioMedical Research (NIBR), Basel, Switzerland. Medical writing support was provided by Russell Huby, Bioscript Science, Macclesfield, UK, and funded by Merck KGaA, Darmstadt, Germany.
Disclosure of conflict of interest
None.
References
- 1.Asami K, Atagi S. Epidermal growth factor receptor tyrosine kinase inhibitors for non-small cell lung cancer. World J Clin Oncol. 2014;5:646–659. doi: 10.5306/wjco.v5.i4.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5:390–401. doi: 10.1016/j.apsb.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan CS, Cho BC, Soo RA. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor-mutant non-small cell lung cancer. Lung Cancer. 2016;93:59–68. doi: 10.1016/j.lungcan.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Toschi L, Cappuzzo F. Clinical implications of MET gene copy number in lung cancer. Future Oncol. 2010;6:239–247. doi: 10.2217/fon.09.164. [DOI] [PubMed] [Google Scholar]
- 8.Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15:2207–2214. doi: 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- 9.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 10.Lee SJ, Lee J, Sohn I, Mao M, Kai W, Park CK, Lim HY. A survey of c-MET expression and amplification in 287 patients with hepatocellular carcinoma. Anticancer Res. 2013;33:5179–5186. [PubMed] [Google Scholar]
- 11.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 12.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 13.Siegfried JM, Weissfeld LA, Luketich JD, Weyant RJ, Gubish CT, Landreneau RJ. The clinical significance of hepatocyte growth factor for non-small cell lung cancer. Ann Thorac Surg. 1998;66:1915–1918. doi: 10.1016/s0003-4975(98)01165-5. [DOI] [PubMed] [Google Scholar]
- 14.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta MG, Buttitta F, Incarbone M, Toschi L, Finocchiaro G, Destro A, Terracciano L, Roncalli M, Alloisio M, Santoro A, Varella-Garcia M. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009;27:1667–1674. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garajova I, Giovannetti E, Biasco G, Peters GJ. c-Met as a target for personalized therapy. Transl Oncogenomics. 2015;7:13–31. doi: 10.4137/TOG.S30534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 17.Okuma HS, Kondo S. Trends in the development of MET inhibitors for hepatocellular carcinoma. Future Oncol. 2016;12:1275–1286. doi: 10.2217/fon.16.3. [DOI] [PubMed] [Google Scholar]
- 18.Bladt F, Faden B, Friese-Hamim M, Knuehl C, Wilm C, Fittschen C, Gradler U, Meyring M, Dorsch D, Jaehrling F, Pehl U, Stieber F, Schadt O, Blaukat A. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin Cancer Res. 2013;19:2941–2951. doi: 10.1158/1078-0432.CCR-12-3247. [DOI] [PubMed] [Google Scholar]
- 19.Bladt F, Friese-Hamim M, Ihling C, Wilm C, Blaukat A. The c-Met inhibitor MSC2156119J effectively inhibits tumor growth in liver cancer models. Cancers (Basel) 2014;6:1736–1752. doi: 10.3390/cancers6031736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Z, Du R, Jiang S, Wu C, Barkauskas DS, Richey J, Molter J, Lam M, Flask C, Gerson S, Dowlati A, Liu L, Lee Z, Halmos B, Wang Y, Kern JA, Ma PC. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99:911–922. doi: 10.1038/sj.bjc.6604559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krumbach R, Schuler J, Hofmann M, Giesemann T, Fiebig HH, Beckers T. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer. 2011;47:1231–1243. doi: 10.1016/j.ejca.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa T, Takeuchi S, Yamada T, Nanjo S, Ishikawa D, Sano T, Kita K, Nakamura T, Matsumoto K, Suda K, Mitsudomi T, Sekido Y, Uenaka T, Yano S. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol Cancer Ther. 2012;11:2149–2157. doi: 10.1158/1535-7163.MCT-12-0195. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Li Q, Takeuchi S, Yamada T, Koizumi H, Nakamura T, Matsumoto K, Mukaida N, Nishioka Y, Sone S, Nakagawa T, Uenaka T, Yano S. Met kinase inhibitor E7050 reverses three different mechanisms of hepatocyte growth factor-induced tyrosine kinase inhibitor resistance in EGFR mutant lung cancer. Clin Cancer Res. 2012;18:1663–1671. doi: 10.1158/1078-0432.CCR-11-1171. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Zhou JY, Zhao J, Chen JJ, Ma SN, Zhou JY. Crizotinib overcomes hepatocyte growth factor-mediated resistance to gefitinib in EGFR-mutant non-small-cell lung cancer cells. Anticancer Drugs. 2013;24:1039–1046. doi: 10.1097/CAD.0000000000000011. [DOI] [PubMed] [Google Scholar]
- 25.Nanjo S, Yamada T, Nishihara H, Takeuchi S, Sano T, Nakagawa T, Ishikawa D, Zhao L, Ebi H, Yasumoto K, Matsumoto K, Yano S. Ability of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitors. PLoS One. 2013;8:e84700. doi: 10.1371/journal.pone.0084700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sano Y, Hashimoto E, Nakatani N, Abe M, Satoh Y, Sakata K, Fujii T, Fujimoto-Ouchi K, Sugimoto M, Nagahashi S, Aoki M, Motegi H, Sasaki E, Yatabe Y. Combining onartuzumab with erlotinib inhibits growth of non-small cell lung cancer with activating EGFR mutations and HGF overexpression. Mol Cancer Ther. 2015;14:533–541. doi: 10.1158/1535-7163.MCT-14-0456. [DOI] [PubMed] [Google Scholar]
- 27.Kunii E, Ozasa H, Oguri T, Maeno K, Fukuda S, Uemura T, Takakuwa O, Ohkubo H, Takemura M, Niimi A. Reversal of c-MET-mediated resistance to cytotoxic anticancer drugs by a novel c-MET inhibitor TAS-115. Anticancer Res. 2015;35:5241–5247. [PubMed] [Google Scholar]
- 28.Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016;82:943–956. doi: 10.1111/bcp.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu PK, Park JI. MEK1/2 inhibitors: molecular activity and resistance mechanisms. Semin Oncol. 2015;42:849–862. doi: 10.1053/j.seminoncol.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soo RA, Kim DW, Yang JCH, Stammberger U, Xiong H, Ihling C, Wu YL. The highly selective c-Met inhibitor tepotinib in combination with gefitinib is active in Asian patients with c-Met-positive EGFR mutant NSCLC. Eur J Cancer. 2015;51 Abstract 3082. [Google Scholar]
- 31.Paik PK, Stammberger U, Bruns R. A phase II trial investigating the highly selective c-Met inhibitor tepotinib in stage IIIB/IV lung adenocarcinomas with MET exon 14 alterations after failure of at least one prior therapy. European Soc for Med Oncol 41st Congress-ESMO 2016 congress. 2016 Abstract 1292TiP. [Google Scholar]