

Abstract
Multiple Endocrine Neoplasia type 1 (MEN1) syndrome is a rare complex tumor-predisposing hereditary disorder, inherited in an autosomal dominant manner (OMIM 131100). MEN1 is characterized by tumors of the parathyroids, the neuroendocrine cells of the gastro-entero-pancreatic tract, and the anterior pituitary. The molecular mechanisms that control parathyroid tumorigenesis are still poorly understood. Here we studied the global microRNAs (miRNAs) expression profile in MEN1 parathyroid adenomas to understand the role of these regulatory factors in MEN1 parathyroid tumorigenesis. miRNA arrays containing 1890 human miRNAs were used to profile seven different MEN1 parathyroid adenomas (four presenting somatic loss of heterozygosity (LOH) at 11q13 and three still retaining one wild type copy of the MEN1 gene). Eight miRNAs in non-LOH MEN1 parathyroid adenomas and two miRNAs in LOH MEN1 parathyroid adenomas resulted to be differentially expressed, with a significant fold change, with respect to the control pool. Six microRNAs also resulted to be differentially expressed between LOH MEN1 parathyroid adenomas and non-LOH MEN1 parathyroid adenomas. Significantly differentially expressed miRNAs were all validated by SYBR green real-time quantitative RT-PCR. Pearson correlation coefficient indicated miR-4258, miR-664 and miR-1301 as the most significant miRNAs. In silico target-prediction and network analysis showed miR-664 and miR-1301 as organized in predicted GRNs with genes interested in parathyroid adenomas and carcinomas. In conclusion, our study identified three new miRNAs involved in the MEN1 parathyroid neoplasia, directly targeting genes associated with the development of different inheritable forms of parathyroid tumors. These identified miRNAs could be revealed as prognostic and diagnostic biomarkers for parathyroid tumors to improve the diagnosis of MEN1 neoplasia and other syndromes.
Keywords: MEN1, neoplasia, parathyroid adenomas, microRNAs, gene regulatory networks, primary hyperparathyroidism, diagnostic biomarkers
Introduction
MicroRNAs (miRNAs) are a family of naturally occurring, evolutionary conserved, small (approximately 19-23 nucleotides), non-protein-coding RNAs that negatively regulate post-transcriptional gene expression. miRNAs are estimated to account for > 3% of all human genes and to control expression of thousands of target mRNAs, with multiple miRNAs targeting for a single mRNA [1]. Recent studies have also supported a role of miRNAs in the initiation and progression of human malignancies [2].
Multiple Endocrine Neoplasia type 1 (MEN1) syndrome is a rare complex tumor-predisposing hereditary disorder, inherited in an autosomal dominant manner (OMIM 131100) [3]. MEN1 is characterized by tumors of the parathyroids, the neuroendocrine cells of the gastro-entero-pancreatic tract, and the anterior pituitary. The responsible gene, MEN1, is a tumor suppressor gene that encodes the menin protein. The first copy of wild type MEN1 gene is inactivated by germinal mutation, inherited by one parent or de novo developed at the embryonic level, while the second copy of the gene is typically lost at somatic level through large deletions and loss of heterozygosity (LOH) at 11q13 locus or, more rarely, through development of an inactivating mutation within the second allele of the MEN1 gene, inducing the development of tissue-specific MEN1 tumors [3]. The complete absence of wild type menin protein is responsible for the induction and development of MEN1-associated tumors; the exact mechanisms of MEN1 tumorigenesis is still not completely understood, and it seems to be highly tissue specific. Our research group has recently shown that, in MEN1 parathyroid adenomas from patients bearing a MEN1 gene mutation, tumor occurrence and development are under the control of an “incoherent feedback loop” between the miR-24-1 and menin protein, which mimics the second hit of Knudson’s hypothesis for tumor suppressor genes [4]. We hypothesized that this gene regulatory network (GRN) between menin and miR-24-1 acts as a “homeostatic regulatory network” that needs to be perturbed to induce the progression of MEN1 neoplasia [4].
In order to further investigate the molecular mechanisms involved in MEN1 parathyroid tumorigenesis, we performed microarray analysis of global miRNA expression profile in seven MEN1 parathyroid adenomas (four with somatic LOH at 11q13 locus and three still retaining, at somatic level, one copy of wild type MEN1 gene).
Materials and methods
Tissue collection
Parathyroid adenoma specimens have been collected from patients who underwent surgical parathyroid excision; all clinical, genetic and molecular data associated to each sample were collected and analyzed anonymously. In particular, we obtained seven parathyroid adenomas from MEN1 patients, bearing different MEN1 gene mutations, and two sporadic parathyroid adenomas from non-MEN1 subjects (negative to MEN1 mutation screening both at blood genomic level and at parathyroid somatic level, and not presenting any other endocrine or non-endocrine manifestation associable to MEN1 syndrome). Two healthy parathyroid tissues were obtained from non-MEN1 patients operated for sporadic thyroid carcinoma (genetic screening of the coding region and intron-exon junctions of the MEN1 gene has been performed at the parathyroid somatic level to exclude the presence of any MEN1 mutation). Histological, genetic and clinical data of the MEN1 samples are illustrated in Table 1. All the tissues used in this study were authorized by the Local Ethical Committee (LEC) of the Azienda Ospedaliero-Universitaria Careggi, Firenze, which approved this study concerning the “parathyroid tumorigenesis” (tumorigenesi della paratiroide). Samples were collected by clinicians and surgeons after having obtained verbal informed consent from the patients. It was clearly explained to the patients that their samples would be used for basic research not related to their specific clinical case and that each sample would be analyzed anonymously by the researchers. After collection, each tissue sample was coded and associated only to data regarding age, sex, and clinical features (i.e. MEN1-related parathyroid adenoma, non-MEN1 parathyroid adenoma or healthy parathyroid gland). At this time, there is no way to associate the sample to the name of the donor in order to ask them for retrospective written informed consent, in agreement with LEC.
Table 1.
Main genetic and histological characteristics of parathyroid samples, and clinical features
| Parathyroid tissue histological analysis | Mutation of the MEN1 gene | LOH at 11q13 (MEN1 locus) | First endocrine manifestation | Endocrine lesions |
|---|---|---|---|---|
| Non-LOH MEN1 PA | g.953delGA | NO | PHPT | Multiple parathyroid adenomas |
| Non-LOH MEN1 PA | g.317insGCCCC | NO | Hypoglycemia | Multiple parathyroid adenomas, pancreatic adenoma |
| Non-LOH MEN1 PA | g.359delGTCT | NO | Kidney stones | Multiple parathyroid adenomas, osteoporosis |
| LOH MEN1 PA | g.953delGA | YES | PHPT | Multiple parathyroid adenomas |
| LOH MEN1 PA | g.335delA | YES | Hypoglycemic crisis | Multiple parathyroid adenomas, microprolactinoma, insulinoma, bilateral surrenalic hyperplasia |
| LOH MEN1 PA | g.1434delC | YES | ZES | Multiple parathyroid adenomas, pancreatic gastrinoma, microprolactinoma |
| LOH MEN1 PA | g.908delGCT | YES | PHPT, kidney stones | Multiple parathyroid adenomas, microprolactinoma, pituitary macroadenoma secreting GH, lung carcinoid, pancreatic adenoma |
PA = parathyroid adenoma; LOH = loss of heterozygosity; NA = Data not available; PHPT = primary hyperparathyroidism; ZES = Zollinger-Ellison syndrome; GH = growth hormone.
MEN1 gene mutational analysis
Genomic DNA of the MEN1 patients and of the patients affected by sporadic parathyroid adenomas was extracted from peripheral blood leukocytes using a microvolume silica membrane-based column system, according to the manufacturer’s instructions (NucleoSpin Blood Quick Pure; Macherey-Nagel, Easton, PA, USA). DNA from all the parathyroid samples included in this study was extracted using the Trizol method, according to the manufacturer’s instructions (Invitrogen, Waltham, MA, USA). Mutational analysis of the encoding regions (exons 2-10) and of the intron-exon junctions of the MEN1 gene was performed by PCR-based direct sequencing both on genomic DNA from blood and somatic DNA from parathyroid samples (Figure 1).
Figure 1.
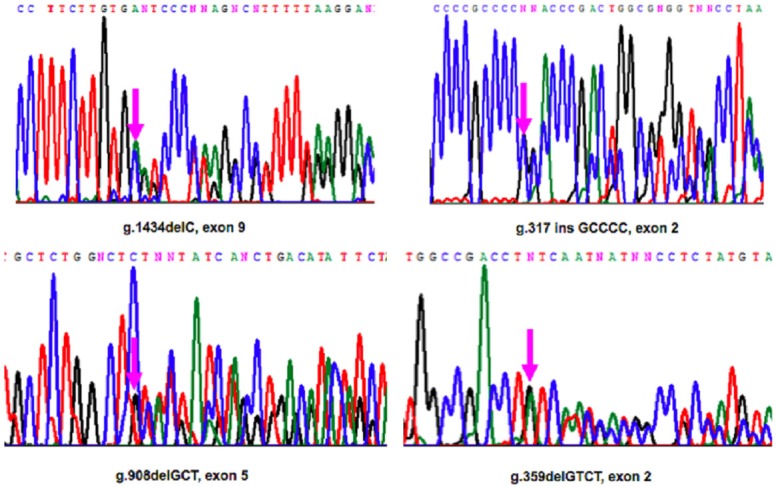
Selection of PCR-based direct sequencing chromatograms of genomic MEN1 mutated sequences in our patients.
Loss of heterozygosity analysis
PCR-based microsatellite analysis for LOH at the 11q13 MEN1 region was performed in parathyroid tissue somatic DNA samples versus blood genomic DNA samples. LOH analysis was performed using four specific microsatellite markers (D11S480, PYGM, D11S449, and D11S913) flanking the 11q12-13 locus. An independent PCR amplification was performed for each microsatellite in a final volume of 12.5 μl using PuReTaq Ready-To-Go PCR beads (GE Healthcare, Buckinghamshire, UK). An aliquot of each amplification product was denatured at 95°C for 5 min in a solution of formamide and GENESCAN 400HD [ROX] size standard (Applied Biosystems, Foster City, CA USA), and then analyzed on the ABI Prism 3100 Genetic Analyzer (Applied Biosystems) by Genescan® analysis software.
RNA extraction and miRNA microarray analysis
Total RNA was extracted from fresh frozen tissue with Trizol (Invitrogen, Waltham, MA, USA). MiRNA microarray profiling was done using the miRCURY LNA array version 19.0 (Exiqon, Vedbaek Denmark). The 7th Gen array contains 3100 unique capture probes covering almost all mature hsa (~1890), mmu (~1140), rno (679) and related viral (hsa 146, mmu, 57) microRNA sequences as annotated in miRBase v19.0, with 4 duplicate probes per miRNA. Sporadic parathyroid adenomas have been used as control pool. One μg of total RNA for each MEN1 sample and pooled normal reference were labeled with Hy3 and Hy5 fluorescent label, respectively, using the miRacULS miRNA labeling kit (Kreatech, Amsterdam, Holland), as described by the manufacturer. The Hy3-labeled samples and a Hy5-labeled reference RNA sample were mixed pair-wise and hybridized to the miRCURY LNA array. The miRCURY LNA array microarray slides were scanned using the PERKIN ELMER ScannerArray (Perkin Elmer, Winter St. Waltham, MA, USA).
Transient transfections
BON1 cells were transfected with siRNAs (200 pmol) against MEN1 mRNA (FlexiTube SiRNA-Hs MEN1 1: SI00630231) and siRNAs negative control by using siPORTTM NeoFX (Ambion, USA) according to the manufacturer’s instructions. RNAs were isolated by using “mirVana PARIS” (Ambion, Austin, TX, USA) and analyzed by semi-quantitative PCR; amplification analysis was performed using miScript/QuantiTect SYBR Green PCR Kit (Qiagen, Germany) and MEN1(Hs_MEN1_1_SG: QT00064848), pre-miR-4258 (MS00021175), 5S (MS00007574), 18S(Hs_RRN18S_1_SG: QT00199367) miScript Primer/QuantiTect assay.
Quantitative real-time RT-PCR
Quantitative real-time RT-PCR was carried out as it follows. Each RNA sample was analyzed in triplicate (including the normalization control 5S). Ten µg of total RNA from each sample were DNAse treated with a DNA-free kit (Ambion, Applied Biosystems, Foster City, CA USA). Two µl of DNA-free RNA were reverse transcribed by using miScript reverse transcription Kit (Qiagen, Hilden, Germany) and amplified using miScript SYBR Green PCR Kit and specific microRNAs and 5S miScript Primer assay (Quiagen)-Hs_miR_4258_1:cod.MS00021175Hs_miR_664_1:cod.MS00014819 Hs_miR_1301_2: cod. MS00031381. RNA expression was quantified with a MX3000P multiplex quantitative PCR instrument (Stratagene, La Jolla, CA, USA), using MXPro software, following a unique three-step protocol: one cycle at 95° for 15 min and 40 amplification cycles (94°C for 15 sec, 1 min at 55°C and 70°C for 30 sec). Sample fluorescence was detected during the annealing step. The fluorescence data were collected continuously to obtain the dissociation curve. Fluorescence was plotted versus the threshold cycle (Ct) based on baseline-corrected, reference dye-normalized fluorescence (dRn) to obtain the standard curve and to measure the initial mRNA quantity. Gene expression was normalized to 5S RNA. miRNA expression level was calculated using delta cycle threshold (ΔCt), which is the Ct of the miRNA of interest subtracted by the Ct value of the endogenous control. Fold difference between samples (non-LOH MEN1 parathyroid adenomas vs LOH MEN1 parathyroid adenomas, non-LOH MEN1 parathyroid adenomas vs control pool, and LOH MEN1 parathyroid adenomas vs control pool) was determined by the formula: fold change = 2-ΔΔct.
Bioinformatic and statistical analyses
The microarray GPR files were loaded into R/Bioconductor using the LIMMA package [5]. The log2 ratio of the intensity of Cy5 to Cy3 signals were calculated for each miRNA on every array and normalized by LOESS normalization. Differentially expressed miRNAs were identified using p-value < 0.05 and log2 fold change (FC) > 1.5, with adjustment for false discovery rate using the Benjamini-Hochberg method. A linear model with one factor, tumor type, with three groups (non-LOH MEN1 parathyroid adenomas, LOH MEN1 parathyroid adenomas, and control pool) was established, followed by linear contrasts (non-LOH MEN1 parathyroid adenomas vs LOH MEN1 parathyroid adenomas; non-LOH MEN1 parathyroid adenomas vs control pool; LOH MEN1 parathyroid adenomas vs control pool). The Pearson coefficient of correlation (r), analyzed by using SPSS18 statistical software, was applied to compare the microarray data with the SYBR-green real-time quantitative RT-PCR data. The Pearson coefficient of correlation (r) was used to measure the linear dependence or correlation between the variables: microarray data and SYBR-green real-time quantitative RT-PCR data. It has a value between +1 to -1. Total positive linear correlation is when r = 1, while there isn’t correlation between variables when r = 0. Finally, total negative linear correlation is when r = 1. The microarray data with r > 0.65 value were considered.
miRNA target gene prediction
The selected miRNAs were further analyzed to identify the target gene/genes and their possible function in parathyroid tumorigenesis. MiRNA target genes were predicted by ComiR, a web tool for combinatorial miRNA target prediction that combines all the four scoring schemes, used by miRanda, PITA, TargetScan and mirSVR, into one, using a support vector machine (SVM) trained on Drosophila AGO1 IP data. The end result is the SVM probability, which represents the likelihood that this mRNA is regulated by the set of miRNA genes [6].
MIR@NT@N computational analysis of networks
We used MIR@NT@N (MIRna @Nd Transcription factor @nalysis Network), based on a graph-theoretical method, to integrate multiple regulation levels into a unified model. MIR@NT@N predicts novel molecular actors and the form of their interplay. Based on these predictions or on lists of known molecular actors, users can generate regulatory networks and extract feedback loop (FBL) and feedforward loop (FFL) sub-networks [7].
miRNAs function inference
We used a network biology approach, miRUPnet, to infer the functions of miRNAs, based on a functional analysis of the interactome of transcription factors (TFs) that regulate each selected miRNA [8], and to identify the enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [9].
Results
Differentially expressed miRNAs
We investigated the expression profile of miRNAs (Figure 2) in seven parathyroid adenomas from MEN1 gene mutation carriers (four presenting LOH at 11q13 locus and three still retaining one wild type copy of the MEN1 gene) (Table 1).
Figure 2.
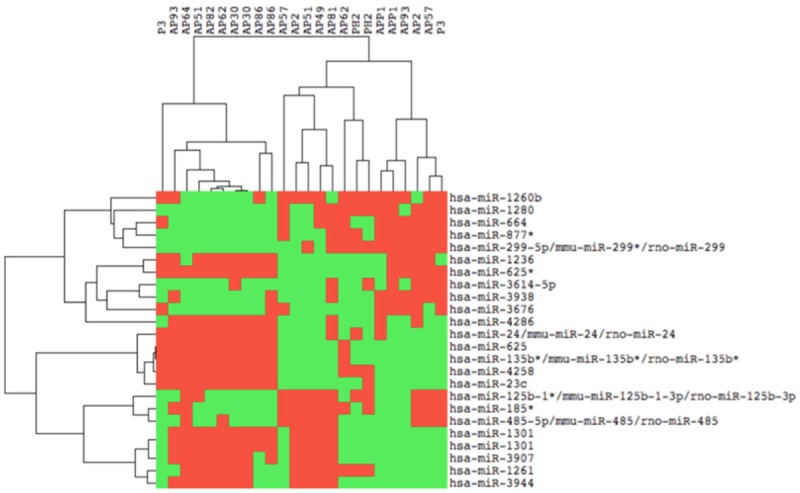
Cluster analysis of miRNA expression. Tree generated by cluster analysis of three different parathyroid adenoma samples based on differentially regulated miRNAs (miRNAs showing a log2 FC ≥ 1.5 and a p value ≤ 0.05).
Eight miRNAs in non-LOH MEN1 parathyroid adenomas and two miRNAs in LOH MEN1 parathyroid adenomas resulted to be significantly differentially expressed (considering a log2 FC > 1.5 and a p-value < 0.05) with respect to the control pool (Figure 3). In addition, six miRNAs resulted to be significantly differentially expressed by comparing LOH MEN1 parathyroid adenomas vs non-LOH MEN1 parathyroid adenomas, using p-value < 0.05 and log2 FC > 1.5 (Figures 3 and 4). All these expression values were validated and confirmed by real-time quantitative RT-PCR (Table 2).
Figure 3.
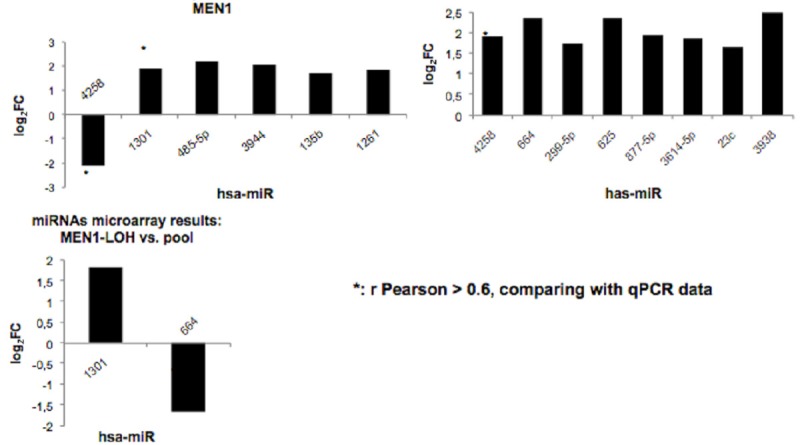
miRNA microarray analysis showing significant differential expression levels between groups. The results are presented as log2 FC and they reported only miRNAs showing a log2 FC > 1.5 and a p value < 0.05.
Figure 4.
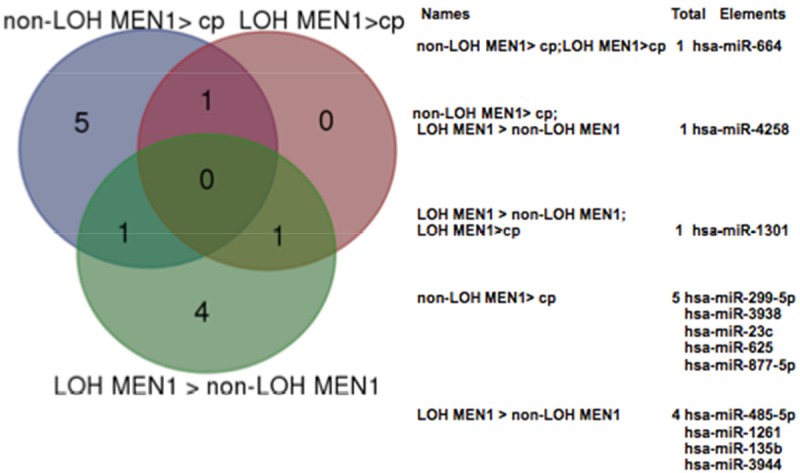
Comparisons of miRNA expression in parathyroid adenoma: the Venn diagram shows the number of miRNAs differentially expressed in LOH MEN1 parathyroid adenomas vs non-LOH/MEN1 parathyroid adenomas, LOH MEN1 parathyroid vs control pool and non-LOH/MEN1 parathyroid adenomas vs control pool. cp = control pool.
Table 2.
Validation of miRNA differential expression levels between groups by real-time quantitative RT-PCR
| miRNAs | LOH MEN1 vs Non-LOH MEN1 |
|
| |
| hsa-miR-4258 | 0.48 |
| hsa-miR-1301 | 0.78 |
| hsa-miR-485-5p | 0.95 |
| hsa-miR-3944 | 0.91 |
| hsa-miR-135b | 11.40 |
| hsa-miR-1261 | 0.59 |
|
| |
| miRNAs | Non-LOH MEN1 vs control pool |
|
| |
| hsa-miR-4258 | 2.35 |
| hsa-miR-664 | 0.42 |
| hsa-miR-299-5p | 0.75 |
| hsa-miR-625 | 2.47 |
| hsa-miR-877-5p | 1.05 |
| hsa-miR-3614-5p | 1.0 |
| hsa-miR-23c | 0.36 |
| hsa-miR-3938 | 3.21 |
|
| |
| miRNAs | LOH MEN1 vs control pool |
|
| |
| hsa-miR-1301 | 0.93 |
| hsa-miR-664 | 0.42 |
The results are presented as FC (2-ΔΔCt).
The r Pearson coefficient of correlation was applied to compare the microarray data with the SYBR-green real-time quantitative RT-PCR data; correlation results are shown in Table 3.
Table 3.
Microarray data validation using real-time quantitative RT-PCR with Pearson Correlation Coefficient
| miRNA | Sample groups | Pearson correlation coefficient (r) |
|---|---|---|
| hsa-miR-4258 | LOH MEN1 vs Non-LOH MEN1 | 0.65 |
| hsa-miR-1301 | LOH MEN1 vs Non-LOH MEN1 | 0.84 |
| hsa-miR-664 | LOH MEN1 vs control pool | 0.92 |
| hsa-miR-4258 | Non-LOH MEN1 vs control pool | 0.84 |
miR-4258, miR-664 and miR-1301 resulted to be significantly differentially expressed in the two groups (LOH MEN1 parathyroid adenomas and non-LOH MEN1 parathyroid adenomas) (Figure 4).
Interestingly, miR-4258 resulted strongly down-regulated in LOH MEN1 parathyroid adenomas with respect to non-LOH MEN1 parathyroid adenomas, thus showing a correlation with the presence of a least one wild type MEN1 allele. MiR-4258 also resulted up-regulated in non-LOH MEN1 parathyroid adenomas with respect to control pool (Figure 3). MiR-664 was up-regulated in non-LOH MEN1 parathyroid adenomas and down-regulated in LOH MEN1 parathyroid adenomas, both with respect to control pool (Figure 3). MiR-1301 resulted up-regulated in LOH MEN1 parathyroid adenomas, both with respect to non-LOH MEN1 parathyroid adenomas and control pool (Figure 3).
These data allow us to postulate that, like miR-24-1 [13], some miRNAs need the presence of wild type menin protein to be expressed (miR-4258, miR-664 and miR-1301).
In order to confirm and to validate this data, we transfected the BON1 cell lines with specific siRNAs against MEN1 mRNA and analyzed the expression of miR-4258 pre-miRNA.
As showed in Figure 5, the silencing of MEN1 mRNA induced the downregulation of pre-miR-4258 thus confirming that the biogenesis of miR-4258 was under the MEN1 expression control.
Figure 5.
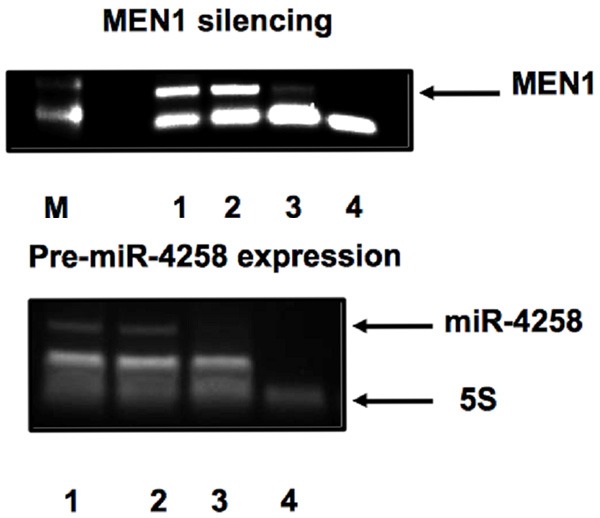
Menin siRNA-silencing. MEN1 mRNA expression was analyzed in total RNA (1), siRNA negative control (2), siRNA-MEN1 (3). (4): no-RT; M, 100-base pair marker lane. Pre-miR-4258 expression after MEN1 mRNA silencing. 4258 pre-miR-RNA expression was analyzed in total RNA (1), siRNA negative control (2), siRNA-MEN1 (3). (4): no-RT.
Target and network analysis for differentially expressed miRNAs
To identify putative gene targets of the differentially expressed miRNAs detected in this study, and potentially involved in parathyroid tumorigenesis, we used the ComiR tool [6], principally focusing on genes known to be involved in parathyroid neoplasia (Lgals3, MKI67, CDKN2A, CDKN2B, CDKN2C, CCND1, CDC73/HRPT2, RET, CDKN1B, MEN1, RB1, CTNNB1, VDR, CaSR, PTH, AP2S1, CDKN1A, CCDN2).
We found that miR-4258 targets the CCND1 gene, miR-1301 targets CDKN1B, RB1, CTNNB1, CCDN2 and RET genes, and MiR-664 targets CDC73/HRPT2 and CDKN2C genes. Predicted GRNs analysis showed a complex network where both miR-1301 and miR-664 play a pivotal role by repressing oncosuppressor genes involved in the familial forms of primary hyperparathyroidism and parathyroid adenomas. In addition, miR-1301 targets RB1 and miR-664 targets CDC73/HRPT2, genes also known to be involved in parathyroid carcinomas (Figure 6).
Figure 6.
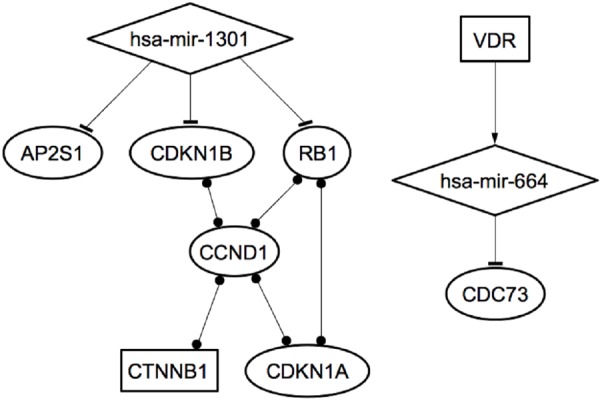
The two microRNAs-Transcription Factors-mRNA networks identified in MEN1 parathyroid tumorigenesis.
Interestingly, miR-664 was involved in a circuit together with vitamin D receptor (VDR), involved in the regulation of parathyroid hormone (PTH) secretion and calcium homeostasis (Figure 6).
We also assessed the over-represented functional pathways derived from the KEGG database for our three selected miRNAs [9]. We detected an enrichment of genes involved in regulation of the cell cycle and in cancer-related pathways. The identified enriched categories (with a false discovery rate (FDR) less than 0.05) are shown in Table 4.
Table 4.
Significant KEGG pathways of genes regulated by our selected miRNAs and their gene regulatory networks
| KEGG | P value | FDR |
|---|---|---|
| Cell cycle | 1.46e-038 | 1.01E-36 |
| ErbB signaling pathway | 1.13e-028 | 2.59E-27 |
| Pathways in cancer | 1.37e-019 | 2.36E-18 |
| Prostate cancer | 3.95e-024 | 2.5E-20 |
| Small cell lung cancer | 9.06e-048 | 7.25E-46 |
| Endocytosis | 4.56e-030 | 1.82E-28 |
| Thyroid cancer | 1.79e-043 | 1.32E-41 |
Discussion
Primary hyperparathyroidism may occur sporadically (without a family history) or as part of some familial inherited syndromes such as MEN1, multiple endocrine neoplasia type 2A (MEN2A), multiple endocrine neoplasia type 4 (MEN4), and isolated familial hyperparathyroidism. Currently, the exact molecular mechanisms governing the parathyroid tumorigenesis are poorly understood.
MiRNA profiling in parathyroid tumor could be useful to identify miRNA diagnostic, prognostic and therapeutic markers of parathyroid tumorigenesis. Rahbari et al. [10] and Corbetta et al. [11,12] have already analyzed miRNA profiling in parathyroid tumors. Rahbari [10] showed three miRNAs (miR-26b, miR-30b, and miR-126*) to be significantly deregulated between parathyroid carcinoma and parathyroid adenoma. Receiver-operating characteristic curve analysis showed mir-126* was the best diagnostic marker, but none of these miRNAs target the CDC73/HRPT2 gene responsible for hereditary parathyroid carcinomas. Corbetta et al. showed that miR-517-c, miR-222 and miR-503 were up-regulated in parathyroid carcinomas while miR-296 and miR-139 were down-regulated [11,12]. We recently showed that, in parathyroid adenomas from MEN1 mutation carriers, the neoplasia development is under the control of an “incoherent feedback loop” between the miR-24-1 and menin protein, that mimics the second hit of Knudson’s hypothesis [4], in a still reversible way. We hypothesized that this GRN between menin and miR-24-1 acts as a “homeostatic regulatory network” that needs to be perturbed to induce the progression of MEN1 neoplasia [4]. We speculated that additional miRNAs could be involved in the homeostasis of MEN1 disease and, probably, organized in GRNs that need to be perturbed or preserved to move from precursor lesions to neoplasm. Therefore, we performed a global microarray analysis of miRNA expression in four MEN1 parathyroid adenomas with LOH at 11q13 (thus completely missing the expression of wild type MEN1 mRNA and menin) and three MEN1 parathyroid adenomas without LOH at 11q13 or somatic mutations of the MEN1 gene (thus still retaining one copy of wild type MEN1 and expressing wild type MEN1 mRNA and menin). Significantly differentially expressed miRNAs from microarray analysis were validated by SYBR Green real-time quantitative RT-PCR. Seven miRNAs in non-LOH MEN1 parathyroid adenomas and two miRNAs in LOH MEN1 parathyroid adenomas resulted to be differentially expressed, with a significant fold change, with respect to the control pool. In addition, six miRNAs resulted differentially expressed between non-LOH MEN1 parathyroid adenomas and LOH MEN1 parathyroid adenomas. Pearson correlation coefficient indicated miR-4258, miR-1301 and miR-664 as the best predictive diagnostic markers in distinguishing MEN1 parathyroid adenomas from non-MEN1 parathyroid adenomas and healthy glands, or in distinguishing between MEN1 parathyroid adenomas with or without 11q13 LOH. In particular, miR-4258 resulted down-regulated in LOH MEN1 parathyroid adenomas with respect to the non-LOH counterparts, while it resulted up-regulated in non-LOH MEN1 parathyroid adenomas with respect to the parathyroid control pool. MiR-664 was up-regulated in non-LOH MEN1 parathyroid adenomas and down-regulated in LOH MEN1 parathyroid adenomas, both with respect to the parathyroid control pool. MiR-1301 resulted up-regulated in LOH MEN1 parathyroid adenomas with respect to both non-LOH MEN1 parathyroid adenomas and the parathyroid control pool. All these data indicate that the expressions of miR-1301, miR-4258 and miR-664 were under the control of menin protein. Indeed, miR-4258 and miR-664 were down-regulated in parathyroid cells that have lost both the wild type MEN1 alleles and up-regulated in parathyroid cells retaining, and expressing, one copy of wild type MEN1 allele, while MiR-1301 is over-expressed after biallelic inactivation of wild type MEN1. We have seen, in the BON1 cell line, derived from a lymphnode metastasis of a human carcinoid tumor of the pancreas, that menin binds specifically to the primary RNA sequence pri-miR-24-1 by promoting the miR-24-1 biogenesis (unpublished data); this data allows us to hypothesize that menin can be involved in the biogenesis of miR-4258, miR-664 and miR-1301.
The most interesting data concern the analysis of the mRNA target of these three miRNAs. Tumor suppressor genes, known to be associated with familial forms of hyperparathyroidism and parathyroid adenoma and carcinoma, resulted to be targeted by our identified miRNAs, presumably confirming the key role of these three miRNAs in the tumorigenesis of parathyroid, and not only in MEN1 syndrome.
In particular, miR-4258 targets and represses the CCND1 gene, encoding for the cyclin D1, a positive regulator of cell cycle progression; thus, the down-regulation of miR-4258, subsequent to biallelic loss of wild type MEN1, could be responsible for the induction of uncontrolled parathyroid cell growth, leading to tumor onset and development.
MiR-1301 targets the RET oncogene (responsible for the development of MEN2A syndrome), the CDKN1B tumor suppressor gene (encoding the p27Kip1 protein, an inhibitor of cell cycle E-CDK2 and D-CDK4 cyclins and responsible for the development of MEN4 syndrome), RB1 tumor suppressor gene (whose inactivation is a common event in parathyroid tumorigenesis and biallelic loss is associated to parathyroid malignancy) [13], VDR gene (a regulator of PTH expression and calcium homeostasis) and PRDM2 gene (whose inactivation, via intragenic allelic loss, is common in parathyroid tumors and may cause parathyroid tumorigenesis via a mechanism in which genetic alteration of the MEN1 gene is redundant) [14]. Therefore, the up-regulation of miR-1301, subsequent to biallelic loss of wild type MEN1, represses the expression of CDKN1B, RB1 and PRDM2, favoring the progression of parathyroid tumorigenesis.
MiR-664 targets the CDKN2C gene and especially the CDC73/HRPT2 tumor suppressor gene, encoding parafibromin, (whose germinal and somatic inactivating mutations have been associated, respectively, with hyperparathyroidism jaw-tumor syndrome (HPT-JT) or sporadic parathyroid carcinomas). MiR-664 is down-regulated in parathyroid adenomas with biallelic loss of wild type MEN1 gene and this fact could explain why the development of parathyroid carcinoma is an extremely rare event in MEN1 syndrome.
In addition, we applied the MIR@NT@N computational approach to our identified miRNAs, to predict GRNs and sub-networks that could be implicated in parathyroid tumorigenesis and carcinogenesis. Thanks to this approach, we were able to identify two GRNs within the biological context of hyperparathyroidism and parathyroid tumorigenesis. The first identified network included beta-catenin, a transcription-activating protein with oncogenic potential, which is known to interact with different nodes that involve CCND1, CDKN1A, RB1 and CDKN1B. The identification of this network is in agreement with the excessive beta-catenin signaling reported as a major alteration in primary hyperparathyroidism and parathyroid tumors, and in secondary hyperparathyroidism associated to hyperplastic non-tumoral parathyroid glands [15].
The second identified network consists of a small circuit involving VDR, miR-664 and CDC73/HRPT2. However, no other genes involved in calcium homeostasis, such as CaSR, PTH, or PTHr, were found to be modulated by our selected miRNAs.
Looking at the cell fate decision during tumorigenesis as a dynamical system where change in the GRNs plays a pivotal role, we can speculate that these two identified dynamic networks could generate a hyperplastic dynamic microenvironment to arrange for the miR-24-menin “homeostatic network” perturbation and progression of MEN1 parathyroid neoplasia. These data also reinforced the “embryonic signature” hypothesis for parathyroid tumor, assuming that the repression of some specific miRNAs in parathyroid cancer cells is representative of the dedifferentiated tumor phenotype [16]. Many cancers have embryonic stem cell-like signatures [17]. Histologically, poorly differentiated tumors show preferential overexpression of genes normally enriched in embryonic stem cells, combined with preferential repression of Polycomb-regulated genes. Moreover, activation targets of Nanog, Oct4, Sox2, and c-Myc are more frequently overexpressed in poorly differentiated tumors than in well-differentiated tumors [17]. Indeed, miRNA-24-1 results to be an essential regulator of self-renewal and embryonic stem cell differentiation, operating as suppressors of embryonic stem cell self-renewal [18]. MiR-664 and miR-1301 are also differentially expressed in human embryonic stem cells [19]. Last, but not least, ComiR analysis showed that miR-664 targets NANOG and miR-1301 targets SOX2, both transcription factors that play an essential role in the growth and self-renewal of undifferentiated embryonic stem cells and neoplastic stem-like cells.
In conclusion, our study identified three new miRNAs involved in MEN1 parathyroid neoplasia, directly targeting genes associated with the development of different inheritable forms of parathyroid tumors. Some of these genes are organized in two GRNs in which both miR-1301 and miR-664 play a pivotal role.
These identified miRNAs could be revealed as prognostic and diagnostic biomarkers for parathyroid tumors to improve the diagnosis of MEN1 neoplasia and other syndromes.
Acknowledgements
The authors thank Dr. Luca Pandolfini (Scuola Normale Superiore, Pisa-Italy) for his help in the bioinformatic study of microarray raw data.
Disclosure of conflict of interest
None.
Authors’ contribution
EL: conceived the study, participated in its design and coordination, carried out the RNA molecular, cellular and bioinformatic studies, designed the experiments, drafted the manuscript. SC carried out qRT-PCR and helped in the draft of the manuscript. FM’s contribution “Performed MEN1 mutation analyses and 11q13 LOH analyses” helped in the draft of the manuscript. CM carried out microarray. GG carried out cellular preparations. MLB coordinated the study and revised the final version of the manuscript.
References
- 1.Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. 2008;9:831–842. doi: 10.1038/nrg2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saini HK, Griffiths-Jones S, Enright AJ. Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci U S A. 2007;104:17719–17724. doi: 10.1073/pnas.0703890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T, Brandi ML. Multiple endocrine neoplasia type 1 (MEN1): not only inherited endocrine tumors. Genet Med. 2009;11:825–835. doi: 10.1097/GIM.0b013e3181be5c97. [DOI] [PubMed] [Google Scholar]
- 4.Luzi E, Marini F, Giusti F, Galli G, Cavalli L, Brandi ML. The negative feedback-loop between the oncomir mir-24-1 and the oncosuppressor menin modulates the MEN1 tumorigenesis by mimicking the “Knudson’s second hit”. PLoS One. 2012;7:e39767. doi: 10.1371/journal.pone.0039767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth GK. Linear models and empirical Bayes for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 6.Coronello C, Benos PV. ComiR: Combinatorial microRNA target prediction tool. Nucleic Acids Res. 2013;41:W159–64. doi: 10.1093/nar/gkt379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Béchec A, Portales-Casamar E, Vetter G, Moes M, Zindy PJ, Saumet A, Arenillas D, Theillet C, Wasserman WW, Lecellier CH, Friederich E. MIR@NT@N: a framework integrating transcription factors, microRNAs and their targets to identify sub-network motifs in a meta-regulation network model. BMC Bioinformatics. 2011;12:67. doi: 10.1186/1471-2105-12-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiu C, Wang D, Wang E, Cui Q. An upstream interacting context based framework for the computational inference of microRNA functions. Mol Biosyst. 2012;8:1492–1498. doi: 10.1039/c2mb05469h. [DOI] [PubMed] [Google Scholar]
- 9.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rahbari R, Holloway AK, He M, Khanafshar E, Clark OH, Kebebew E. Identification of differentially expressed microRNA in parathyroid tumors. Ann Surg Oncol. 2011;18:1158–1165. doi: 10.1245/s10434-010-1359-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaira V, Elli F, Forno I, Guarnieri V, Verdelli C, Ferrero S, Scillitani A, Vicentini L, Cetani F, Mantovani G, Spada A, Bosari S, Corbetta S. The microRNA cluster C19MC is deregulated in parathyroid tumours. J Mol Endocrinol. 2012;49:115–124. doi: 10.1530/JME-11-0189. [DOI] [PubMed] [Google Scholar]
- 12.Corbetta S, Vaira V, Guarnieri V, Scillitani A, Eller-Vainicher C, Ferrero S, Vicentini L, Chiodini I, Bisceglia M, Beck-Peccoz P, Bosari S, Spada A. Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue. Endocr Relat Cancer. 2010;17:135–146. doi: 10.1677/ERC-09-0134. [DOI] [PubMed] [Google Scholar]
- 13.Cetani F, Pardi E, Viacava P, Pollina GD, Fanelli G, Picone A, Borsari S, Gazzerro E, Miccoli P, Berti P, Pinchera A, Marcocci C. A reappraisal of the Rb1 gene abnormalities in the diagnosis of parathyroid cancer. Clin Endocrinol (Oxf) 2004;60:99–106. doi: 10.1111/j.1365-2265.2004.01954.x. [DOI] [PubMed] [Google Scholar]
- 14.Carling T, Du Y, Fang W, Correa P, Huang S. Intragenic allelic loss and promoter hypermethylation of the RIZ1 tumor suppressor gene in parathyroid tumors and pheochromocytomas. Surgery. 2003;134:932–940. doi: 10.1016/s0039-6060(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 15.Bjorklund P, Akerstrom G, Westin G. Accumulation of nonphosphorylated bcatenin and cmyc in primary and uremic secondary hyperparathyroid tumors. J Clin Endocrinol Metab. 2007;92:338–344. doi: 10.1210/jc.2006-1197. [DOI] [PubMed] [Google Scholar]
- 16.Verdelli C, Forno I, Vaira V, Corbetta S. MicroRNA deregulation in parathyroid tumours suggests an embryonic signature. J Endocrinol Invest. 2015;38:383–388. doi: 10.1007/s40618-014-0234-y. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Y, Yao N, Liu G, Dong L, Liu Y, Zhang M, Wang F, Wang B, Wei X, Dong H, Wang L, Ji S, Zhang J, Wang Y, Huang Y, Yu J. Functional screen reveals essential roles of miR-27a/24 in differentiation of embryonic stem cells. EMBO J. 2015;34:361–378. doi: 10.15252/embj.201489957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morin RD, O’Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, Eaves CJ, Marra MA. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]