

Abstract
This study tested the hypothesis that erythropoietin (EPO) and cyclosporine (CsA) could effectively reduce brain infarct area (BIA) in rat after acute ischemic stroke (AIS) through regulating inflammation, oxidative stress, MAPK family signaling and microRNA (miR-223/miR-30a/miR-383). Adult male Sprague-Dawley rats (n = 48) were equally divided into group 1 (sham control), group 2 (AIS), group 3 [AIS+EPO (5,000 IU/kg at 0.5/24/48 h, subcutaneous)] and group 4 [AIS+CsA (20.0 mg/kg at 0.5/24/48 h, intra-peritoneal)]. By 72 h, histopathology showed that BIA was largest in group 2 and smallest in group 1, and significantly larger in group 4 than group 3 (all P<0.0001). The three microRNAs expressed were higher in group 2 than in the other three groups (all P<0.04); between these three latter groups there were no significant differences. The protein expressions of MAPK family [phosphorylated (p)-ERK1/2, p-p38/p-JNK], inflammatory (iNOS/MMP-9/TNF-α/NF-κB/IL-12/MIP-1α/CD14/CD68/Ly6g), apoptotic (caspase-3/PARP/mitochondrial-Bax), oxidative-stress (NOX-1/NOX-2/oxidized protein) and mitochondrial-damaged (cytosolic cytochrome-C) biomarkers exhibited an identical pattern to BIA findings (all P<0.0001). The cellular expressions of brain edema (AQP4+), inflammation (CD11+/glial-fibrillary-acid protein+), and cellular damage (TUNEL assay/positive Periodic acid-Schiff stain) biomarkers exhibited an identical pattern, whereas the cellular-integrity markers (neuN+/MAP2+/doublecorin+) exhibited an opposite pattern to BIA (all P value <0.001). EPO-CsA therapy markedly reduced BIA mainly by suppressing the innate immune response to inflammation, oxidative stress, microRNAs (miR-223/miR-30a/miR-383) and MAPK family signaling.
Keywords: Acute ischemic stroke, erythropoietin, cyclosporine, inflammation, oxidative stress, microRNAs, MAPK signaling
Introduction
Despite decades of research increasing our knowledge regarding acute ischemic stroke (IS) incidence, epidemiology, etiologies, prognoses, classification, diagnostics and pharmacological refinements [1-13], the morbidity and residual severe disability following IS have remained largely unchanged over this time period [1,2,14].
It is well established that acute organ ischemia/necrosis elicits an acute and rigorous inflammatory reaction [15-19]. It is known that the nonspecific innate immune reaction responds rapidly to inflammatory stimulation [20-22]. Inflammation and the innate immune response are closely related and can act together to cause organ damage in the setting of acute organ ischemia/tissue necrosis [20-22]. Numerous studies [18,23-27] have demonstrated that acute IS elicits a vigorous inflammatory reaction, augments the generation of cytokines and oxidative stress, and initiates the complement cascade. These factors, in turn, further aggravate brain damage after acute IS [18,25-27], causing irreversible and neurological sequelae [18,25-27]. However, the role of innate immune reaction in response to inflammatory stimulation in the setting of acute IS has been not been reported.
The Mitogen-activated protein kinase (MAPK)signaling pathway is activated in response to environmental and oxidative stresses, cellular proliferation and apoptosis, as well as inflammation and cytokine stimulations [21,28-31]. Additionally, it has recently been identified that miR-223 participates directly in regulating the inflammatory response after acute spinal cord injury in rat [32]. Furthermore, miR-30a has recently been found to play an important role in regulating post-SAH cerebrovascular changes [33]. These findings implicate that micro-RNAs may play a crucial role during the initiation and propagation of brain damage. However, how inflammation, the innate immune reaction and MAPK signaling work together, and whether micro-RNAs may also act critically in the pathogenic aftermath of acute IS have not been investigated.
Studies have previously shown that erythropoietin (EPO) therapy alleviated ischemia-related organ dysfunction through anti-ischemic and cellular protective effects [34-37]. Our experimental study has previously further shown that EPO therapy remarkably reduced brain infarct size and improved neurological impairment in a rat acute IS model mainly through inhibiting the inflammatory response and cellular-molecular perturbations [27]. Additionally, our and other experimental studies have revealed that cyclosporine A (CsA) therapy significantly protected the organ from ischemia-reperfusion and permanent ischemic injury mainly though inhibiting mitochondrial permeability transition pores (mPTP)and via an immunosuppressive effect [16,18,27,38-40].
Accordingly, based on the above, the present study used a rat acute IS model to assess IS-elicited activations of innate immune/MAPK/microRNAs signaling and particularly attempted to determine the role of EPO-CsA in inhibiting inflammation and oxidative stress as well as preserving brain integrity.
Materials and methods
Ethics
All animal experimental protocols and procedures were approved by the Institute of Animal Care and Use Committee at Kaohsiung Chang Gung Memorial Hospital (Affidavit of Approval of Animal Use Protocol No. 2010092302) and performed in accordance with the Guide for the Care and Use of Laboratory Animals [The Eighth Edition of the Guide for the Care and Use of Laboratory Animals (NRC 2011)].
Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-approved animal facility in our hospital (IACUC protocol no. 101008) with controlled temperature and light cycles (24°C and 12/12 light cycles).
Experimental model of acute ischemic stroke
The protocol and procedure of the rodent model of acute ischemic stroke (IS) have previously been described in detail [18,27,41,42]. Adult male Sprague-Dawley rats, weighing 325-350 g, were utilized (Charles River Technology, BioLASCOTaiwan Co., Ltd., Taiwan). Each animal was anesthetized by 2% inhalational isoflurane in a supine position on a warming pad (37°C). After exposure of the left common carotid artery (LCCA) through a transverse neck incision, a small incision was made on the LCCA through which a nylon filament (0.28 mm in diameter) was carefully advanced into the distal left internal carotid artery for occlusion of the left middle cerebral artery (LMCA) to cause brain ischemia and infarction of its supplied area. The nylon filament was removed 50 min after occlusion, followed by closure of the muscle and skin in layers. The rats were then placed in a portable animal intensive care unit (ThermoCare®) for 24 hours.
Animal grouping and the treatment protocol
Forty-eight adult male Sprague-Dawley rats were equally divided into group 1 [sham control (SC), i.e., by cutting open the neck skin and exploring the LCCA only], group 2 (IS only), group 3 [IS+EPO (5,000 IU/kg at 0.5/24/48 h, subcutaneous)] and group 4 [IS+CsA (20.0 mg/kg at 0.5/24/48 h, intra-peritoneal)].
The dosages of EPO and cyclosporine to be utilized in the current study were based on our previous report [27,42].
Specimen collection and preparation for individual study
For examination of protein expression, animals in all groups (n = 6) were sacrificed on day 3 after brain acute IS procedure, and the brain of each rat was promptly removed, immersed in cold saline, snap-frozen in liquid nitrogen and then stored at -80°C for individual study. For immunofluorescent (IF) and immunohistochemical (IHC) staining studies, the brains of 6 other animals in each group were reperfused with normal saline via the carotid artery, removed, fixed with 4% paraformaldehyde in 1×PBS (pH7.4), and soaked in 20% sucrose in 1×PBS (freshly prepared) until the brain took on a completely sunken appearance. The sucrose was then discarded and the brainsoaked in 30% sucrose in 1×PBS (freshly prepared) for 48 h. The infarcted and non-infarcted parts were then collected. Finally, the OCT block (Tissue-Tek, Sakura, Netherlands) was prepared for IHC and IF staining.
Measurement of brain infarct area (BIA)
The brain was immediately removed from each rat following anesthetic overdose. Repeated flushing of the carotid artery with normal saline to washout the red blood cells was performed immediately after brain removal. To evaluate the impact of CsA and EPO treatment on brain infarction, three coronal sections (1 cm in thickness) of the brain were obtained from six animals in each group. Each cross section was then stained with 2% 3,5-Triphenyl-2H-Tetrazolium Chloride (TTC) (Alfa Aesar) for BIA analysis as previously described [41,42]. Briefly, all brain sections were placed on a tray with a scaled vertical bar to which a digital camera was attached. The sections were photographed from directly above at a fixed height. The images obtained were then analyzed using Image Tool 3 (IT3) image analysis software (University of Texas, Health Science Center, San Antonio, UTHSCSA; Image Tool for Windows, Version 3.0, USA). The BIA was identified as either whitish or pale yellowish regions and confirmed by microscopic examination. The percentages of infarct area were then calculated by dividing the area with total cross-sectional area of the brain.
IF and IHC staining of brain specimens
The procedure and protocol of IF and IHC were based on our previous reports [18,27,41,42]. In detail, frozen sections (4 µm thick) were obtained from BIA of each animal, permeated with 0.5% Triton X-100, and incubated with antibodies against NeuN (1:1000, Millipore, Billerica, MA, USA), glial fibrillary acid protein (GFAP; 1:500, DAKO, Carpinteria, CA, USA), aquaporin4 (AQP4; 1:200, Abcam, Cambridge, MA, USA), CXCR4 (1:100, Abcam, Cambridge, MA, USA), CD11 (1:200, Abcam, Cambridge, MA, USA), CD68 (1:100, Abcam, Cambridge, MA, USA), microtubule associated protein 2 (MAP-2) (1:400, Abcam, Cambridge, MA, USA), periodic acid-Schiff (PAS) (ScyTekLaboratories, Logan, UT, USA) and dobulecorin (1:200, Santa cruz, Texas, USA), at 4°C overnight. Alexa Fluor488, Alexa Fluor568, or Alexa Fluor594-conjugated goat anti-mouse or rabbit IgG were used to localize signals. Sections were finally counterstained with DAPI and observed with a fluorescent microscope equipped with epifluorescence (Olympus IX-40).
Three brain sections were analyzed for each rat. For quantification, three randomly selected high-power fields (HPFs; ×400 for IHC and IF studies) were analyzed in each section. The mean number of positively-stained cells per HPF for each animal was then determined by summation of all numbers divided by 9.
Western blot analysis of brain specimens
The procedure and protocol of Western blot were based on our previous reports [18,27,41,42]. Equal amounts (50 μg) of protein extracts were loaded and separated by SDS-PAGE using 12% acrylamide gradients. After electrophoresis, the separated proteins were transferred electrophoretically to a polyvinylidene difluoride (PVDF) membrane (Amersham Biosciences, Amersham, UK). Nonspecific sites were blocked by incubation of the membrane in blocking buffer [5% nonfat dry milk in T-TBS (TBS containing 0.05% Tween 20)] overnight. The membranes were incubated with monoclonal antibodies against NOX-1 (1:1500, Sigma-Aldrich), NOX-2 (1:500, Sigma-Aldrich), matrix metalloproteinase (MMP)-9 (1:2000, Abcam), tumor necrosis factor alpha (TNF-α) (1:1000, Cell Signaling), inducible nitric oxide synthase (iNOS) (1:200, Abcam), Cytochrome C (1:2000, BD Bioscience, San Jose, CA, USA), Aquaporin 4 (1:750, Abcam), nuclear factor (NF)-κB (1:1000, Abcam), JNK (1:500, Abcam), phosphorylated (p)-JNK (1:1000, Abcam), P38 (1:10000, Sigma-Aldrich), p-p38 (1:1000, Cell Signaling), Bax (1:1000, Abcam), Poly (ADP-ribose) polymerase (PARP) (1:1000, Cell Signaling), Akt (1:1000, Cell Signaling), p-Akt (1:1000, Cell Signaling), ERK1/2 (1:1000, Calbiochem), p-ERK1/2 (1:1000, Calbiochem), CD14 (1:1000, Abcam), CD68 (1:500, Abcam), Ly6g (1:1000, Abcam), interleukin (IL)-12 (1:500, Abcam), macrophage inflammatory protein (MIP)-1α (1:2000, Abcam), Caspase 3 (1:1000, Cell Signaling) for 1 hr at room temperature. Horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin IgG (1:2000, Cell Signaling) was used as a second antibody for 1 hr at room temperature. The washing procedure was repeated eight times within 1 h, and immunoreactive bands were visualized by enhanced chemiluminescence (ECL; Amersham Biosciences) and exposure to Medical X-ray film (FUJI). For quantification, ECL signals were digitized using Labwork software (UVP, Waltham, MA, USA). A standard control sample was loaded on each gel.
MicroRNAs extraction and quantification
Total RNA were extracted from the brain of each animal using the miRNeasy kit (Qiagen) following the protocol of the manufacturer. For mature miRNA quantification, cDNA were generated with reverse transcription performed by the miScript II RT kit (Qiagen) and cDNA were further utilized as a template for real-time PCR. Expressions of rodent mature miR-223, miR-30a and miR-383 were quantified by miScript SYBR Green PCR assay (Qiagen) and normalized by small nucleolar RNA, RNU6 (Qiagen). Triplicate assays were performed for each sample on Step One-Plus machine (ABI).
TUNEL assay for apoptotic nuclei
For each rat, six sections of BIA were analyzed by an in situ Cell Death Detection Kit, AP (Roche) according to the manufacturer’s guidelines. Three randomly chosen high-power fields (HPFs) (×400) were observed for terminal deoxynucleotidyl transferase-mediated 2’-deoxyuridine 5’-triphosphate nick-end labeling (TUNEL)-positive cells for each section. The mean number of apoptotic nuclei per HPF for each animal was obtained by dividing the total number of cells by 18.
Assessment of oxidative stress
The procedure and protocol for evaluating the protein expression of oxidative stress have been described in detail in our previous reports [16,18,19]. The Oxyblot Oxidized Protein Detection Kit was purchased from Chemicon, Billerica, MA, USA (S7150). DNPH derivatization was carried out on 6 μg of protein for 15 minutes according to the manufacturer’s instructions. One-dimensional electrophoresis was carried out on 12% SDS/polyacrylamide gel after DNPH derivatization. Proteins were transferred to nitrocellulose membranes that were then incubated in the primary antibody solution (anti-DNP 1:150) for 2 hours, followed by incubation in secondary antibody solution (1:300) for 1 hour at room temperature. The washing procedure was repeated eight times within 40 minutes. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL; Amersham Biosciences, Amersham, UK) which was then exposed to Biomax L film (Kodak, Rochester, NY, USA). For quantification, ECL signals were digitized using Labwork software (UVP, Waltham, MA, USA). For oxyblot protein analysis, a standard control was loaded on each gel.
Statistical analyses
Quantitative data are expressed as mean ± SD. Statistical analysis was performed by ANOVA followed by Bonferroni multiple-comparison post hoc test. All analyses were conducted using SAS statistical software for Windows version 8.2 (SAS institute, Cary, NC). A probability value <0.05 was considered statistically significant.
Results
Effect of CsA and EPO on reducing the brain infarct area (BIA) and apoptotic nuclei and preserving the integrity of neuron cytoskeletal by day 3 after acute IS (Figure 1)
Figure 1.
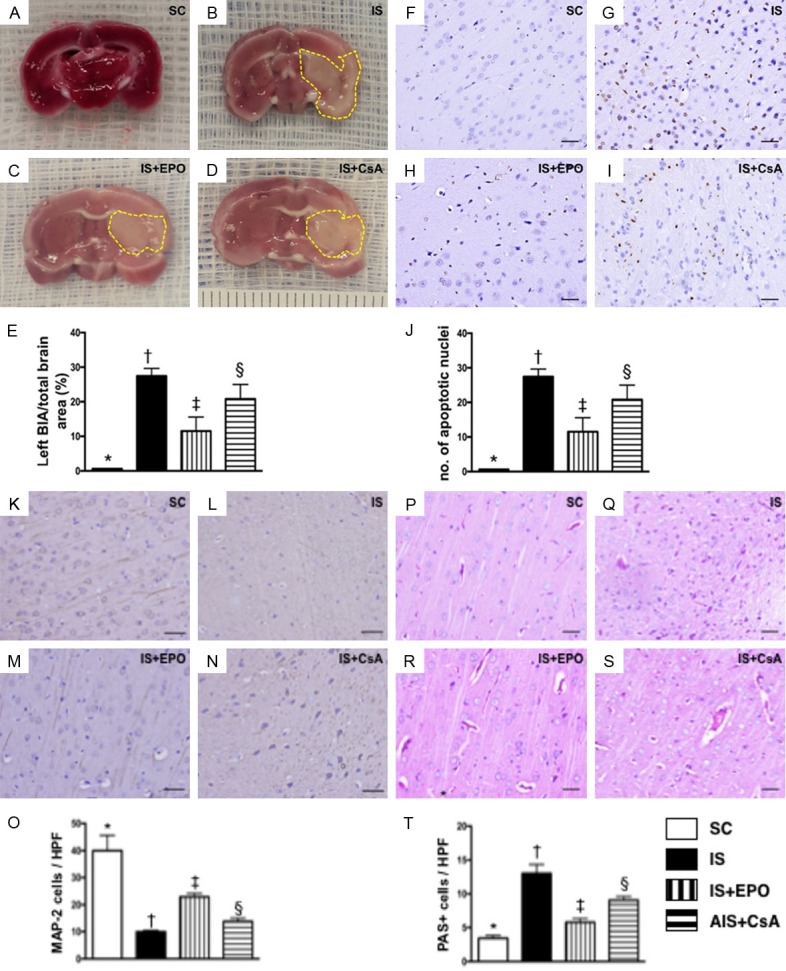
3,5-Triphenyl-2H-Tetrazolium Chloride (TTC) staining and TUNEL assay for assessment of brain damage, and identification of integrity of neuron cytoskeletal protein and expression of glycogen/glycoprotein by day 3 after acute IS. A-D. Illustration of TTC staining of brain tissues for identification of brain infarct area (BIA) (whitish color identified by yellow dotted line). E. BIA/total brain area (TBA), * vs. other groups with different symbols (†, ‡, §), P<0.0001. F-I. Illustrating microscopy (400×) with immunohistochemical staining (i.e., by TUNEL assay) of apoptotic nuclei (gray color). J. Number of apoptotic nuclei in BIA, * vs. other groups with different symbols (†, ‡, §), P<0.0001. Scale bars in right lower corner represent 20 µm. K-N. Microscopy (200×) of microtubule associated protein 2 (MAP-2) staining for identification of neuron-specific cytoskeletal protein (spindle shaped with gray color). O. Number of positively stained neuron-specific cytoskeletal protein, * vs. other groups with different symbols (†, ‡, §), P<0.0001. P-S. Microscopy (200×) for periodic acid-Schiff (PAS) for detecting the expression of glycogen/glycoprotein in BIA (pink color). T. Number of PAS+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. Scale bars in the right lower corner represent 50 µm. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡, §) indicate significance at the 0.05 level. SC = sham control; IS = ischemic stroke; EPO = erythropoietin; CsA = cyclosporine; BIA = brain infarct area.
As expected, TTC staining (Figure 1A-D) of brain tissues on day 3 after acute IS showed that the BIA was largest in group 2 (IS only), smallest in group 1 (SC), and significantly larger in group 4 (IS-CsA) than in group 3 (IS-EPO) (Figure 1E). Additionally, the number of apoptotic nuclei assessed by TUNEL assay (Figure 1F-I) showed an identical pattern to BIA among the four groups (Figure 1J).
Detection of microtubule associated protein 2 (MAP2) (Figure 1K-N), a neuron-specific cytoskeletal protein that is enriched in dendrites, implicating a role in determining and stabilizing dendritic shape during neuron development, was significantly lower in group 2 than in the other groups, significantly lower in groups 3 and 4 than in group 1, and significantly lower in group 4 than in group 3 (Figure 1O).
Conversely, Periodic acid-Schiff (PAS) (Figure 1P-S), a staining method for detecting the expression of glycogen/glycoprotein, displayed an opposite pattern to MAP2 among the four groups (Figure 1T), implicating the pathogenic change of neuron/myelin sheath in the BIA.
Effect of CsA and EPO on preserving the integrity of neurons and inhibiting the expressions of AQP4 and GFPA in BIA by day 3 after acute IS (Figure 2)
Figure 2.
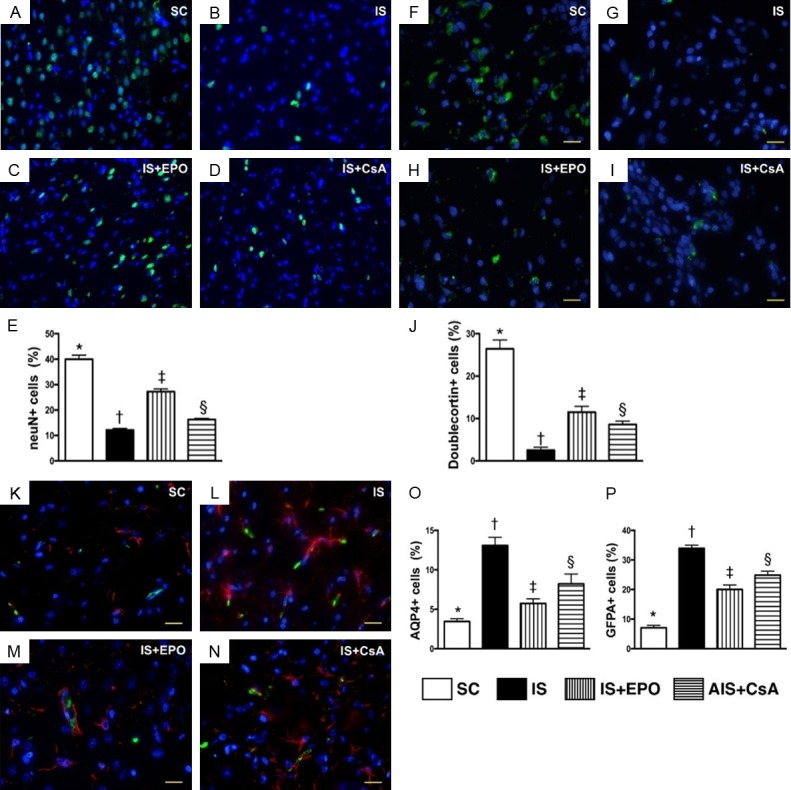
Assessment of integrity of neurons and neurogenesis, and cellular expressions of AQP4 and GFPA in BIA by day 3 after acute IS. A-D. Immunofluorescent (IF) microscopy (400×) for neuN+ cells (red color). E. Analytical result of number of neuN+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. F-I. IF microscopy (400×) of dobulecorin+ cells (green color). J. Number of dobulecorin+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. K-N. IF microscopy (400×) of aquaporin 4 (AQP4)+ cells (green color). O. Analytical result of number of AQP4+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. K-N. IF microscopy (400×) for glial fibrillary acid protein (GFAP)+ cells (red color). P. Analytical result of number of GFAP + cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. Scale bars in the right lower corner represent 20 µm. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡, §) indicate significance at the 0.05 level. SC = sham control; IS = ischemic stroke; EPO = erythropoietin; CsA = cyclosporine; BIA = brain infarct area.
The IF stain demonstrated that the number of neuN+ cells (Figure 2A-D) was lowest in group 2, highest in group 1, and significantly lower in group 4 than in group 3 (Figure 2E). Additionally, the number of dobulecorin cells (Figure 2F-I), an index of neurogenesis, exhibited an identical pattern to neuN+ cells among the four groups (Figure 2J).
IF microscopy identified that the number of AQP4+ cells (Figure 2K-N), an indicator of brain edema, was significantly higher in group 2 than in the other groups, significantly higher in groups 3 and 4 than in group 1, and significantly higher in group 4 than in group 3 (Figure 2O). IF microscopy consistently showed that the number of GFPA+ cells (Figure 2P), an indicator of inflammation, displayed an identical pattern to AQP4+ cells among the four groups.
Effect of CsA and EPO on suppressing the infiltrations of inflammatory cells in BIA by day 3 after acute IS (Figure 3)
Figure 3.

Infiltration of inflammatory cells in BIA by day 3 after acute IS. A-D. IF microscopy (400×) of CD68+ cells (green color). E. Analytical result of number of F4/80+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. F-I. IF microscopy (400×) of CD11+ cells (green color). J. Analytical result of number of CD11+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. Scale bars in right lower corner represent 20 µm. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡, §) indicate significance at the 0.05 level. SC = sham control; IS = ischemic stroke; EPO = erythropoietin; CsA = cyclosporine; BIA = brain infarct area.
IF staining displayed that the numbers of CD68+ cells (Figure 3A-D), an index of macrophages, was significantly higher in group 2 than in the other groups, significantly higher in groups 3 and 4 than in group 1, and significantly higher in group 4 than in group 3 (Figure 3E). Additionally, IF staining demonstrated that the number of CD11+ cells (Figure 3F-I), another indicator of inflammatory cells, exhibited an identical pattern to CD68+ cells among the four groups (Figure 3J).
Protein expressions of innate inflammatory reaction, oxidative stress, and brain edema in BIA by day 3 after IS induction (Figures 4 and 5)
Figure 4.
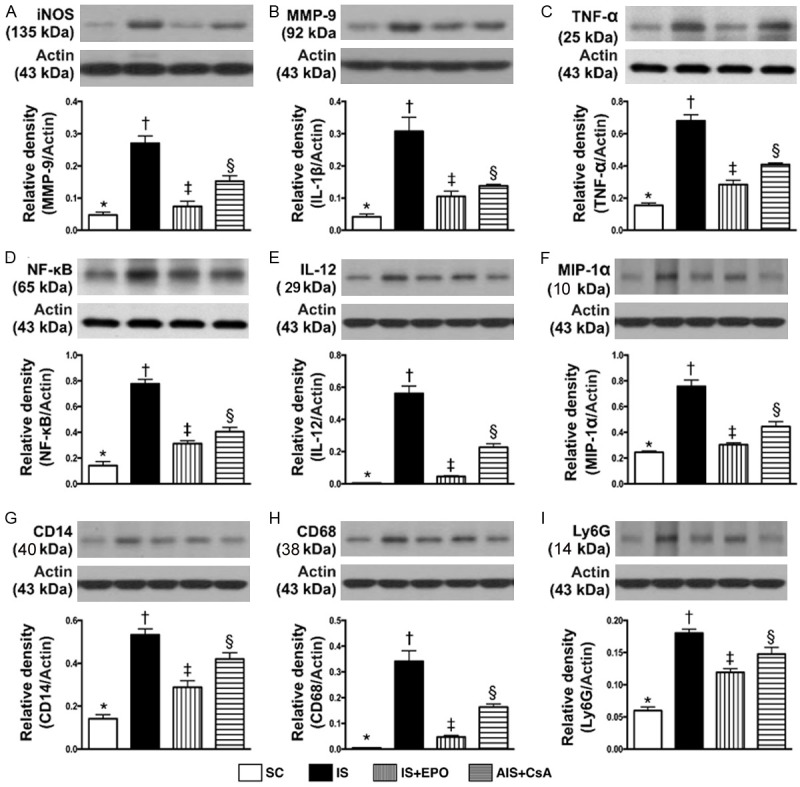
Protein expressions of innate inflammatory reactionin BIA by day 3 after IS induction. A. The protein expression of inducible nitric oxide (iNOS), * vs. other groups with different symbols (†, ‡, §), P<0.0001. B. Protein expression of matrix metalloproteinase (MMP)-9, * vs. other groups with different symbols (†, ‡, §), P<0.0001. C. Protein expression of tumor necrosis factor (TNF)-α, * vs. other groups with different symbols (†, ‡, §), P<0.0001. D. Protein expression of Nuclear factor (NF)-κB, * vs. other groups with different symbols (†, ‡, §), P<0.0001. E. Protein expression of interleukin (IL)-12, * vs. other groups with different symbols (†, ‡, §), P<0.0001. F. Protein expression of macrophage inflammatory protein (MIP)-1α, * vs. other groups with different symbols (†, ‡, §), P<0.0001. G. Protein expression of CD14, * vs. other groups with different symbols (†, ‡, §), P<0.0001. H. Protein expression of CD68, * vs. other groups with different symbols (†, ‡, §), P<0.0001. I. Protein expression of Ly6G, * vs. other groups with different symbols (†, ‡, §), P<0.0001. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡, §) indicate significance at the 0.05 level. SC = sham control; IS = ischemic stroke; EPO = erythropoietin; CsA = cyclosporine; BIA = brain infarct area.
Figure 5.
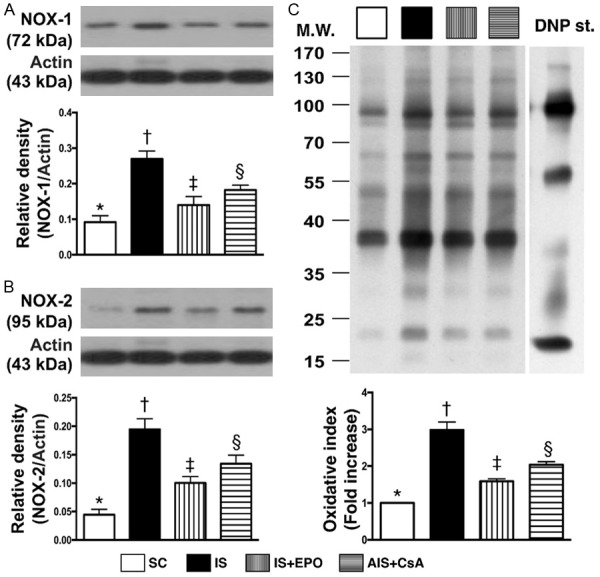
Protein expressions of oxidative stress and brain edema biomarkers in BIA by day 3 after IS induction. A. Protein expression of NOX-1, * vs. other groups with different symbols (†, ‡, §), P<0.001. B. Protein expression of NOX-2, * vs. other groups with different symbols (†, ‡, §), P<0.0001. C. Protein expression of oxidized protein, *vs. other groups with different symbols (†, ‡, §), P<0.0001. (Note: left and right lanes shown on the upper panel represent protein molecular weight marker and control oxidized molecular protein standard, respectively). M.W = molecular weight; DNP = 1-3 dinitrophenylhydrazone. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡, §) indicate significance at the 0.05 level. SC = sham control; IS = ischemic stroke; EPO = erythropoietin; CsA = cyclosporine; BIA = brain infarct area.
Protein expressions of iNOS, MMP-9, TNF-α, NF-κB, IL-12, MIP-1α, CD14, CD68 and Ly6g, nine indicators of innate inflammatory reaction, were significantly higher in group 2 than in the other groups, significantly higher in groups 3 and 4 than in group 1, and significantly higher in group 4 than in group 3 (Figure 4). The protein expressions of NOX-1, NOX-2 and oxidized protein, three indicators of oxidative stress, and protein expression of AQP4, an indicator of brain edema, revealed an identical pattern of innate inflammation among the four groups (Figure 5).
Protein expression of apoptotic and mitochondrial biomarkers as well as micro-RNA expressions in BIA by day 3 after IS induction (Figure 6)
Figure 6.
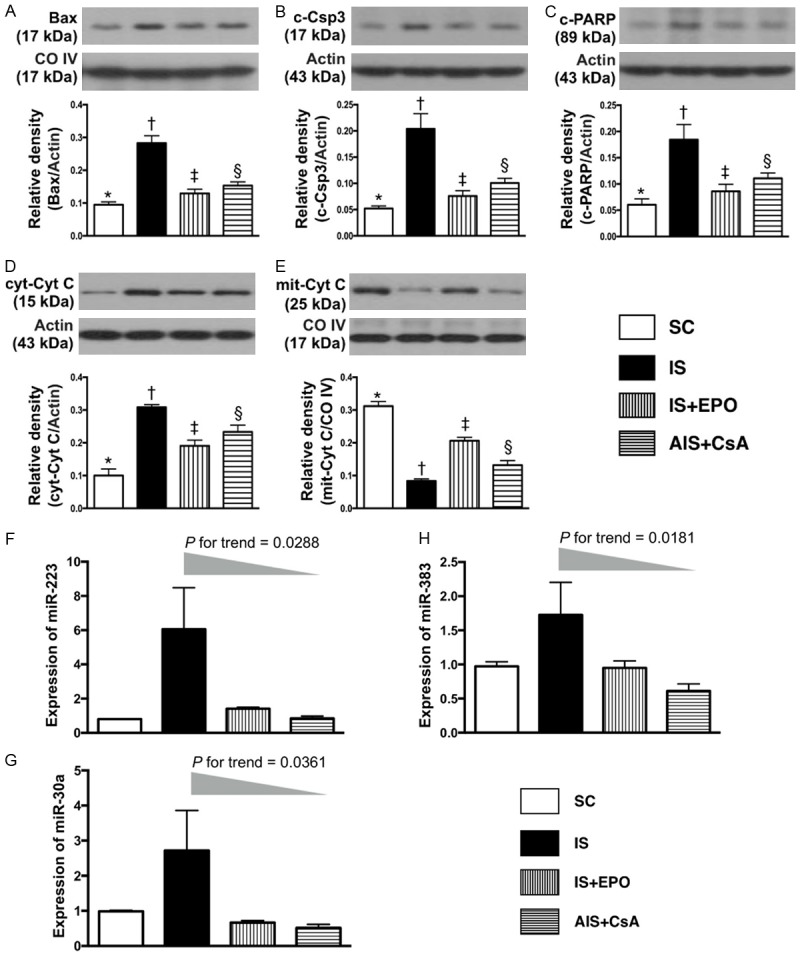
Protein expression of apoptotic and mitochondrial integrity biomarkers, and expression of Micro-RNAs in BIA by day 3 after IS induction. A. Protein expression of mitochondrial Bax (mit-Bax), * vs. other groups with different symbols (†, ‡, §), P<0.0001. B. Protein expression of cleaved caspase 3 (c-Casp 3), * vs. other groups with different symbols (†, ‡, §), P<0.0001. C. Protein expression of cleaved Poly (ADP-ribose) polymerase (c-PARP), * vs. other groups with different symbols (†, ‡, §), P<0.001. D. Protein expression of cytosolic cytochrome C (cyt-Cyto C), * vs. other groups with different symbols (†, ‡, §), P<0.0001. E. Protein expression of mitochondrial cytochrome C (mito-Cyto C), * vs. other groups with different symbols (†, ‡, §), P<0.0001. F. Expression of miR-223, P value for trend = 0.0288. G. Expression of miR-30a, P value for trend = 0.0361. H. Expression of miR-383, P value for trend = 0.0181. EPO = erythropoietin. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡, §) indicate significance at the 0.05 level. SC = sham control; IS = ischemic stroke; EPO = erythropoietin; CsA = cyclosporine; BIA = brain infarct area.
The protein expression of mitochondrial Bax, cleaved (c)-caspase 3 and c-PARP, three indicators of apoptosis, were highest in group 2, lowest in group 1, and significantly higher in group 4 than group 3 (Figure 6). Additionally, the protein expression of cytosolic cytochrome C, an indicator of mitochondrial damage, showed an identical pattern to apoptosis among the four groups (Figure 6). Conversely, the protein expression of mitochondrial cytochrome C, an indicator of mitochondrial integrity, exhibited an opposite pattern to apoptosis among the four groups (Figure 6).
The gene expressions of miR-223 and miR-30a, two indicators of response to acute inflammatory stimulation, expressed an identical pattern to apoptosis among the four groups (Figure 6). Additionally, the gene expression of miR-383, a regulator of cell apoptosis, displayed an identical pattern to miR-223 and miR-30a among the four groups (Figure 6).
Protein expressions of MAPK family and Akt signaling, and expressions of endothelial cell and endothelial progenitor cell biomarkers in BIA by day 3 after IS induction (Figure 7)
Figure 7.
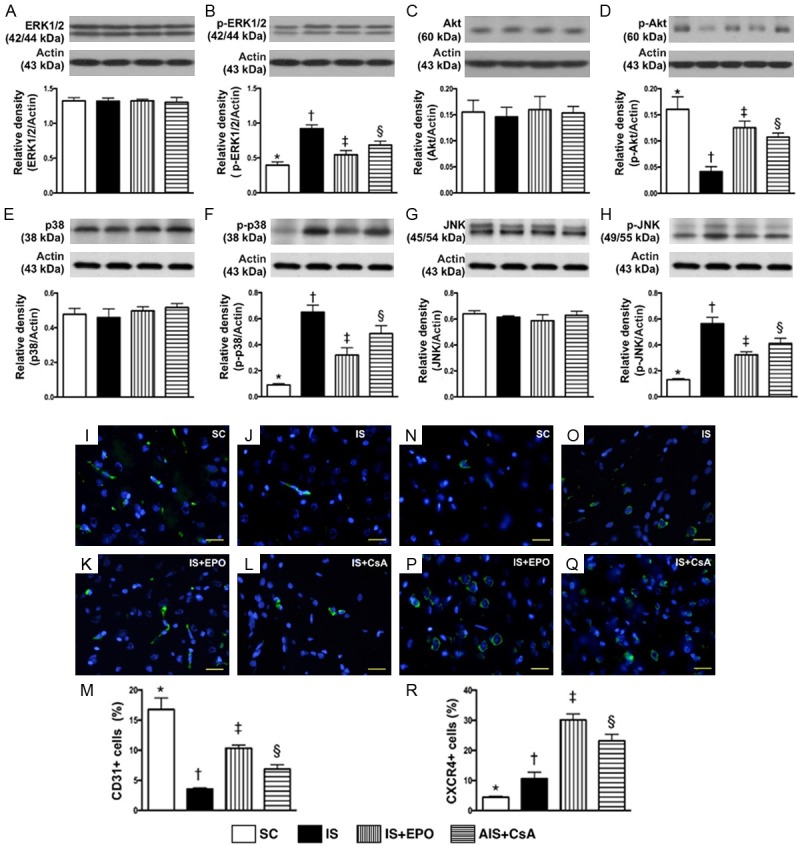
Protein expressions of MAPK family, and Angiogenesis cell expressions in BIA by day 3 after IS induction. A. Total protein expression of ERK1/2, P>0.5. B. Protein expression of phosphorylated (p)-p-ERK1/2, * vs. other groups with different symbols (†, ‡, §), P<0.001. C. Total protein expression of Akt, P>0.8. D. Protein expression of p-Akt, * vs. other groups with different symbols (†, ‡, §), P<0.0001. E. Total protein expression of p38, P>0.8. F. Protein expression of p-p38, * vs. other groups with different symbols (†, ‡, §), P<0.0001. G. Total protein expression of JNK, P>0.8. H. Total protein expression of p-JNK, * vs. other groups with different symbols (†, ‡, §), P<0.0001. I-L. IF microscopy (400×) of CD31+ cells (green color). M. Analytical result of number of CD31+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. N-Q. IF microscopy (400×) of CXCR4+ cells (green color). R. Analytical result of number of CXCR4+ cells, * vs. other groups with different symbols (†, ‡, §), P<0.0001. Scale bars in right lower corner represent 20 µm. All statistical analyses were performed by one-way ANOVA, followed by Bonferroni multiple comparison post hoc test (n = 6 for each group). Symbols (*, †, ‡, §) indicate significance at the 0.05 level. SC = sham control; IS = ischemic stroke; EPO = erythropoietin; CsA = cyclosporine; BIA = brain infarct area.
The protein expressions of p-ERK1/2, p-P38 and p-JNK, three indicators of MAPK family signaling, were significantly higher in group 2 than in other groups, significantly higher in groups 3 and 4 than in group 1, and significantly higher in group 4 than in group 3 (Figure 7). On the other hand, the protein expression of p-Akt, a signal transduction pathway that promotes survival and growth in response to extracellular signals, exhibited an opposite pattern of MAPK family (Figure 7).
IF staining exhibited that the number of CD31+ cells, an index of endothelial cell marker, was highest in group 1, lowest in group 2, and significantly higher in group 3 than in group 4 (Figure 7). Additionally, IF staining identified that the number of CXCR4+ cells, an indicator of endothelial progenitor cell (EPC), was highest in group 3, lowest in group 1, and significantly higher in group 4 than in group 2, suggesting an intrinsic response to ischemic stimulation (Figure 7).
Discussion
The most important finding in the present study was that BIA was markedly attenuated in acute IS animals with, than in without, EPO-CsA treatment. Additionally, histopathology demonstrated that the numbers of apoptotic nuclei and AQP4 cells (i.e., a brain edema biomarkers) were notably reduced and the integrities of neurons, myelin sheath and neurogenesis were better preserved in acute IS animals with, than in without, EPO-CsA therapy. Intriguingly, our previous experimental studies have also shown that EPO-CsA therapy significantly reduced BIA, notably through protecting neurons from ischemia-related damage, and significantly improved neurological recovery in rodents after acute IS [27,42]. Moreover, experimentally [27,37,42] and clinically [43,44], we found that EPO therapy significantly improved heart function in mini-pig after myocardial infarction and clinical outcomes in patients following acute IS, suggesting an important role for enhancing circulating numbers of endothelial progenitor cells (EPCs) that participate in angiogenesis in the ischemic region to restore blood flow. The present study also consistently displayed that EPO-CsA therapy enhanced the expression of endothelial cells/ECPs in the BIA. Accordingly, our findings reinforced those of our previous studies [27,37,42-44] and highlighted that EPO-CsA may be considered as an alternative candidate combination therapy for clinical application for those patients who have sustained acute IS but have responded poorly to conventional medications.
Although the results of our previous studies [27,37,42-44] were promising, the limitations of these studies included that the exact underlying mechanisms of EPO-CsA therapy on reducing BIA and improving neurological and cardiac function have not been fully investigated [27,37,42-44]. An essential finding in the present study was that abundant acute innate immune-inflammatory reactions, at both protein and cellular levels, were rapidly and substantially upregulated in animals after acute IS. In fact, our previous studies have shown that the acute inflammatory response was always augmented in different organs/tissues after acute ischemic/ischemia-reperfusion injury [16,18-27,37,41-43]. Additionally, this inflammatory response has been clarified to participate directly in cellular/tissue injury [16,20-22]. In this way, the results of our present study corroberated those of our previous studies [16,18-27,37,41-43]. Importantly, the results of the present study revealed that EPO-CsA therapy significantly reduced BIA, suggesting a role for suppressing the innate immune response to acute inflammation in animals after acute IS.
Another essential finding in the present study was that expressions of the MAPK family signaling pathway (i.e., p-ERK1/2, p-P38, p-JNK) were remarkably increased in acute IS animals as compared with those of SC animals. Interestingly, a strong association between increased MAPK signaling and myocardial infarction size has been identified by our mini-pig model of acute myocardial infarction [21]. In this way, our finding was consistent with the results of our previous study [21], highlighting that the MAPK pathway might participate in regulating BIA and outcome in rat after acute IS. Of importance was that EPO-CsA therapy could ameliorate the expression of MAPK signaling and BIA in acute IS animals.
Recent studies [32,33] have shown that miR-223 and miR-30a played a crucial role in augmenting the inflammatory response and innate immune cell infiltration in spinal cord injury and brain hemorrhagic area. A principal finding in the present study was that the gene expressions of miR-223 and miR-30a were significantly increased in acute IS animals compared to SC animals. Our findings, therefore, in addition to strengthening the findings of two recent studies [32,33], suggest that these two microRNAs are essential for neurological damage through initiating and propagating the inflammatory reaction. Of particular importance was that the levels of these two microRNAs in BIA were significantly suppressed in IS animals after receiving EPO-CsA therapy. This could, partially, explain why the BIA area was notably attenuated by EPO-CsA therapy in acute IS animals.
It is well recognized that oxidative stress plays a crucial role in cellular apoptosis and mitochondrial damage [16,18-20]. Intriguingly, a study has previously found that miR-383 was a contributor to cell apoptosis [45]. One important finding in the present study was that the protein of oxidative stress was remarkably higher in acute IS animals than in SC animals. Additionally, the gene level of miR-383 was notably increased in the acute IS group than in the SC group. Our findings, in addition to being consistent with the findings of previous studies [16,18-20,45], could partially explain why the protein expressions of apoptosis and cytosolic cytochrome C (i.e., a mitochondrial damage marker) as well as BIA were significantly higher in acute IS animals without, than with, EPO-CsA treatment. It is well recognized that Akt which is a signal transduction pathway promotes cell proliferation, growth, and survival in response to extracellular signals, including hypoxia, oxidative-stress and ischemic stimulations. In the present study, we found that p-Akt was significantly increased in IS animals than in control animals, implicating an intrinsic response to ischemic stimulation. Of importance was that p-Akt was more significantly increased in IS animals after receiving EPO-CsA treatment. This finding could once again explain why the BIA was notably reduced in acute IS animals with, than without, EPO-CsA treatment.
Study limitations
This study has limitations. First, for the purpose of detecting change in the acute phase of innate immune-inflammatory response after IS, the animals were euthanized at day 3 after the acute IS procedure. Therefore, the potential benefit of EPO-CsA therapy on neurological functional recovery could not be provided due to the short time interval. Second, this study did not test the optimal dosage of EPO and CsA for protecting the BIA. Therefore, whether EPO therapy is superior to CsA, or vice versa, for attenuating BIA is still unclear.
In conclusion, the current study highlighted that the innate immune response to inflammatory stimulation, oxidative stress, micro-RNAs and MAPK pathway were quickly upregulated after acute IS, and that the BIA was remarkably reduced by EPO-CsA therapy.
Acknowledgements
This study was supported by a program grant from Chang Gung Memorial Hospital, Chang Gung University (grant no. CMRPG891521; CMRPG8B1352; CRRPG8B1353).
Disclosure of conflict of interest
None.
References
- 1.Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA. Global burden of stroke: an underestimate-Authors’ reply. Lancet. 2014;383:1205–1206. doi: 10.1016/S0140-6736(14)60596-1. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O’Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA 3rd, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muir KW, Weir CJ, Murray GD, Povey C, Lees KR. Comparison of neurological scales and scoring systems for acute stroke prognosis. Stroke. 1996;27:1817–1820. doi: 10.1161/01.str.27.10.1817. [DOI] [PubMed] [Google Scholar]
- 4.Adams HP Jr, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993;24:35–41. doi: 10.1161/01.str.24.1.35. [DOI] [PubMed] [Google Scholar]
- 5.Chen ZM, Sandercock P, Pan HC, Counsell C, Collins R, Liu LS, Xie JX, Warlow C, Peto R. Indications for early aspirin use in acute ischemic stroke: A combined analysis of 40 000 randomized patients from the chinese acute stroke trial and the international stroke trial. On behalf of the CAST and IST collaborative groups. Stroke. 2000;31:1240–1249. doi: 10.1161/01.str.31.6.1240. [DOI] [PubMed] [Google Scholar]
- 6.Grau AJ, Weimar C, Buggle F, Heinrich A, Goertler M, Neumaier S, Glahn J, Brandt T, Hacke W, Diener HC. Risk factors, outcome, and treatment in subtypes of ischemic stroke: the German stroke data bank. Stroke. 2001;32:2559–2566. doi: 10.1161/hs1101.098524. [DOI] [PubMed] [Google Scholar]
- 7.Yip HK, Liou CW, Chang HW, Lan MY, Liu JS, Chen MC. Link between platelet activity and outcomes after an ischemic stroke. Cerebrovasc Dis. 2005;20:120–128. doi: 10.1159/000086802. [DOI] [PubMed] [Google Scholar]
- 8.Johnston SC, Mendis S, Mathers CD. Global variation in stroke burden and mortality: estimates from monitoring, surveillance, and modelling. Lancet Neurol. 2009;8:345–354. doi: 10.1016/S1474-4422(09)70023-7. [DOI] [PubMed] [Google Scholar]
- 9.Lewsey JD, Gillies M, Jhund PS, Chalmers JW, Redpath A, Briggs A, Walters M, Langhorne P, Capewell S, McMurray JJ, Macintyre K. Sex differences in incidence, mortality, and survival in individuals with stroke in Scotland, 1986 to 2005. Stroke. 2009;40:1038–1043. doi: 10.1161/STROKEAHA.108.542787. [DOI] [PubMed] [Google Scholar]
- 10.Granziera C, Daducci A, Meskaldji DE, Roche A, Maeder P, Michel P, Hadjikhani N, Sorensen AG, Frackowiak RS, Thiran JP, Meuli R, Krueger G. A new early and automated MRIbased predictor of motor improvement after stroke. Neurology. 2012;79:39–46. doi: 10.1212/WNL.0b013e31825f25e7. [DOI] [PubMed] [Google Scholar]
- 11.Siemonsen S, Lobel U, Sedlacik J, Forkert ND, Mouridsen K, Ostergaard L, Thomalla G, Fiehler J. Elevated T2-values in MRI of stroke patients shortly after symptom onset do not predict irreversible tissue infarction. Brain. 2012;135:1981–1989. doi: 10.1093/brain/aws079. [DOI] [PubMed] [Google Scholar]
- 12.Pan Y, Wang A, Liu G, Zhao X, Meng X, Zhao K, Liu L, Wang C, Johnston SC, Wang Y, Wang Y CHANCE Investigators. Cost-effectiveness of clopidogrel-aspirin versus aspirin alone for acute transient ischemic attack and minor stroke. J Am Heart Assoc. 2014;3:e000912. doi: 10.1161/JAHA.114.000912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Wang Y, Zhao X, Liu L, Wang D, Wang C, Wang C, Li H, Meng X, Cui L, Jia J, Dong Q, Xu A, Zeng J, Li Y, Wang Z, Xia H, Johnston SC CHANCE Investigators. Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med. 2013;369:11–19. doi: 10.1056/NEJMoa1215340. [DOI] [PubMed] [Google Scholar]
- 14.Guerini F, Frisoni GB, Morghen S, Speciale S, Bellelli G, Trabucchi M. Clinical instability as a predictor of negative outcomes among elderly patients admitted to a rehabilitation ward. J Am Med Dir Assoc. 2010;11:443–448. doi: 10.1016/j.jamda.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Cha J, Wang Z, Ao L, Zou N, Dinarello CA, Banerjee A, Fullerton DA, Meng X. Cytokines link Toll-like receptor 4 signaling to cardiac dysfunction after global myocardial ischemia. Ann Thorac Surg. 2008;85:1678–1685. doi: 10.1016/j.athoracsur.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheu JJ, Chua S, Sun CK, Chang LT, Yen CH, Wu CJ, Fu M, Yip HK. Intra-coronary administration of cyclosporine limits infarct size, attenuates remodeling and preserves left ventricular function in porcine acute anterior infarction. Int J Cardiol. 2011;147:79–87. doi: 10.1016/j.ijcard.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Varda-Bloom N, Leor J, Ohad DG, Hasin Y, Amar M, Fixler R, Battler A, Eldar M, Hasin D. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J Mol Cell Cardiol. 2000;32:2141–2149. doi: 10.1006/jmcc.2000.1261. [DOI] [PubMed] [Google Scholar]
- 18.Yuen CM, Chung SY, Tsai TH, Sung PH, Huang TH, Chen YL, Chen YL, Chai HT, Zhen YY, Chang MW, Wang CJ, Chang HW, Sun CK, Yip HK. Extracorporeal shock wave effectively attenuates brain infarct volume and improves neurological function in rat after acute ischemic stroke. Am J Transl Res. 2015;7:976–994. [PMC free article] [PubMed] [Google Scholar]
- 19.Chen HH, Chen YT, Yang CC, Chen KH, Sung PH, Chiang HJ, Chen CH, Chua S, Chung SY, Chen YL, Huang TH, Kao GS, Chen SY, Lee MS, Yip HK. Melatonin pretreatment enhances the therapeutic effects of exogenous mitochondria against hepatic ischemia-reperfusion injury in rats through suppression of mitochondrial permeability transition. J Pineal Res. 2016;61:52–68. doi: 10.1111/jpi.12326. [DOI] [PubMed] [Google Scholar]
- 20.Chua S, Leu S, Sheu JJ, Lin YC, Chang LT, Kao YH, Yen CH, Tsai TH, Chen YL, Chang HW, Sun CK, Yip HK. Intra-coronary administration of tacrolimus markedly attenuates infarct size and preserves heart function in porcine myocardial infarction. J Inflamm (Lond) 2012;9:21. doi: 10.1186/1476-9255-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang CH, Sheu JJ, Tsai TH, Chua S, Chang LT, Chang HW, Lee FY, Chen YL, Chung SY, Sun CK, Leu S, Yen CH, Yip HK. Effect of tacrolimus on myocardial infarction is associated with inflammation, ROS, MAP kinase and Akt pathways in mini-pigs. J Atheroscler Thromb. 2013;20:9–22. doi: 10.5551/jat.14316. [DOI] [PubMed] [Google Scholar]
- 22.Sheu JJ, Sung PH, Leu S, Chai HT, Zhen YY, Chen YC, Chua S, Chen YL, Tsai TH, Lee FY, Chang HW, Ko SF, Yip HK. Innate immune response after acute myocardial infarction and pharmacomodulatory action of tacrolimus in reducing infarct size and preserving myocardial integrity. J Biomed Sci. 2013;20:82. doi: 10.1186/1423-0127-20-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuen CM, Chiu CA, Chang LT, Liou CW, Lu CH, Youssef AA, Yip HK. Level and value of interleukin-18 after acute ischemic stroke. Circ J. 2007;71:1691–1696. doi: 10.1253/circj.71.1691. [DOI] [PubMed] [Google Scholar]
- 24.Yeh KH, Tsai TH, Chai HT, Leu S, Chung SY, Chua S, Chen YL, Lin HS, Yuen CM, Yip HK. Comparison of acute versus convalescent stage high-sensitivity C-Reactive protein level in predicting clinical outcome after acute ischemic stroke and impact of erythropoietin. J Transl Med. 2012;10:6. doi: 10.1186/1479-5876-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai TH, Chen YL, Lin HS, Liu CF, Chang HW, Lu CH, Chang WN, Chen SF, Wu CJ, Leu S, Ko SF, Yip HK. Link between lipoprotein-associated phospholipase A2 gene expression of peripheral-blood mononuclear cells and prognostic outcome after acute ischemic stroke. J Atheroscler Thromb. 2012;19:523–531. doi: 10.5551/jat.10751. [DOI] [PubMed] [Google Scholar]
- 26.Lin HS, Tsai TH, Liu CF, Lu CH, Chang WN, Chen SF, Huang CW, Huang CR, Tsai NW, Huang CC, Liou CW, Lin TK, Lan MY, Yip HK. Serum level and prognostic value of neopterin in patients after ischemic stroke. Clin Biochem. 2012;45:1596–1601. doi: 10.1016/j.clinbiochem.2012.07.113. [DOI] [PubMed] [Google Scholar]
- 27.Yuen CM, Sun CK, Lin YC, Chang LT, Kao YH, Yen CH, Chen YL, Tsai TH, Chua S, Shao PL, Leu S, Yip HK. Combination of cyclosporine and erythropoietin improves brain infarct size and neurological function in rats after ischemic stroke. J Transl Med. 2011;9:141. doi: 10.1186/1479-5876-9-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 29.Ortega-Perez I, Cano E, Were F, Villar M, Vazquez J, Redondo JM. c-Jun N-terminal kinase positively regulates NFATc2 transactivation through phosphorylation within the N-terminal regulatory domain. J Biol Chem. 2005;280:20867–20878. doi: 10.1074/jbc.M501898200. [DOI] [PubMed] [Google Scholar]
- 30.Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K, Tamaki T. Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smooth muscle cells. Hypertens Res. 2001;24:251–261. doi: 10.1291/hypres.24.251. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence MC, Naziruddin B, Levy MF, Jackson A, McGlynn K. Calcineurin/nuclear factor of activated T cells and MAPK signaling induce TNF-α gene expression in pancreatic islet endocrine cells. J Biol Chem. 2011;286:1025–1036. doi: 10.1074/jbc.M110.158675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi LB, Tang PF, Zhang W, Zhao YP, Zhang LC, Zhang H. Naringenin inhibits spinal cord injury-induced activation of neutrophils through miR-223. Gene. 2016;592:128–133. doi: 10.1016/j.gene.2016.07.037. [DOI] [PubMed] [Google Scholar]
- 33.Muller AH, Povlsen GK, Bang-Berthelsen CH, Kruse LS, Nielsen J, Warfvinge K, Edvinsson L. Regulation of microRNAs miR-30a and miR-143 in cerebral vasculature after experimental subarachnoid hemorrhage in rats. BMC Genomics. 2015;16:119. doi: 10.1186/s12864-015-1341-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, Cerami A, Brines M. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci U S A. 2003;100:4802–4806. doi: 10.1073/pnas.0630444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, Talan MI. Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc Natl Acad Sci U S A. 2003;100:11612–11617. doi: 10.1073/pnas.1930406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirata A, Minamino T, Asanuma H, Fujita M, Wakeno M, Myoishi M, Tsukamoto O, Okada K, Koyama H, Komamura K, Takashima S, Shinozaki Y, Mori H, Shiraga M, Kitakaze M, Hori M. Erythropoietin enhances neovascularization of ischemic myocardium and improves left ventricular dysfunction after myocardial infarction in dogs. J Am Coll Cardiol. 2006;48:176–184. doi: 10.1016/j.jacc.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Chua S, Leu S, Lin YC, Sheu JJ, Sun CK, Chung SY, Chai HT, Lee FY, Kao YH, Wu CJ, Chang LT, Ko SF, Yip HK. Early erythropoietin therapy attenuates remodeling and preserves function of left ventricle in porcine myocardial infarction. J Investig Med. 2011;59:574–586. doi: 10.2310/JIM.0b013e31820877dc. [DOI] [PubMed] [Google Scholar]
- 38.Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, Ovize M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transitiondependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 40.Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Bejui F, Robert D, Ovize M. Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc Res. 2004;61:115–122. doi: 10.1016/j.cardiores.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Leu S, Lin YC, Yuen CM, Yen CH, Kao YH, Sun CK, Yip HK. Adipose-derived mesenchymal stem cells markedly attenuate brain infarct size and improve neurological function in rats. J Transl Med. 2010;8:63. doi: 10.1186/1479-5876-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuen CM, Leu S, Lee FY, Yen CH, Lin YC, Chua S, Chung SY, Chai HT, Sheu JJ, Ko SF, Sun CK, Yip HK. Erythropoietin markedly attenuates brain infarct size and improves neurological function in the rat. J Investig Med. 2010;58:893–904. doi: 10.231/JIM.0b013e3181e80c40. [DOI] [PubMed] [Google Scholar]
- 43.Yip HK, Tsai TH, Lin HS, Chen SF, Sun CK, Leu S, Yuen CM, Tan TY, Lan MY, Liou CW, Lu CH, Chang WN. Effect of erythropoietin on level of circulating endothelial progenitor cells and outcome in patients after acute ischemic stroke. Crit Care. 2011;15:R40. doi: 10.1186/cc10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai TH, Lu CH, Wallace CG, Chang WN, Chen SF, Huang CR, Tsai NW, Lan MY, Sung PH, Liu CF, Yip HK. Erythropoietin improves longterm neurological outcome in acute ischemic stroke patients: a randomized, prospective, placebo-controlled clinical trial. Crit Care. 2015;19:49. doi: 10.1186/s13054-015-0761-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao L, Gu H, Chang J, Wu J, Wang D, Chen S, Yang X, Qian B. MicroRNA-383 regulates the apoptosis of tumor cells through targeting Gadd45g. PLoS One. 2014;9:e110472. doi: 10.1371/journal.pone.0110472. [DOI] [PMC free article] [PubMed] [Google Scholar]