

Abstract
Till now, no functional studies for FGF-16 in diabetic heart have been reported. Therefore, this study aims to evaluate the potential function of FGF-16 in inhibiting adverse cardiac remodeling in post myocardial infarction (MI) of diabetic heart. We investigated the role of fibroblast growth factor-16 (FGF-16) in post-MI remodeling and role of cardio-protection in the diabetic infarct heart. Adult db/db diabetic mice were assigned to sham group, MI group and MI+FGF-16 group, respectively. MI group was induced by permanent coronary artery ligation, and the mice were subjected to 2D trans-thoracic echocardiography 2-4 weeks post-surgery. The results showed that the infiltration of monocytes, the associated pro-inflammatory cytokines were significantly increased, and the adverse cardiac remodeling and left ventricular dysfunction were observed in MI group. FGF-16 treatment protected against apoptosis, cardiac dysfunction and chamber dilatation post-MI, and decreased monocyte infiltration and cardiomyocyte hypertrophy/apoptosis. Meanwhile, the FGF-16 treatment also attenuated interstitial fibrosis and myocardial inflammation post-MI, increased M2 macrophage differentiation and associated anti-inflammatory cytokines, reduced adverse remodeling, and improved cardiac function. In conclusion, our results suggest that the heart appears to be a target of systemic and possibly locally generated FGF-16, which plays a therapeutic role in cardiac protection in the post-MI diabetic heart.
Keywords: FGF-16, cardiac remodeling, myocardial infarction, diabetes
Introduction
Diabetes mellitus is a metabolic disorder and characterized by hyperglycemia consequent to the body’s inability to either produce any or sufficient quantities of insulin, either of which leads to extensive cardiovascular complications, such as impairment of angiogenesis [1]. Diabetic patients, with chronic uncontrolled glycemic indexes, are at a higher risk for developing secondary disorders, including cardiovascular diseases, retinopathy, nephropathy, and neuropathy [2]. Coronary artery disease is one of the major complications of diabetes mellitus [3]. Furthermore, myocardial ischaemia/infarction is the leading cause of morbidity and mortality in the patients with diabetes mellitus [4].
Fibroblast growth factors (FGFs) are polypeptide growth factors with diverse biological activities [5]. FGFs widely express in developing and adult tissues and play important roles in development and metabolism [6]. FGF-16 is a member of the fibroblast growth factor family. FGF-16 was originally identified in the heart by a homology-based polymerase chain reaction [7]. FGF-16 is a 207 amino acid protein sharing 75% and 62% amino acid sequence similarity to the FGF-9 and FGF-20, respectively. Among postnatal tissues, FGF-16 predominantly expresses in the heart [7,8]. Recent studies showed the function of FGF-16 in cardiac myocyte proliferation and the development of coronary vasculature in the embryonic mouse [9]. However, evidence for the role of FGF-16 in cardiovascular system is also indirect. FGF-16 expression increases in the myocardium at birth where this heparin-binding protein can be produced and released in a glycosylated form, and which could modify the signaling environment [10]. The expression of FGF-16 in the heart correlates with the change in the proliferative potential of cardiac myocytes, which is also suggested to interfere with the proliferation potential of neonatal cardiac myocytes and to affect the cell cycle-related gene expression [11]. Recently, a study reported that the conditional expression of FGF-16 in post-MI heart promotes vascularization and left ventricular (LV) hypertrophy, improves systolic function, and reduces subsequent death [12]. So far, the functions of the FGF-16 in diabetic heart have not been fully reported. The current study aims to evaluate the potential role of FGF-16 in inhibiting adverse cardiac remodeling in the post-MI diabetic heart.
Materials and methods
Experimental model and FGF-16 treatment
Male and female db/db diabetic mice (8-10 weeks old), were divided into 3 groups (n>5 animals/group), including Sham group, MI group and MI+FGF-16 group (1 ng/10 μl 0.1% BSA in 1×PBS). Mice were randomized and anesthetized by using endo-tracheal delivered isoflurane and subjected to a left thoracotomy. The left anterior descending coronary artery was permanently ligated by using a 7.0 polypropylene suture in the MI and MI+FGF-16 groups. The surgery in Sham group involved an identical procedure with the exception of coronary artery ligation. Subsequent analyses were performed up to 4 weeks after surgery. Buprenorphine analgesia (0.05 mg/kg i.m.) was administered prior to and after surgery, as required. To investigate the effects of FGF-16 on post-MI remodeling, MI or sham-operated mice were randomly assigned to be chronically infused with FGF-16. In details, following ligation, the MI+FGF-16 group was injected with FGF-16 into two independent sites (1 ng FGF-16/injection prepared as aforementioned for a total dose of 2 ng FGF-16) in the peri-infarct region using a 29-guage floating needle. For ex vivo analysis of infarct size and gene/protein expression, animals were sacrificed using 4% inhalatory isoflurane for 10 min followed by cervical dislocation, 2 to 4 weeks post-surgery. Hearts from all groups were removed, washed in phosphate buffered saline (PBS), and preserved in formalin solution for future studies. All animal protocols were reviewed and approved by Animal Welfare and Ethical Committee of Shandong University.
Tissue section and infarct size
Excised hearts were initially embedded in paraffin, then sliced into 5 μm serial sections and placed on Colorfrost plus-plus slides. After de-paraffinization and rehydration, heart sections were stained with Masson’s Trichrome to visualize fibrosis (blue area). Infarct area, area at risk and total LV area from each section were measured by using computerized planimetry (Image J), and totaled for all sections. Infarct size was calculated as a percentage of area at risk by measuring (infarct area (mm2)/total area (mm2) ×100. Interstitial fibrosis (IF) was obtained by evaluating the total blue area per mm2 with NIH Image J software. Infarct size (%) and interstitial fibrosis were measured in 2-3 sections (5 images captured per section and calculated data averaged per section) from each animal.
Echocardiography and cardiac function analysis
Three weeks after surgery, mice were anaesthetized with 1.5% isoflurane/oxygen, placed on a warming pad. Transthoracic 2D echocardiography was performed by using the 5500 Ultrasound System. M-mode parasternal short-axis scans of left ventricle at papillary muscle level were used to quantify left ventricular internal dimension-diastole (LVIDd), left ventricular internal dimension-systole (LVIDs), shortening percentage ((LVIDd-LVIDs)/LVIDd×100), left ventricular volume at end diastole (EDV), left ventricular volume at end systole (ESV), and ejection fraction ((EDV-ESV)/EDV×100).
Analysis of post-MI re-modeling
Hearts were weighed and then normalized to total body weight after mice killing. We performed all histological analyses on fixed (10% neutral-buffered formalin), paraffin-embedded LV sections (5 mm). HE staining was used to determine the cardio-myocyte cross-sectional area, followed by further analyzing cells with centrally located nuclei. Picrosirius staining (0.1% w/v) was used to assess the cardiac interstitial fibrosis, excluding coronary vessels and perivascular regions.
Immunohistochemistry
The rabbit anti-mouse CD45 polyclonal antibody (Santa Cruz), rabbit anti-mouse F4/80 polyclonal antibody (Santa Cruz), rat anti-mouse Mac-2 polyclonal antibody (BD Biosciences), rat anti-mouse p-P65 polyclonal antibody (BD Biosciences), rat anti-mouse p-P38 monoclonal antibody (Abcam), rat anti-mouse p-JNK1/2 monoclonal antibody (Abcam), and rat anti-mouse IL-6 monoclonal antibody (Abcam) were used in this study. For immunohistochemistry, we used two-step assay in accordance with PV-9000 kit instruction of the standard immunohistochemical staining, with using PBS as a negative control. Positive cell percentage (%) was quantified by digital image analysis (Image J).
Cardiomyocyte apoptosis
Cardiomyocyte apoptosis was examined by using the TUNEL staining (Roche). TUNEL-positive cardiomyocyte nuclei were counted as the percentage (%) of total nuclei identified by DAPI staining in the same sections, i.e. Apoptotic rate = apoptotic myocardial cells/total number of cardiomyocytes ×100%.
Real-time RT-PCR
Total RNA was extracted from heart homogenates according to the manufacturer’s instructions using TRI reagent RNA isolation reagent (Sigma-Aldrich). RNA was reversely transcribed using iScript select cDNA synthesis kit (Bio-Rad) with random primers, and real-time PCR was carried out using a C1000 Thermal Cycler (Bio-Rad). The mRNA expression of pro-collagen II, pro-collagen III, connective tissue growth factor (CTGF), fibronectin, and TGF-3 were analyzed by using real-time reverse transcription-PCR (RT-PCR) and fluorescent SYBR Green (Bio-Rad). Meanwhile, the β-actin was used as the internal control to normalize the mRNA expression. Relative expression was evaluated by using the comparative CT method correcting for amplification efficiency of the primers. Primer sequences are shown as shown in Table 1.
Table 1.
Primer sequences for real-time RT-PCR assay
| Gene | Primer sequences | |
|---|---|---|
| TNF-α | Forward | 5’-CACAGAAAGCATGATCCGCGACGT-3’ |
| Reverse | 5’-CGGCAGAGAGGAGGTTGACTTTCT-3’ | |
| Procollagen II | Forward | 5’-CCTCAGGGTATTGCTGGACAAC-3’ |
| Reverse | 5’-TTGATCCAGAAGGACCTTGTTTG-3’ | |
| Procollagen III I | Forward | 5’-AGGAGCCAGTGGCCATAATG-3’ |
| Reverse | 5’-TGACCATCTGATCCAGGGTTTC-3’ | |
| CTGF | Forward | 5’-GCTGCCTACCGACTGGAAGAC-3’ |
| Reverse | 5’-CCTAATGGCTTCCACCCTCTTC-3’ | |
| Fibronectin | Forward | 5’-CCGGTGGCTGTCAGTCAGA-3’ |
| Reverse | 5’-CCGTTCCCACTGCTGATTTATC-3’ | |
| TGF-3 | Forward | 5’-GGAGAGAGTCCAACTGGGTCTG-3’ |
| Reverse | 5’-ACATTTTCCAGTATGTCTCCATTGG-3’ | |
| β-actin | Forward | 5’-CGTGAAAAGATGACCCAGATCA-3’ |
| Reverse | 5’-TGGTACGACCAGAGGCATACAG-3’ | |
Western blotting
Western blot analyses were performed by using antibodies against total glycogen synthase kinase 3β GSK3) phospho-GSK3, total Akt, phospho-Akt (Cell Signaling Technology, Inc, Beverly, MA, USA). αα-smooth muscle actin antibody was used at 1:200 to visualize actin myofilament differentiation with DAPI nuclear counterstaining imaged on a fluorescence microscope at 200× magnification (Nikon Eclipse). CTGF mRNA expression was evaluated by using real-time RT-PCR and fluorescent SYBR Green. Meanwhile, the GAPDH was used as the internal control to normalize the protein expression. Primer sequences are shown above.
Statistical analysis
Statistical analysis of data was performed using one-way analysis of variance (ANOVA) followed by the Tukey test. Data is presented as a mean ± SEM with P<0.05 considered statistically significant.
Results
FGF-16 decreases infarct size and fibrosis in post-MI diabetic myocardium
To study the effects of FGF-16 on infarct size and interstitial fibrosis in the post-MI diabetic heart, we performed Masson’s trichrome staining on heart sections in all study groups. As shown in Figures 1, 2, quantitative analysis indicated a significant increase in infarcted myocardium in the MI group relative to the sham group (P<0.05, Figure 1A, 1B). However, when administered FGF-16 post-MI, infarct size was significantly decreased compared to the MI group (P<0.05, Figure 1A, 1B). Interstitial fibrotic area, quantified in the left ventricular myocardium by direct measurement of the blue area by using Image J software, was significantly elevated in the MI group relative to the sham group (P<0.05, Figure 1A, 1B). Markedly, the interstitial fibrotic region was significantly reduced following FGF-16 treatment (P<0.05, Figure 1A, 1B). Collectively, our data suggested that treatment with FGF-16 reduced infarct size as well as inhibited fibrosis formation in the post-MI diabetic myocardium.
Figure 1.
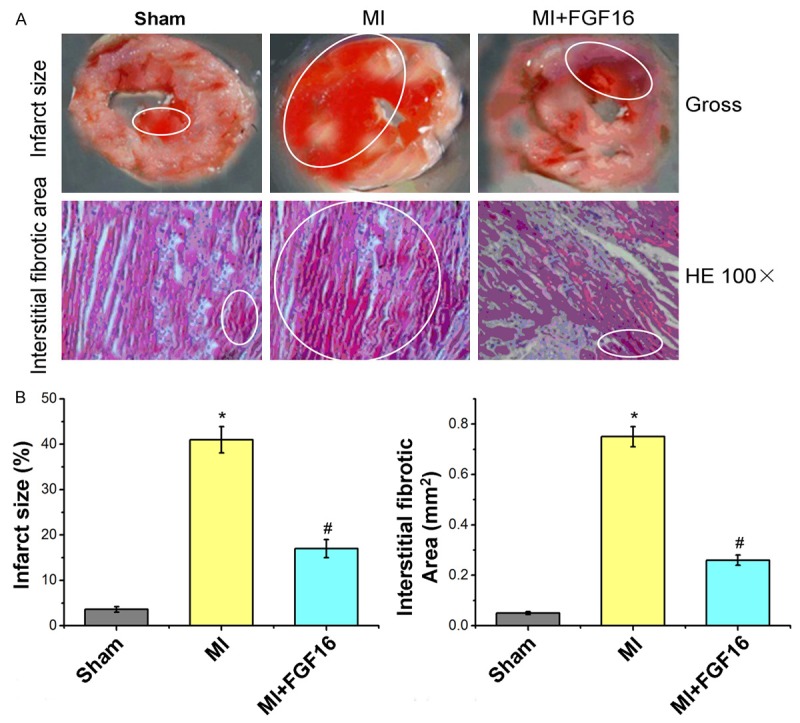
Effect of FGF-16 on infarct size and interstitial fibrotic areas after MI. A. Images for infract size areas and interstitial fibrotic areas (%; n=15); B. Statistical analysis for infarct size and Interstitial fibrotic area (mm2; n=15). P<0.05 represents the infarct size or interstitial fibrotic areas in MI group compared to Sham group or in MI+FGF16 group compared to MI group. *P<0.05 represents infarct size or interstitial fibrotic area in MI group compared to Sham group. #P<0.05 represents infarct size or interstitial fibrotic area in MI+GFG16 compared to MI group.
Figure 2.
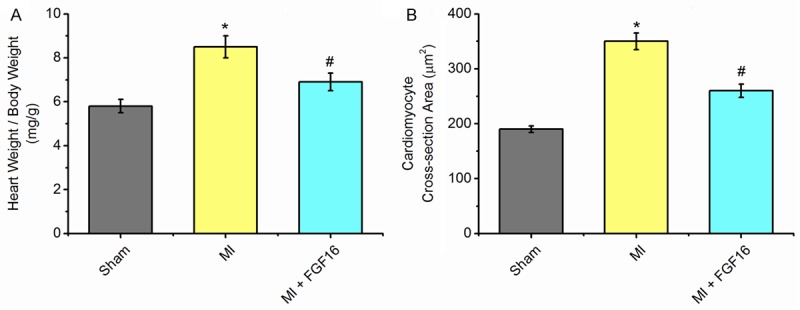
Effect of FGF-16 on heart weight/body weight and cardiomyocyte cross-sectional areas after MI. A. Heart weight/ body weight (HW/BW; n=15), B. Cardiomyocyte cross-sectional area (mm2; n=15). P<0.05 represents the heart weight/body weight or cardiomyocyte cross-sectional areas in MI group compared to Sham group or in MI+FGF16 group compared to MI group. *P<0.05 represents the values in MI group compared to Sham group. #P<0.05 represents values in MI+GFG16 compared to MI group.
Effect of FGF-16 on cardiomyocyte remodeling
FGF-16 treatment reduced MI-induced myocardial hypertrophy, as indicated by heart/body weight ratio and cardiomyocyte cross-section area (Figure 2A, 2B). Quantitative analysis indicated a significant up-regulation of heart/body weight ratio and cardiomyocyte area following MI relative to the sham group (P<0.05, Figure 2A, 2B). Conversely, heart/body weight ratio and cardiomyocyte area were significantly reduced upon FGF-16 treatment compared to the MI group (P<0.05, Figure 2A, 2B).
Effect of FGF-16 on extra-cellular matrix remodeling
As shown in Figure 1, interstitial fibrotic area was significantly decreased in FGF-16-treated MI mice compared to MI controls. We further quantify the mRNA expression of cardiomyocyte genes by real-time RT-PCR as shown in Figure 3. Consistent with cardiomyocyte remodeling data, MI caused the significant increases of several key extra-cellular matrix genes, such as TNF-α, procollagen II, procollagen IIII, CTGF, fibronectin and TGF-3, and which were significantly reduced in MI+FGF-16 groups compared to the MI groups (Figure 3A-F).
Figure 3.
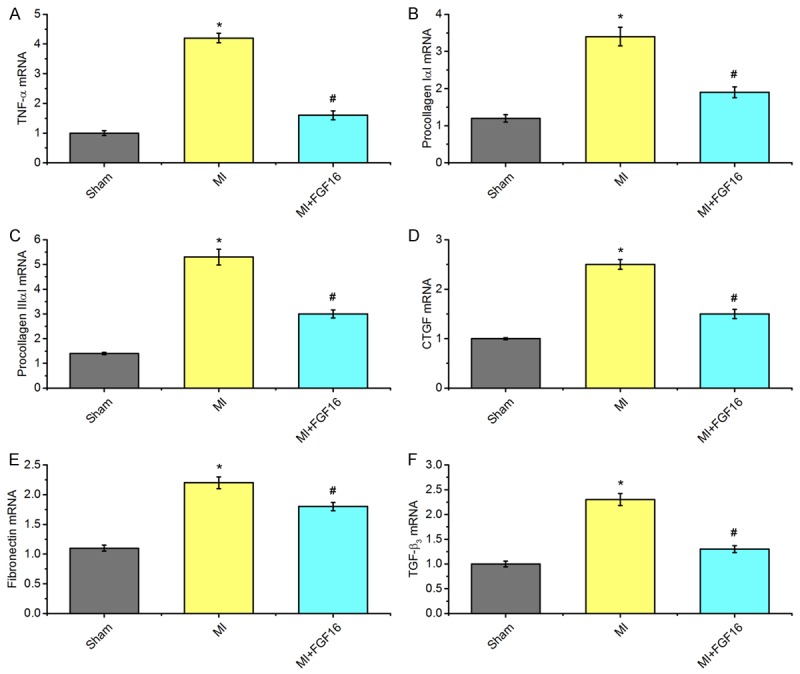
Effect of FGF-16 on cardiomyocyte remodelling after MI. A-F. quantification data of mRNA expression of cardiomyocyte genes by real-time RT-PCR (n=15). P<0.05 represents the gene levels in MI group compared to Sham group or in MI+FGF16 group compared to MI group. *P<0.05 represents the mRNA levels in MI group compared to Sham group. #P<0.05 represents mRNA levels in MI+GFG16 compared to MI group.
FGF-16 prevents myocardial cell injury and inflammation
After HE staining, myocardial fibers were arranged in rows in sham group without myocardial interstitial inflammatory cell infiltration. However, in MI group, myocardial cell swelling was seen in myocardial infarct border zone fiber, a qualitative as well as a large number of small perivascular inflammatory cell infiltration were disordered, and visible focal necrosis was also observed. In MI + FGF16 group, infarct border zone of myocardial damage became less significant, and there were significant differences (P<0.05) compared with the MI group (Figure 4). Myocardial infiltration of CD45-positive leukocytes and F4/80-positive macrophages after MI was significantly prevented by FGF-16 treatment (Figure 4A, 4B). CD45+cell number declined from 496 ± 42 cells/mm2 in MI group to 248 ± 29 cells/mm2 in MI+FGF-19 group (Figure 4A). And F4/80+cells decreased from 496 ± 42 cells/mm2 in MI group to 248 ± 29 cells/mm2 in MI+FGF-19 group (Figure 4B). Consistent with this, there are a lot of Mac-2 positive cell infiltration in the infarcted area and the border area in MI group. In sham group, 151.2 ± 16.3 cells/mm2 positive cells was found. In MI group, Mac-2 positive cells density was 456.6 ± 36.2 cells/mm2, while in the MI+FGF16 group, the density of Mac-2 positive cells was 212.5 ± 15.9 cells/mm2, and the difference was statistically significant (P<0.01, Figure 4C). Experimental results showed that in terms of quality and peri-vascular myocardial infarction junction region, 70.3 ± 3.2 cells/mm2 in sham group were IL-6 positive. In MI group, the cell density was (248.7 ± 26.5 cells/mm2) significantly decreased compared to the MI+FGF16 group with IL-6 positive cells (cell density: 103.5 ± 6.6 cells/mm2) (P<0.01, Figure 4D).
Figure 4.
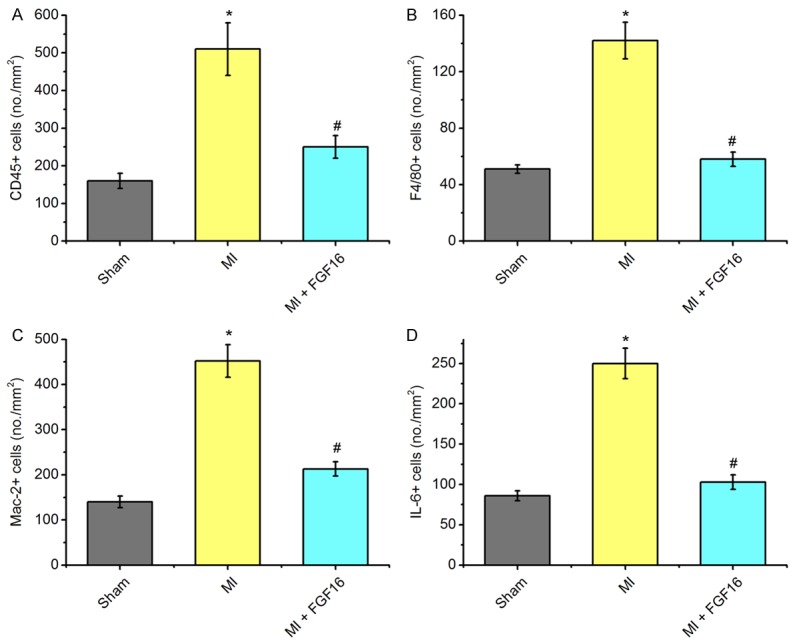
Effect of FGF-16 on myocardial inflammation after MI. A-D. Quantification data CD45, F4/80, Mac-2, and IL-6 staining to assess leukocyte and macrophage infiltration from sham, MI, and MI+FGF16 groups (n=15). P<0.05 represents the cell levels in MI group compared to Sham group or in MI+FGF16 group compared to MI group. *P<0.05 represents the values in MI group compared to Sham group. #P<0.05 represents values in MI+GFG16 compared to MI group.
FGF-16 involves in signaling pathways related to MI inflammation and apoptosis
According to the TUNEL staining results, we found that the TUNEL positive cells in FGF-16 treated group was lower significantly compared to the MI group (Figure 5A, P<0.05). From immunohistochemical staining, we found that the phosphorylated NF-B p65, phosphorylated JNK1/2, and phosphorylated p38 MAPK proteins mainly locate in the cell nucleus or cytoplasm. From the quantification of phosphorylated NF-B p65 staining, there were 151.2 ± 6.3 cells/mm2 in sham group, 496.6 ± 36.2 cells/mm2 in MI group, and 192.7 ± 12.9 cells/mm2 in MI+FGF16 group (Figure 5B). For phosphorylated p38 MAPK staining, there were 236.4 ± 16.5 cells/mm2 in sham group, 909.1 ± 58.7 cells/mm2 in MI group, and 510.9 ± 40.1 cells/mm2 in MI+FGF16 group (Figure 5C). For phosphorylated JNK1/2 staining, there were 123.5 ± 7.5 cells/mm2 in sham group, 493.1 ± 28.6 cells/mm2 in MI group, and 150.4 ± 10.8 cells/mm2 in MI+FGF16 group (Figure 5D). The difference was statistical significant in all tests (P<0.01).
Figure 5.
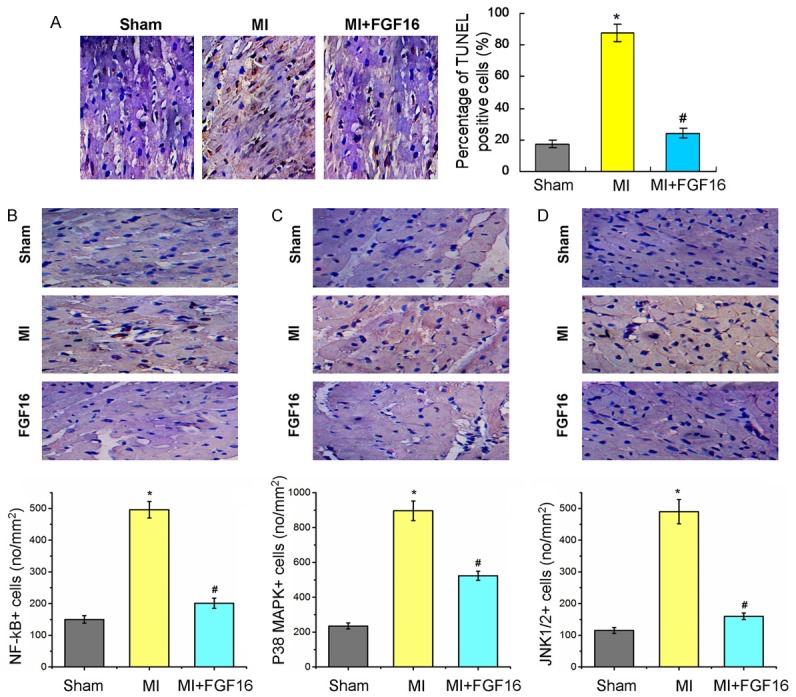
Quantification analysis of TUNEL assay for apoptosis and immunohistochemical staining showing effect of FGF-16 on NF-B p65, p38 MAPK, and JNK1/2 in post-MI diabetic myocardium. (A) TUNEL staining and statistical analysis for the apoptosis cells. The levels of phosphorylated NF-κB p65 (B), p38 MAPK (C), and JNK1/2 (D) in the MI+FGF16 group are significantly reduced compared to those in the MI group (n=15). P<0.05 represents the cytokine levels in MI group compared to Sham group or in MI+FGF16 group compared to MI group. *P<0.05 represents the values in MI group compared to Sham group. #P<0.05 represents values in MI+GFG16 compared to MI group.
Study of potential cardio-protective signaling mechanisms
We next investigated possible candidate pathways which mediate the protective effects of FGF-16 on post-MI remodeling. Phosphorylation of the pro-survival kinase Akt was significantly increased by FGF-16 treatment in post-MI diabetic myocardium (Figure 6A), in association with a significant increase in total Akt protein expression (Figure 6B). Furthermore, both phosphorylated and total GSK-3 were significantly increased undergoing the FGF-16 treatment (Figure 7A, 7B), which suggests that the FGF-16 treatment deactivates the GSK-3 signaling in post-MI diabetic myocardium.
Figure 6.
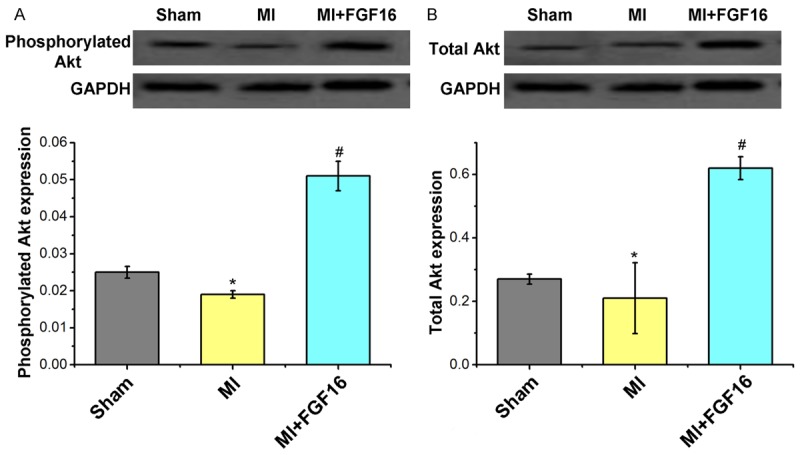
Effect of FGF-16 on Akt expression in post-MI diabetic myocardium. A. Western blot assay for the phosphorylated Akt expression and the statistical analysis. B. Western blot assay for total Akt expression and the statistical analysis. P<0.05 represents the protein levels in MI group compared to Sham group or in MI+FGF16 group compared to MI group. *P<0.05 represents the protein expression in MI group compared to Sham group. #P<0.05 represents protein expression in MI+GFG16 compared to MI group.
Figure 7.
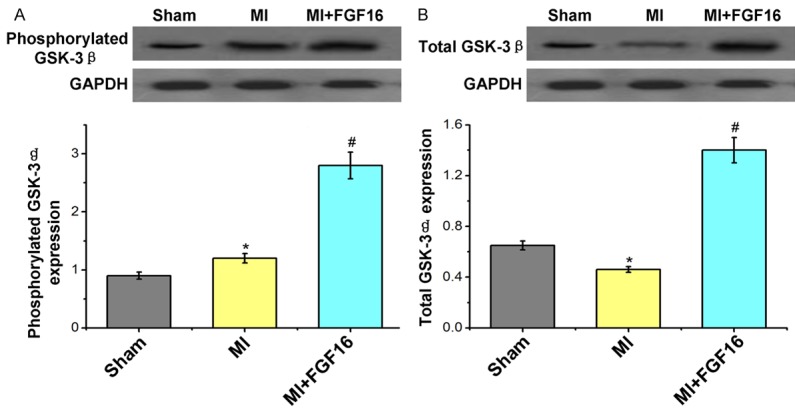
Effect of FGF-16 on GSK-3β expression in post-MI diabetic myocardium. A. Western blot assay for the phosphorylated GSK-3β expression and the statistical analysis. B. Western blot assay for the total GSK-3β expression and the statistical analysis. P<0.05 represents the protein levels in MI group compared to Sham group or in MI+FGF16 group compared to MI group. *P<0.05 represents the protein expression in MI group compared to Sham group. #P<0.05 represents protein expression in MI+GFG16 compared to MI group.
Exogenous FGF-16 improves cardiac function in post-MI diabetic mice
To determine the effect of FGF-16 on left ventricular function post-MI, we performed 2D transthoracic echocardiography on both control and experimental mice at 3 weeks post MI. Data were analyzed and quantified as shown in Figure 8. Three weeks post-MI in MI group, LVIDd, LVIDs, EDV and ESV were significantly increased (n=15, P<0.05, Figure 8A, 8B, 8D, 8F, respectively), whereas shortening percentage and ejection percentage were significantly decreased compared to the sham mice (P<0.05, Figure 6C, 6F, resepctively). Notably, LVIDd (Figure 8A), LVIDs (Figure 8B), EDV (Figure 8D), and ESV (Figure 8E) were significantly decreased (n=15, P<0.05). However, shortening percentage (Figure 8C) and ejection percentage (Figure 8F) were significantly increased (n=15, P<0.05) in the FGF-16 treated mice compared to the MI alone mice. The echocardiographic data suggests that FGF-16 preserves systolic and diastolic function and protects the diabetic heart from cardiac dysfunction after MI.
Figure 8.
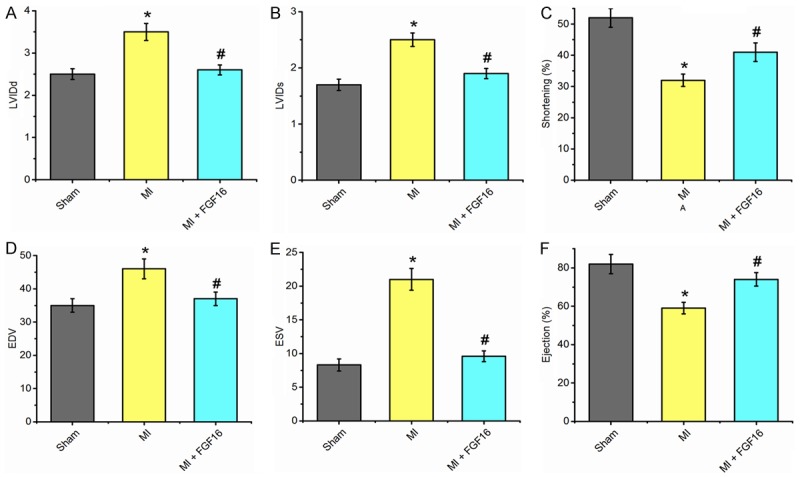
Exogenous FGF-16 Treatment Improves Cardiac Function in post-MI diabetic myocardium. Quantitative analyses are shown for (A) left ventricularinternal dimension-diastole (LVIDd); (B) left ventricular internal dimension-systole (LVIDs); (C) shortening percentage (%); (D) left ventricular volume at enddiastole (EDV), (E) left ventricular volume at end systole (ESV), and (F) ejection percentage (%). (n=15, P<0.05). *P<0.05 represents the values in MI group compared to Sham group. #P<0.05 represents values in MI+GFG16 compared to MI group.
Discussion
Although the expression profile of FGF-16 and the activity of recombinant FGF-16 in vitro suggest potential roles for FGF-16 in the heart, the actual roles of FGF-16 in vivo remain unclear. Myocardial infarction (MI) is associated with adverse cardiac remodeling with an attempt to rescue endogenous myocardial architecture with consequential loss of normal left ventricular function [13]. It is well known that the infarct size in diabetic hearts is larger compared to non-diabetic hearts [14]. Moreover, myocardial protection by preconditioning and post-conditioning is hampered in diabetic animals [15]. So far, no studies have been reported concerning the function of FGF-16 in the diabetic post-MI heart. Here, the results showed that FGF-16 limits infarct size in diabetic hearts for the first time. Following the MI and subsequent loss of cardiac cell types, the fibrosis formation processes are triggered to rebuild and restore infarcted tissue and maintain the architectural integrity of the myocardium. Fibrosis leads to an infarct scar, which lacks contractile capabilities with consequential loss of normal left ventricular function. Two weeks following permanent coronary artery ligation, cardiac myocytes in the infarcted myocardium appeared dystrophic, enlarged, and hypertropic in nature compared to those in sham operated animals. Treatment with ex vivo FGF-16 significantly reduced the infarcted and interstital fibrotic area, and showed a significant decrease in interstitial fibrosis post-MI after FGF-16 treatment. The myocardial filtrate is mainly comprised of monocytes and their macrophage descendants. Monocytic infiltration was significantly increased in hearts of diabetic MI mice, as evidenced by CD45, Mac-2, and IL-6 staining. Our data showed that monocyte infiltration was dramatically reduced after treated with FGF-16, which suggests that FGF-16 plays a role in the inflammatory response following MI by inhibiting infiltration of cardiac monocytic populations. FGF-16 decreased cardiomyocyte hypertrophy/apoptosis but significantly attenuated interstitial fibrosis and myocardial inflammation post-MI. It was associated with altered extra-cellular matrix (procollagen II/IIII, connective tissue growth factor, fibronectin (TGF-3) and inflammatory (IL-6) gene expression in FGF-16 treated mice, together with modulation of both Akt/GSK-3 signaling. The above results suggest that FGF-16 protects the diabetic heart from post-MI remodeling by directing the differentiation of monocytes into M2 macrophages and ultimately minimizing the adverse endogenous inflammatory response. Next, we found that FGF-16 treatment significantly improved left ventricular function as evidenced by enhanced shortening percentage and ejection percentage compared to the MI group, which suggests that FGF-16 treatment provides cardio-protection both structurally and functionally by minimizing adverse inflammatory-induced cardiac remodeling in the diabetic heart.
In conclusions, our data showed that there was a significant increase in infarct size in the diabetic heart at 2-4 weeks post-MI. After the FGF-16 treatment, we found that FGF-16 reduces infarct size and interstitial fibrosis, reduces myocardial monocyte infiltration, and significantly decreases damaged cell populations and pro-inflammatory cytokine expression. Furthermore, the FGF-16 also improves cardiac function in post-MI diabetic mice. All of which suggests that FGF-16 may play a therapeutic role in decreasing and preventing the post-MI adverse remodeling. Collectively, our data suggests that FGF-16 is responsible to the cardio-protective potential in the post-MI diabetic heart.
Disclosure of conflict of interest
None.
References
- 1.Jiao Y, Zhu M, Mao X, Long M, Du X, Wu Y, Abudureyimu K, Zhang C, Wang Y, Tao Y, Luo X, Li L. MicroRNA-130a expression is decreased in Xinjiang uygur patients with type 2 diabetes mellitus. Am J Transl Res. 2015;7:1984–1991. [PMC free article] [PubMed] [Google Scholar]
- 2.Ghaderian SB, Hayati F, Shayanpour S, Beladi Mousavi SS. Diabetes and end-stage renal disease; a review article on new concepts. J Renal Inj Prev. 2015;4:28–33. doi: 10.12861/jrip.2015.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan IA. Coronary artery disease and diabetes-management during ramadan. J Pak Med Assoc. 2015;65(Suppl):S62–64. [PubMed] [Google Scholar]
- 4.Rahim MA, Rahim ZH, Ahmad WA, Hashim OH. Can saliva proteins be used to predict the onset of acute myocardial infarction among high-risk patients? Int J Med Sci. 2015;12:329–335. doi: 10.7150/ijms.11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seeger MA, Paller AS. The roles of growth factors in keratinocyte migration. Adv Wound Care (New Rochelle) 2015;4:213–224. doi: 10.1089/wound.2014.0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu JJ, Foo JP, Liu S, Lim SC. The role of fibroblast growth factor 21 in diabetes and its complications: a review from clinical perspective. Diabetes Res Clin Pract. 2015;108:382–389. doi: 10.1016/j.diabres.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 7.Miyake A, Konishi M, Martin FH, Hernday NA, Ozaki K, Yamamoto S, Mikami T, Arakawa T, Itoh N. Structure and expression of a novel member, FGF-16, on the fibroblast growth factor family. Biochem Biophys Res Commun. 1998;243:148–152. doi: 10.1006/bbrc.1998.8073. [DOI] [PubMed] [Google Scholar]
- 8.Sontag DP, Cattini PA. Cloning and bacterial expression of postnatal mouse heart FGF-16. Mol Cell Biochem. 2003;242:65–70. [PubMed] [Google Scholar]
- 9.Hotta Y, Sasaki S, Konishi M, Kinoshita H, Kuwahara K, Nakao K, Itoh N. Fgf16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev Dyn. 2008;237:2947–2954. doi: 10.1002/dvdy.21726. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Sontag D, Cattini PA. Heart-specific expression of FGF-16 and a potential role in postnatal cardioprotection. Cytokine Growth Factor Rev. 2015;26:59–66. doi: 10.1016/j.cytogfr.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Sofronescu AG, Detillieux KA, Cattini PA. FGF-16 is a target for adrenergic stimulation through NF-kappaB activation in postnatal cardiac cells and adult mouse heart. Cardiovasc Res. 2010;87:102–110. doi: 10.1093/cvr/cvq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sontag DP, Wang J, Kardami E, Cattini PA. FGF-2 and FGF-16 protect isolated perfused mouse hearts from acute doxorubicin-induced contractile dysfunction. Cardiovasc Toxicol. 2013;13:244–253. doi: 10.1007/s12012-013-9203-5. [DOI] [PubMed] [Google Scholar]
- 13.Xu D, Wang A, Jiang F, Hu J, Zhang X. Effects of interleukin-37 on cardiac function after myocardial infarction in mice. Int J Clin Exp Pathol. 2015;8:5247–5251. [PMC free article] [PubMed] [Google Scholar]
- 14.Wider J, Przyklenk K. Ischemic conditioning: the challenge of protecting the diabetic heart. Cardiovasc Diagn Ther. 2014;4:383–396. doi: 10.3978/j.issn.2223-3652.2014.10.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin X, Zheng Y, Zhai X, Zhao X, Cai L. Diabetic inhibition of preconditioning- and postconditioning-mediated myocardial protection against ischemia/reperfusion injury. Exp Diabetes Res. 2012;2012:198048. doi: 10.1155/2012/198048. [DOI] [PMC free article] [PubMed] [Google Scholar]