

Abstract
Pulmonary fibrosis is a complex pathological process characterized by massive destruction of the structure of lung tissues and aggravated pulmonary function impairment. The underlying mechanisms of pulmonary fibrosis are incompletely understood and therefore limited treatment options are available currently. Here, we report that MLN4924, an NEDD8 activation enzyme (NAE) activity-inhibiting molecule, blocks the maintenance and progression of established pulmonary fibrosis. We found that MLN4924 acts against bleomycin-induced pulmonary fibrosis mainly at the early inflammatory stage. Pharmacologically targeting the neddylation of Cullin-Ring E3 ligase (CRL) by MLN4924, significantly abrogated NF-κB responses, suppressed MAPK activity, and reduced secretion of TNF-α-elicited pro-inflammatory cytokines and MCP1-induced chemokines. MLN4924 inhibited pro-inflammatory responses while maintaining or increasing the production of the anti-inflammatory mediators such as anti-inflammatory interleukins (ILs) following bleomycin administration, which is closely correlated to its blocking NF-κB-mediated signaling. Consistently, our studies identified MLN4924 as a promising therapeutic drug for pulmonary fibrosis and suggested a potential role of MLN4924 that fine tunes the MAPK signaling pathway controlling the inflammatory reactions at the early stages of pulmonary fibrosis. In addition, our findings may broaden the potential practical application of MLN4924 as an effective therapeutic strategy against other inflammation-associated diseases.
Keywords: MLN4924, bleomycin, pulmonary fibrosis, inflammation
Introduction
Pulmonary fibrosis is a fatal form of progressive and irreversible lung damage with excessive accumulation of the extracellular matrix and striking destruction of normal pulmonary structures concomitant with pathological tissue reconstruction. Pulmonary fibrosis is generally caused by severe injury, infection, spontaneous immune reaction, toxic substances or adverse responses to drugs [1,2]. The lungs of patients with pulmonary fibrosis lose their function due to the break-down of normal tissue structure, leading to release of detrimental proteins/cytokines that accelerate the progression of pulmonary fibrosis in a bad feedback loop [2]. Pulmonary fibrosis is a complex pathological process that involves numerous cell types and cytokines [2,3], and the underlying mechanisms of such pathogenesis haven’t been fully understood [4].
Previous studies concerning the pathogenesis of pulmonary fibrosis focused on the regulation of proliferation, differentiation and collagen secretion [4,5]. Recently, a large body of clinical and experimental evidences have shown that the number of inflammatory cells, proteins and cytokines in broncho alveolar lavage fluid (BALF) abnormally increase during the progression of pulmonary fibrosis, suggesting a close correlation between inflammatory mediators and pulmonary fibrosis [1,6-9]. In addition, one of the striking hallmarks of pulmonary fibrosis is loss of endothelial function and subsequent activation of the immune system, which is majorly considered as a direct outcome affected by intracellular pro-inflammatory responses [10]. Thus, inhibition of the early inflammation could be an intriguing alternative approach for blocking the formation of pulmonary fibrosis.
The NF-κB pathway has been well established as one of the key pathways for such pro-inflammatory signaling. NF-κB is regulated through IκB kinases and its activation relies on IκB kinase degradation through the 26S proteasome [10,11]. The ubiquitination of IκB kinase is mediated through Cullin (Cul) proteins, which requires a multistep process called Neddylation [10].
Neddylation is an important post-translational modification involved in the regulation of protein degradation, trafficking and tumorigenesis [12]. Neddylation is an enzymatic cascade catalyzed by E1 (NEDD8-activating enzyme (NAE1/UBA3)), E2 (NEDD8-conjugating enzyme (UBC12)), and E3 NEDD8 ligases [12]. However, the Cullin-Ring E3 ligases (CRLs), which are involved in the degradation of various proteins, are the most important targets of Neddylation [13].
MLN4924, an AMP analogue, is a specific inhibitor of the NEDD8 activation enzyme (NAE) [14]. By forming a complex with NAE, MLN4924 blocks the neddylation of CRLs, leading to its inactivation [14-16] and the resultant accumulation of multiple CRL substrates including DNA replication licensing proteins CDT1 and ORC1 as well as the cell cycle inhibitors p21, p27 and Weel [15]. However, most studies of MLN4924 focused on evaluating its antitumor activities [17] while its role in inflammation is still elusive. Here we report that by testing multiple types of small molecules, we identified MLN4924 could efficiently improve the lung function against bleomycin-induced pulmonary fibrosis at early inflammatory stage. We analyzed the effect of MLN4924 on certain pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) and the NF-κB signaling pathway in LPS-stimulated macrophages. We also examined endothelial function such as iNOS enzyme activity to assess the role of this molecular process during LPS-induced cytokine responsiveness. Our results demonstrate that pharmacologically targeting neddylation of Culs by MLN4924 abrogates the release of certain pro-inflammatory cytokines like MCP1 secreted from macrophages in response to LPS. In addition to the well-known NF-κB transduction pathway, our data show that MLN4924 inhibits LPS-induced cytokine up-regulation at translational level, albeit through a different molecular mechanism underlying the feedback activation of MAPK signaling, suggesting a plausible mechanism for its corresponding down-regulation of cell proliferation and subsequent abnormal tissue reconstruction. Thus, MLN4924 offers a novel treatment strategy for pulmonary fibrosis, which represents unique advantages on initiating the inhibition control of pro-inflammatory mediators and the resultant tissue lesions. Our study also sheds light on how to target specific small molecules towards the up-regulation of pro-inflammatory cytokines and the disease processes associated with such up-regulation.
Materials and methods
Cell lines
A549, RAW264.7 and 3T3 cells were purchased from the Shanghai cell bank of the Chinese academy of sciences. RAW264.7 and 3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal calf serum (Gemini) and penicillin-streptomycin antibiotics (Sangon Biotech). A549 cells were cultured in RPMI-1640 medium (Gibco) supplemented with 10% fetal calf serum (Gemini).
Immunoblotting
Immunoblotting was performed as we have previous described [18]. Briefly, cells were washed using PBS, lysed with SDS lysis buffer and boiled at 100°C for 15 min. Proteins were resolved by SDS/PAGE and transferred onto nitrocellulose membranes (Millipore). Endogenous IκBα, p-P38, P38, p-ERK, ERK, p-JNK and JNK were detected using antibodies purchased from Cell Signaling Technology (1:1000 dilution). Other antibodies were used as follows: Mcl-1 (Santa Cruz, 1:1000 dilution), α-Tubulin (Sigma, 1:3000) and cullin 1/3 (Epitomics, 1:1000 dilution). Immunoblots were analyzed using the Odyssey system (LI-COR Biosciences).
Quantitative real-time PCR
Total cellular RNA was isolated using the TRIzol reagent (Takara) according to the manufacturer’s instructions. cDNA was then synthesized from 500 ng of RNA using the cDNA Reverse Transcription Kit (Takara). Quantitative RT-PCR was performed with SYBR Premix Ex Taq (Takara) using the TaqMan Universal Master Mix II (Applied Biosystems). The PCR primers are in Supplementary Table 1. All reactions were performed in duplicate, and the experiments were repeated at least three times.
Mice
Mice were purchased from the National Rodent Laboratory Animal Resources, Shanghai Branch (Shanghai, China) and maintained in a laminar airflow cabinet under specific pathogen free conditions according to NIH standards established in the Guidelines for the Care and Use of Laboratory Animals. All protocols were approved by East China Normal University.
Bleomycin-induced lung fibrosis and acute lung injury (ALI)
To induce lung fibrosis, C57BL/6J male mice (8-12 weeks old) were anesthetized and subjected to intratracheal infusion of saline or a 5 mg/kg bleomycin sulfate saline solution. For acute lung injury (ALI), the indicated mice were treated with a single intratracheal dose of saline or LPS (5 mg/kg) for 48 hours.
MLN4924 treatment
For lung fibrosis, a dosage of 10 mg/kg MLN4924 or an equal volume of PBS was injected intraperitoneally into mice everyday 28 days after bleomycin administration and mice were sacrificed at the 35th day. For acute lung injury, the same dosage of MLN4924 or PBS was injected intraperitoneally everyday for 7 days as well as LPS administration.
Bronchoalveolar lavage fluid cell count
At the indicated day after intratracheal administration of bleomycin or LPS with or without MLN4924, the mice were euthanized, and BALF was collected by cannulating the trachea and lavaging the lung with 1 ml of sterile PBS three times to recover a final volume of 1 ml. The total cell number was determined using a hemocytometer. More than 200 cells were counted for each sample.
Hydroxyproline and NO measurement
Certain lung tissues were triturated and filtered through a 200-μm pore mesh to remove debris or cell clusters and subsequently used to analyze hydroxyproline levels. Lung hydroxyproline content is considered a biochemical index for parenchymal collagen content and was measured in the lung tissue homogenate with the iagnostic reagent kit (Nanjing Jiancheng Bioengineering) using a colorimetric method at 550 nm. NO content was measured by using Nitric Oxide Assay Kit (Beyotime). The residual lung tissue homogenates were stored at -80°C.
Flow cytometry of apoptotic cells
Total apoptotic cell percentage was evaluated using the PE-Annexin-V Apoptosis staining kit I (BD Pharmingen, San Diego, CA). After treatment with MLN4924 at the indicated doses for 18 hours, mice neutrophils were stained for 30 minutes using the kit. Positive cells were sorted using a FACSCalibur and analyzed by FlowJo software.
Histology
At the indicated days, mice were euthanized and their lungs were inflated and fixed with 4% paraformaldehyde. The lungs were surgically resected and embedded into paraffin wax. Lung sections (3 mm) were cut and stained with hematoxylin and eosin or Masson’s trichrome. The extent of lung injury and fibrosis was graded by a pathologist, blinded to the treatment groups, on a scale of 0 (for normal lung) to 8 (or severe distortion of structure and large tissue areas [19]. The major criteria examined included interstitial thickening of alveolar or bronchiolar walls, hydroxyproline, broncho alveolar lavage fluid cell count, collagen deposition, and inflammatory cell infiltration.
Statistical analysis
Numerical data and histograms were expressed as the mean ± SD. Comparisons between 2 groups were analyzed using 2-tailed unpaired Student’s t test. P-values < 0.05 were considered statistically significant. Analysis of mice was litter-based and at least three litters were analyzed for every parameter. All the experiments were repeated three to four times.
Results
MLN4924 suppressed pulmonary fibrosis lesions in mice induced with bleomycin
To examine whether MLN4924 has any effect on the progression of pulmonary fibrosis, we used the bleomycin-induced pulmonary fibrosis mouse model, which has similar pathological process to that of human pulmonary fibrosis [1,20-22]. The data of HE and Masson’s trichrome staining show that bleomycin treatment significantly destroys mouse lung structure accompanied by pulmonary interstitial collagen hyperplasia and deposition compared with the control group treated with saline (Ctrl) (Figure 1A), while treatment with MLN4924 significantly protected mice from the pulmonary damage by bleomycin (Figure 1B). Bleomycin treatment played an active role in upregulating hydroxyproline level in BALF and enhanced the expression level of collagen 1 and 3 [23,24]. Our data indicate that MLN4924 also significantly inhibits the bleomycin-induced pulmonary collagen expression and hydroxyproline production (Figure 1C and 1D). These results suggest that MLN4924 may block bleomycin-induced pulmonary fibrosis in mice.
Figure 1.
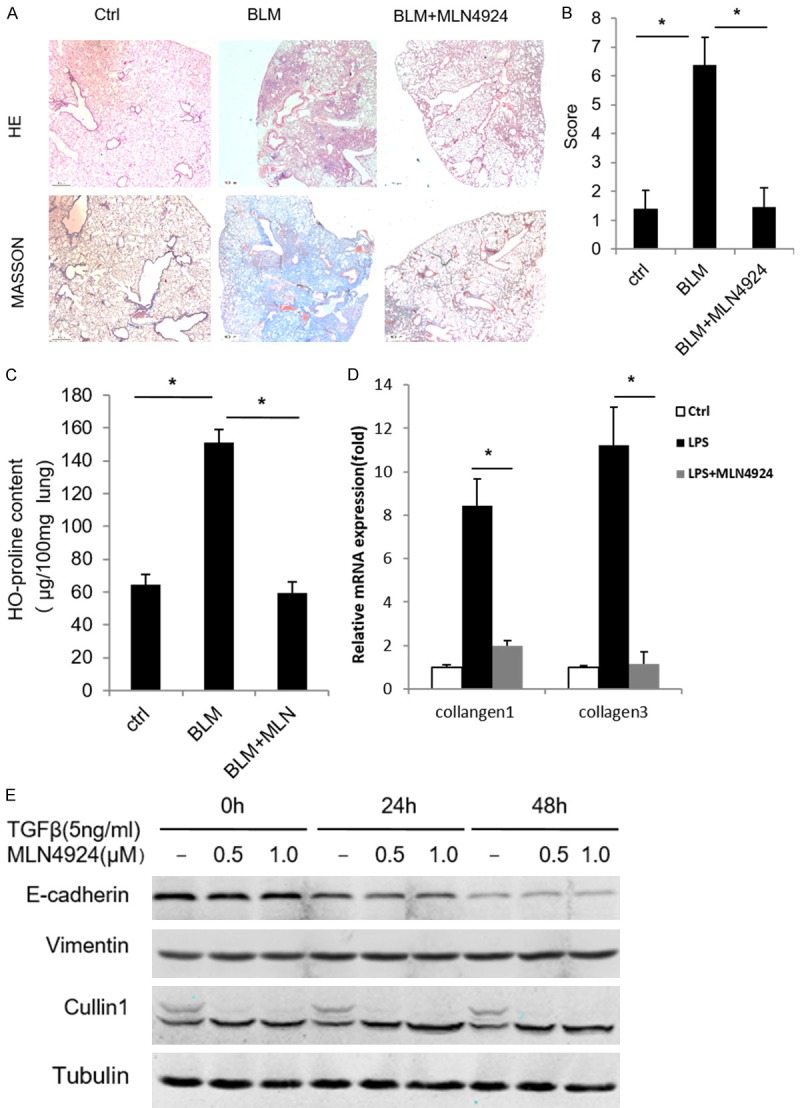
MLN4924 ameliorates bleomycin-induced pulmonary fibrosis. (A) Representative histology of HE and Masson’s trichrome staining of lungs from mice treated with one intratracheal dose of PBS or bleomycin (15 mg/kg), or with a intraperitoneal injection of MLN4924 (10 mg/kg) after bleomycin treatment, respectively (n=8). (B) Fibrosis scores based on histopathological assessment as in (A). (C, D) Hydroxyproline content and relative mRNA expression of mice collagen 1 and collagen 3 from lungs treated as in (A), respectively. Error bars represent the mean ± SD, representative of three experiments, *P < 0.05. (E) Western blotting of A549 cell lines treated with TGF (5 ng/ml) and MLN4924 (0.5 μM or 1 μM) for the indicated times, respectively.
MLN4924 ameliorates acute lung injury in the early stages of pulmonary fibrosis
The hyperproliferation of myofibroblasts is one of the major pathological features of pulmonary fibrosis [1]. Myofibroblasts that are critical for pulmonary fibrosis are mainly produced through Epithelial-Mesenchymal Transitions (EMT) [1,20-22,25]. Thus we examine whether MLN4924 has effect on EMT. To this end, we induce the EMT of A549, a human lung adenocarcinoma epithelial cell line, through treatment with TGFβ (5 ng/ml). EMT is examined by measuring the expression level of EMT markers. It shows that MLN4924 has no significant effect on TGFβ-induced EMT in A549 cells (Figure 1E), suggesting that MLN4924 has no functional impact on the TGFβ-induced EMT.
It has been well established that inflammation is intimately involved in the initiation and progression of pulmonary fibrosis [1,6]. Since MLN4924 had little impact on the EMT process, we investigate whether it functions actively at the early inflammatory stage of pulmonary fibrosis. A LPS-induced acute lung injury (ALI) mouse model is used. Treatment with LPS for two days significantly induced disrupted alveoli, thickened alveoli septum and infiltration of inflammatory cells in murine lungs, which can be remarkably reversed by additional treatment with MLN4924 (Figure 2A). The wet-to-dry ratio and the total protein content in BALF, the markers representing the severity of acute lung injury, are changed in a consistent way (Figure 2B and 2C). Moreover, the expression level of cytokines (IL-1β, IL-6 and TNFα) in murine lung lysates treated with LPS are reduced by MLN4924 treatment (Figure 2D-F). Similar results are observed from the accumulation of neutrophils, macrophages as well as the total cell numbers in BALF throughout all the stages (days 1, 3 and 7) of ALI (Figure 2G-I). Taken together, these results suggest that MLN4924 protects against pulmonary fibrosis through attenuation of cytokine-mediated inflammation at early stages of ALI-related pulmonary fibrosis.
Figure 2.
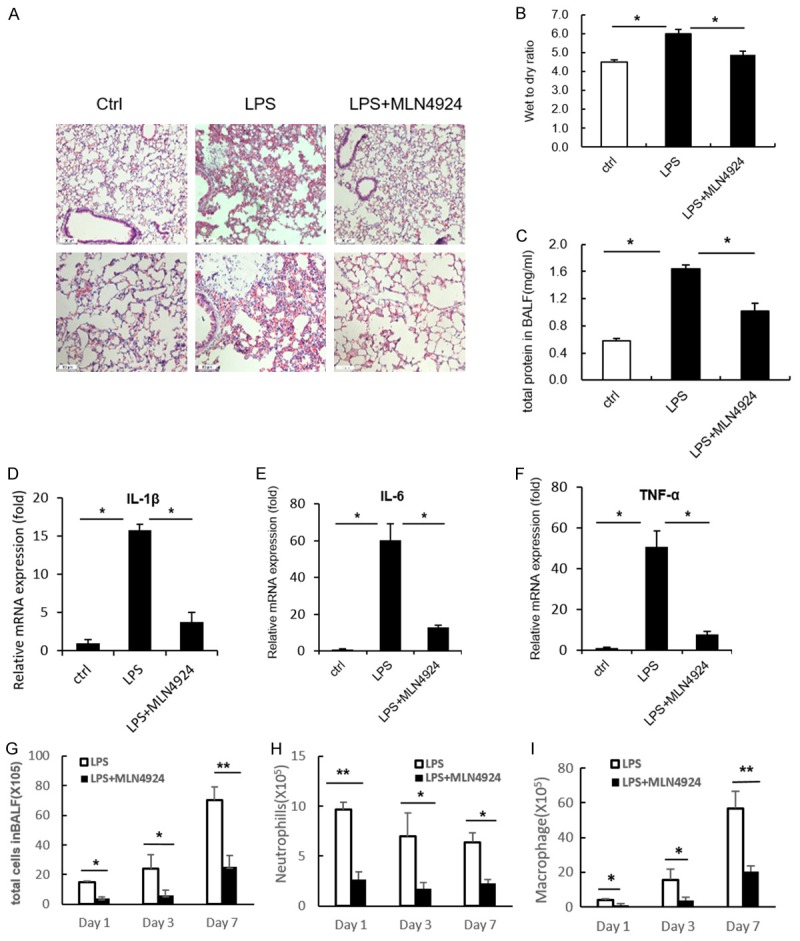
MLN4924 ameliorates LPS-induced acute lung injury. (A) Representative histology of HE staining of lungs from mice treated with a single intratracheal dose of PBS or LPS (5 mg/kg), or with intraperitoneal injection of MLN4924 (10 mg/kg) after LPS treatment, respectively (n=8). (B) Determination of lung wet-to-dry ratio for mice treated as in (A). (C) Protein concentrations of broncho alveolar lavage fluid in lungs of mice treated as in (A). (D-F) Relative mRNA levels of IL-6, IL-1β and TNFα in lung lysates of mice treated as in (A). (G-I) Total cell counts and neutrophil and macrophage numbers in the broncho alveolar lavage fluid of lungs in mice treated with PBS or MLN4924 after LPS intratracheal injection. Error bars represent the mean ± SD, representative of three experiments, *P < 0.05.
MLN4924 inhibits the expression of pro-inflammatory cytokines and chemokines in vitro
Pulmonary macrophages are important indicatives for the initiation of pulmonary fibrosis. They are responsible for releasing pro-inflammatory mediators at early stages of inflammation [1,26,27]. To test whether MLN4924 takes a part in mediating the inflammation-induced effects on macrophage, we investigate whether MLN4924 affects the production of pro-inflammatory mediators. Our data show that treatment with MLN4924 dramatically reduces LPS-induced iNOS expression and NO production in RAW264.7 cells (Figure 3A and 3B). Since it has been reported that MLN4924 inhibits pro-inflammatory cytokines (IL-6 and TNFα) in macrophages [11], we check LPS-induced expression of cytokines and chemokines such as IL-1β, TNFα and MCP-1. Their expressions are also suppressed by MLN4924 treatment (Figure 3C-E). Collectively, MLN4924 may protect against pulmonary fibrosis by inhibiting the production of cytokine, chemokines and NO from macrophages and thus suppressing macrophage-mediated excessive inflammation.
Figure 3.
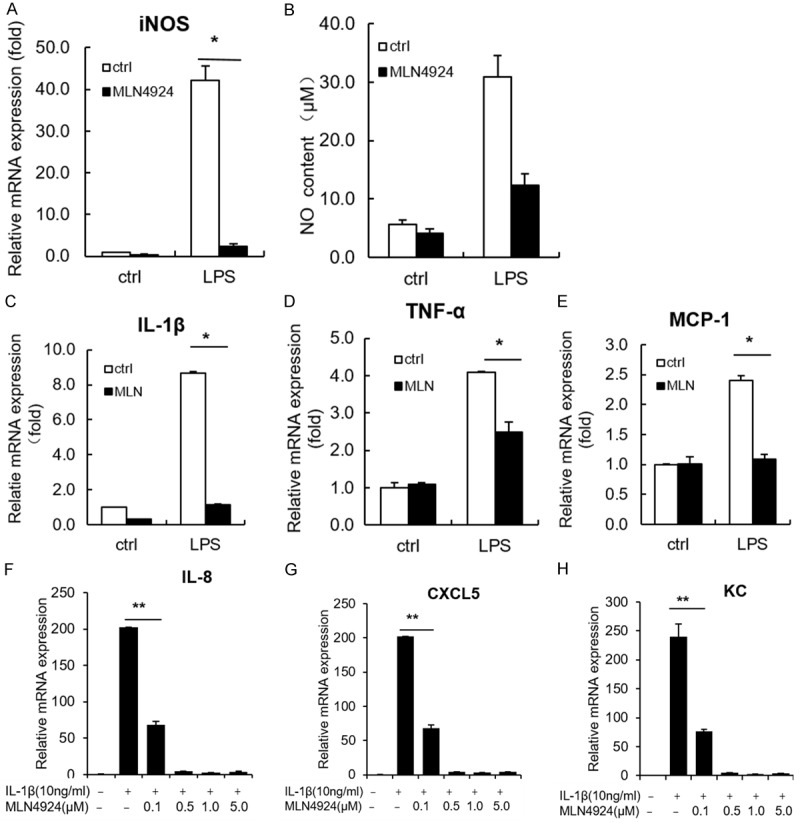
MLN4924 inhibits the expression of pro-inflammatory cytokines and chemokines in vitro. A, B. Determination of iNOS mRNA expression and NO production in LPS (1 μg/ml)-treated RAW264.7 cells, together with MLN4924 (0.5 μM) or PBS treatment for 24 hrs. C-E. Relative mRNA levels of IL-1β, TNFα and MCP-1 in LPS (1 μg/ml)-treated RAW264.7 cells, together with MLN4924 (0.5 μM) or PBS treatment for 12 hrs. F-H. Relative mRNA levels of IL-8, CXCL5 and KC in an A549 cell line treated with IL-1β (10 ng/ml) and MLN4924 at the indicated doses for 24 hrs. Error bars represent the mean ± SD, representative of three experiments, *P < 0.05.
Neutrophils are also involved in the progression of ALI [28]. During the early stages of pulmonary fibrosis, accumulation and transfer of neutrophils to the alveolar space are considered as major mediators for excessive pulmonary inflammation [29]. In the later stages, neutrophils are removed predominantly through apoptosis [30]. As we observed that MLN4924 reduced the accumulation of neutrophils during ALI, we examined whether MLN4924 affects neutrophil apoptosis. Neutrophils isolated from mouse bone marrow were treated with MLN4924 for 18 hours and apoptosis is analyzed using flow cytometry. It shows that MLN4924 had no significant effect on neutrophil apoptosis except at a high dosage (5 μM) (Supplementary Figure 1A). In addition, MLN4924 treatment had no effect on the expression level of Mcl-1, a marker of apoptosis (Supplementary Figure 1B). These results indicate that MLN4924 does not affect neutrophil apoptosis.
At the initial stages of ALI or pulmonary fibrosis, alveolar epithelial cells are damaged upon infection or injury [31,32]. Damaged epithelia produce additional chemokines that recruit pro-inflammatory cells to lung and trigger further immune responses [1]. We thus investigated whether MLN4924 affects the chemokine production of alveolar epithelia. It shows that MLN4924 significantly inhibits IL-1β-induced production of IL-8, CXCL5 and KC in A549 cells (Figure 3F-H), suggesting that MLN4924 can block inflammation-triggered diseases by reducing the chemokine production of alveolar epithelia.
Taken together, our data indicate that MLN4924 suppresses the immune responses of ALI by orchestrating the expression of various cytokines and chemokines produced by both alveolar epithelia and inflammatory cells such as macrophages.
MLN4924 functions through both the NF-κB and MAPK signaling pathways
Next, we investigate the underlying mechanisms by which MLN4924 blocks the production of chemokines and cytokines in both macrophages and epithelial cells. NF-κB pathway is well known in producing cytokines such as IL-1β, IL-6 and TNFα and releasing ROS and NO [33,34]. Therefore, we investigate whether MLN4924 blocks the NF-κB signaling pathway in macrophages. Our data show that MLN4924 significantly blocks the LPS-induced degradation of IκBα even at low concentrations (0.5 μM). The activity of MLN4924 is examined by Cullin1 neddylation (Figure 4A). Similarly, MLN4924 also blocks IκBα degradation in A549 cells (Figure 4B). Therefore, we conclude that MLN4924 efficiently inhibits IκBα degradation in both macrophages and alveolar epithelial cells in vitro.
Figure 4.
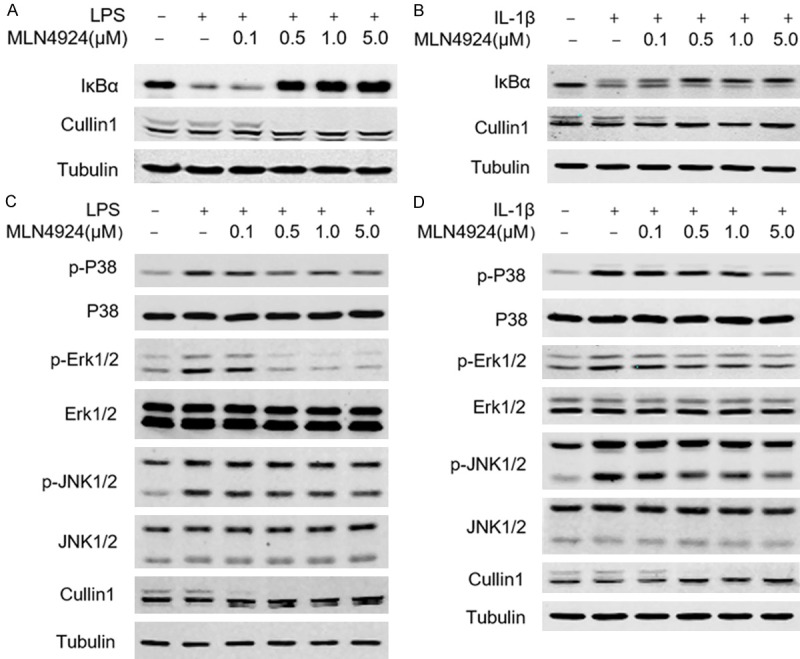
MLN4924 functions through the NF-κB signaling pathway. A, B. Western blotting analysis of NF-κB and MAPK signaling in RAW264.7 cells under LPS (1 μg/ml) and MLN4924 treatment at the indicated doses. C, D. Western blotting analysis of NF-κB and MAPK signaling in an A549 cell line under IL-1β (10 ng/ml) and MLN4924 treatment at the indicated doses.
It has been reported that mitogen-activated protein kinase (MAPK) pathway regulates both innate and acquired immune responses, as well as pulmonary fibrosis [35-37]. Consistently, we find that MLN4924 significantly inhibited LPS-induced phosphorylation of P38, Erk1/2 and Jnk1/2 in RAW264.7 cells (Figure 4C). Similar results are obtained in A549 cells (Figure 4D). These results indicate that MLN4924 blocks the activation of MAPK and NF-κB pathways in both macrophages and alveolar epithelia.
Discussion
Pulmonary fibrosis with persistent lung destruction, accumulation of the extracellular matrix and intemperate lung rebuilding, is refractory to treatment and has a high mortality [1]. The pathological process of pulmonary fibrosis is still elusive and no effective treatment is available [4]. In this study, we found that MLN4924, a small anticancer molecule that inactivates the Cullin-Ring E3 ligase through specific inhibition of the Nedd8-activating enzyme (NAE) [2,3], is a promising therapeutic candidate for bleomycin-induced pulmonary fibrosis (Figure 1).
Although pulmonary fibrosis is a complex process, we found that MLN4924 mainly protects against pulmonary fibrosis at early stage. EMT is one of the most important sources of ECM-produced myofibroblasts and vital for pulmonary fibrosis progression [1,25]. However, we did not find any effect of MLN4924 on EMT of lung epithelial cells whereas we did found that MLN4924 significantly blocked production of the pro-inflammatory cytokines and chemokines produced by both epithelial cells and macrophages. Thus, our data show that MLN4924 significantly blocked acute lung injury at the early inflammatory stage.
Several studies have shown that the NF-κB and MAPK signaling pathways play important roles in immune responses [33,37,38]. We found that MLN4924 blocked the expression of inflammatory cytokines and chemokines in macrophages and epithelial cells through the inhibition of NF-κB and MAPK signal pathways. Because MLN4924 inhibits NAE activity that is critical for numerous Cullin-Ring E3s, it is difficult to determine which E3s are inhibited by MLN4924. Several studies have shown that MLN4924 blocks the degradation of IκBα in different cells [11]. Interestingly, we found that MNL4924 can also significantly block the activation of MAPK by LPS. Since MLN4924 is known as a selective inhibitor of the NAE and CULs, and TRAF6 is the upstream signal of both NF-κB and MAPK pathways, we investigated the neddylation and ubiquitination of TRAF6 with stimulation of MLN4924. However, no significant modification changes were found (data not shown). Thus, it will be of great interest to examine the mechanism by which MLN4924 inhibits the activation of the MAPK pathways.
As an inhibitor of neddylation, accumulating data have shown that MLN4924 has the potential for various cancer treatments. Interestingly, different mechanisms underlying the change of cancer cell proliferation by MLN4924 have been reported. For example, in myeloma cells, MLN4924 has been shown to induce apoptosis via inhibition of Akt and mTOR through upregulation of REDD1 [14]. MLN4924 also induces the apoptosis of chronic lymphocytic leukemia (CLL) B cells by blocking the activation of both the canonical and noncanonical NF-κB pathways [39]. MLN4924 may also suppress liver cancer cell growth by inhibiting the mTOR pathway [40]. In this study, we identified a novel potential role of MLN4924 in treating acute lung injury and pulmonary fibrosis. As MLN4924 significantly blocks the production of pro-inflammatory mediators, ur results may implicate the potential role of MLN4924 as an effective treatment strategy for other inflammatory diseases, such as Rheumatoid Arthritis (RA) and Osteo Arthritis (OA).
In summary, our data support MLN4924 as a novel potential drug for the clinical treatment of pulmonary fibrosis, in addition to its current application in anti-cancer therapy (Figure 5).
Figure 5.
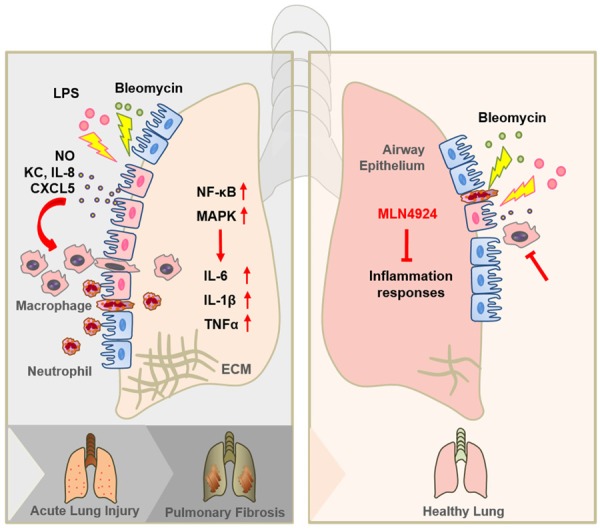
Graphic abstract of MLN4924 protects against bleomycin-induced pulmonary fibrosis by inhibiting the early inflammatory process. When lung injury occurs, the airway epithelia firstly detect the injury and recruit inflammatory cells by secreting chemokines, such as KC, IL-8 and CXCL5. Recruited inflammatory cells, including macrophages and neutrophils, trigger inflammation by producing NO and several cytokines, including IL-6, IL-1β and TNFα, and thus contributing to acute lung injury. When the injury proceeds, extracellular matrix (ECM) begins to form and deposit, which at last leads to pulmonary fibrosis. MLN4924 can effectively block the production of chemokines from the airway epithelia as well as NO and cytokines from the inflammatory cells. By this mechanism MLN4924 suppresses the recruitment of inflammatory cells and the initiation of inflammation responses.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loomis-King H, Flaherty KR, Moore BB. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr Opin Pharmacol. 2013;13:377–385. doi: 10.1016/j.coph.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, Desai L, Bernard K, Thannickal VJ. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123:1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND, Hart WK, Pardo A, Blackwell TS, Xu Y, Chun J, Luster AD. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 7.Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, Couillin I. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179:903–913. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- 8.Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009–1017. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- 9.Heijerman H. Infection and inflammation in cystic fibrosis: a short review. J Cyst Fibros. 2005;4(Suppl 2):3–5. doi: 10.1016/j.jcf.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Ehrentraut SF, Kominsky DJ, Glover LE, Campbell EL, Kelly CJ, Bowers BE, Bayless AJ, Colgan SP. Central role for endothelial human deneddylase-1/SENP8 in fine-tuning the vascular inflammatory response. J Immunol. 2013;190:392–400. doi: 10.4049/jimmunol.1202041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang FM, Reyna SM, Granados JC, Wei SJ, Innis-Whitehouse W, Maffi SK, Rodriguez E, Slaga TJ, Short JD. Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem. 2012;287:35756–35767. doi: 10.1074/jbc.M112.397703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kandala S, Kim I, Su H. Neddylation and deneddylation in cardiac biology. Am J Cardiovasc Dis. 2014;4:140–158. [PMC free article] [PubMed] [Google Scholar]
- 13.Rabut G, Peter M. Function and regulation of protein neddylation. ‘protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, Xiong X, Jia L, Sun Y. Targeting cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012;3:e386. doi: 10.1038/cddis.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 16.Wei D, Morgan MA, Sun Y. Radiosensitization of cancer cells by inactivation of cullin-RING E3 ubiquitin ligases. Transl Oncol. 2012;5:305–312. doi: 10.1593/tlo.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Claperon A, Chretien Y, Rey C, Scatton O, Soubrane O, Conti F, Praz F, Housset C, Rosmorduc O, Desbois-Mouthon C. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013;73:3974–3986. doi: 10.1158/0008-5472.CAN-12-3824. [DOI] [PubMed] [Google Scholar]
- 18.Xiao N, Li H, Luo J, Wang R, Chen H, Chen J, Wang P. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFalpha-induced cancer cell migration. Biochem J. 2012;441:979–986. doi: 10.1042/BJ20111358. [DOI] [PubMed] [Google Scholar]
- 19.Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan X, Wang J, Wang C, Sommer E, Kozasa T, Srinivasula S, Alessi D, Offermanns S, Simon MI, Wu D. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat Cell Biol. 2012;14:686–696. doi: 10.1038/ncb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, Minoo P. Epithelium-specific deletion of TGFbeta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011;121:277–287. doi: 10.1172/JCI42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flechsig P, Dadrich M, Bickelhaupt S, Jenne J, Hauser K, Timke C, Peschke P, Hahn EW, Grone HJ, Yingling J, Lahn M, Wirkner U, Huber PE. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGFbeta and BMP-associated proinflammatory and proangiogenic signals. Clin Cancer Res. 2012;18:3616–3627. doi: 10.1158/1078-0432.CCR-11-2855. [DOI] [PubMed] [Google Scholar]
- 23.Westergren-Thorsson G, Hernnäs J, Särnstrand B, Oldberg Å, Heinegård D, Malmström A. Altered expression of small proteoglycans, collagen, and transforming growth factor-beta 1 in developing bleomycin-induced pulmonary fibrosis in rats. J Clin Invest. 1993;92:632–7. doi: 10.1172/JCI116631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giri SN, Hyde DM, Hollinger MA. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax. 1993;48:959–966. doi: 10.1136/thx.48.10.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, Graham GJ, Kurowska- Stolarska M, Liew FY, McSharry C, Xu D. IL- 33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134:1422–1432. e1411. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang S, Park DW, Tadie JM, Gregoire M, Deshane J, Pittet JF, Abraham E, Zmijewski JW. Human resistin promotes neutrophil proinflammatory activation and neutrophil extracellular trap formation and increases severity of acute lung injury. J Immunol. 2014;192:4795–4803. doi: 10.4049/jimmunol.1302764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Bordon J, Aliberti S, Fernandez-Botran R, Uriarte SM, Rane MJ, Duvvuri P, Peyrani P, Morlacchi LC, Blasi F, Ramirez JA. Understanding the roles of cytokines and neutrophil activity and neutrophil apoptosis in the protective versus deleterious inflammatory response in pneumonia. Int J Infect Dis. 2013;17:e76–83. doi: 10.1016/j.ijid.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc. 2005;2:206–213. doi: 10.1513/pats.200501-009AC. [DOI] [PubMed] [Google Scholar]
- 32.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 33.Zhu T, Wang DX, Zhang W, Liao XQ, Guan X, Bo H, Sun JY, Huang NW, He J, Zhang YK, Tong J, Li CY. Andrographolide protects against LPS-induced acute lung injury by inactivation of NF-kappaB. PLoS One. 2013;8:e56407. doi: 10.1371/journal.pone.0056407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida K, Kuwano K, Hagimoto N, Watanabe K, Matsuba T, Fujita M, Inoshima I, Hara N. MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J Pathol. 2002;198:388–396. doi: 10.1002/path.1208. [DOI] [PubMed] [Google Scholar]
- 36.Underwood DC, Osborn RR, Bochnowicz S, Webb EF, Rieman DJ, Lee JC, Romanic AM, Adams JL, Hay DW, Griswold DE. SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L895–L902. doi: 10.1152/ajplung.2000.279.5.L895. [DOI] [PubMed] [Google Scholar]
- 37.Gao M, Chen L, Yu H, Sun Q, Kou J, Yu B. Diosgenin down-regulates NF-kappaB p65/p50 and p38MAPK pathways and attenuates acute lung injury induced by lipopolysaccharide in mice. Int Immunopharmacol. 2013;15:240–245. doi: 10.1016/j.intimp.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Langereis JD, Raaijmakers HA, Ulfman LH, Koenderman L. Abrogation of NF-κB signaling in human neutrophils induces neutrophil survival through sustained p38-MAPK activation. J Leukoc Biol. 2010;88:655–664. doi: 10.1189/jlb.0809544. [DOI] [PubMed] [Google Scholar]
- 39.Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, Danilov AV. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironmentdriven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20:1576–1589. doi: 10.1158/1078-0432.CCR-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang D, Zhao Y, Liu J, Sun Y, Jia L. Protective autophagy induced by RBX1/ROC1 knockdown or CRL inactivation via modulating the DEPTOR-MTOR axis. Autophagy. 2012;8:1856–1858. doi: 10.4161/auto.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.