

Abstract

17β-Hydroxysteroid dehydrogenase type 2 (17β-HSD2) converts the active steroid hormones estradiol, testosterone, and 5α-dihydrotestosterone into their weakly active forms estrone, Δ4-androstene-3,17-dione, and 5α-androstane-3,17-dione, respectively, thereby regulating cell- and tissue-specific steroid action. As reduced levels of active steroids are associated with compromised bone health and onset of osteoporosis, 17β-HSD2 is considered a target for antiosteoporotic treatment. In this study, a pharmacophore model based on 17β-HSD2 inhibitors was applied to a virtual screening of various databases containing natural products in order to discover new lead structures from nature. In total, 36 hit molecules were selected for biological evaluation. Of these compounds, 12 inhibited 17β-HSD2 with nanomolar to low micromolar IC50 values. The most potent compounds, nordihydroguaiaretic acid (1), IC50 0.38 ± 0.04 μM, (−)-dihydroguaiaretic acid (4), IC50 0.94 ± 0.02 μM, isoliquiritigenin (6), IC50 0.36 ± 0.08 μM, and ethyl vanillate (12), IC50 1.28 ± 0.26 μM, showed 8-fold or higher selectivity over 17β-HSD1. As some of the identified compounds belong to the same structural class, structure–activity relationships were derived for these molecules. Thus, this study describes new 17β-HSD2 inhibitors from nature and provides insights into the binding pocket of 17β-HSD2, offering a promising starting point for further research in this area.
17β-Hydroxysteroid dehydrogenase type 2 (17β-HSD2) belongs to a large family of short-chain dehydrogenase/reductase (SDR) enzymes with the systematic name SDR9C2.1 It is mainly expressed in the placenta, endometrium, breast, prostate, small intestine, liver, and bone.2−5 This NAD+-dependent enzyme converts active sex steroid hormones such as estradiol, testosterone, and 5α-dihydrotestosterone into their respective inactive forms, namely, estrone, Δ4-androstene-3,17-dione (androstenedione), and 5α-androstane-3,17-dione (androstanedione), thereby protecting tissues from excessive sex steroid hormone action (Figure 1).6,7 Furthermore, 17β-HSD2 catalyzes the oxidation of Δ5-androstene-3β,17β-diol (androstenediol) to dehydroepiandrosterone (DHEA). The enzyme shares considerable structural and functional similarity with other extensively studied SDR enzymes such as 17β-HSD1 and 17β-HSD3.8 In contrast to 17β-HSD2, the enzymes 17β-HSD1, 17β-HSD3, and the aldo-keto-reductase 17β-HSD5 (also known as AKR1C3) are oxidoreductases converting the weak estrogen estrone to the potent estradiol and the weak androgens androstenedione and androstanedione to testosterone and 5α-dihydrotestosterone, respectively.9−11 Whereas 17β-HSD3 is responsible for the last step of testosterone synthesis in the testes, 17β-HSD5 is responsible for the production of extratesticular testosterone and plays a crucial role in androgen maintenance in the elderly.9,10
Figure 1.

Enzymatic reactions catalyzed by 17β-HSD2 and reverse reactions catalyzed by other HSD enzymes.
Owing to its favorable localization and its role as a main contributor to the inactivation of estradiol, testosterone, and 5α-dihydrotestosterone in bone cells,2 17β-HSD2 has been proposed as a promising target for the treatment of osteoporosis.12 This condition, where decreased bone density leads to an increased fracture risk, is in the majority of cases linked with the age-related decrease of sex steroid hormones.13 The age-related onset of osteoporosis in postmenopausal women14 and men with low testosterone levels15 can be explained, at least in part, by a decline in the concentrations of estradiol and testosterone, which inhibit bone degradation.16 Thus, by inhibiting 17β-HSD2, the amount of active steroids can be locally increased in the bones, thereby improving bone health. This hypothesis is supported by an in vivo study, where a 17β-HSD2 inhibitor was administered to ovariectomized cynomolgus monkeys.17 In this study, the 17β-HSD2 inhibitor was shown to improve bone strength by increasing bone formation and decreasing bone resorption, although the effects were rather weak and only observed at the highest dose of 25 mg/kg/day.
Although multiple synthetic 17β-HSD2 inhibitors have already been reported,18−21 natural products inhibiting this enzyme are currently underexplored. There are only a few reports on natural product inhibitors of 17β-HSD2 and other steroid-metabolizing enzymes, and the majority of these compounds are flavonoids.22−24 Flavonoids share certain functional similarities with steroids and can be considered as steroid mimetics (Figure S1, Supporting Information). However, most of these compounds are not selective. They also inhibit other members of the SDR enzyme family, and, additionally, they frequently show activity toward estrogen and androgen receptors. Nevertheless, natural compounds play an important role in providing new structures as potential lead candidates in drug discovery, and hence they are of high general interest.25,26 Remarkably, from 1999 to 2008, 28% of all new FDA-approved, first-in-class small-molecule drugs were natural products or compounds derived thereof.27
Despite the fact that osteoporosis is not well represented among the conditions treated with plants and phytotherapy,28 there are many other conditions related to bone homeostasis and fractures that are reported in the literature on ethnopharmacology. Interestingly, an ethnopharmacological study has been reported that shows that plants such as Pholidota articulate Lindl. and Coelogyne cristata Lindl. (both of the Orchidaceae family) contain several flavonoids that are used to treat bone fractures in India.29 Even though part of the observed effects of these compounds may be due to direct modulation of estrogen and androgen receptor activities, the mechanism of action of these compounds in the treatment of bone-related conditions is largely unknown. Accordingly, 17β-HSD2 inhibition might well contribute to the effects of these herbal remedies.
As natural compounds represent a rich source of potential lead structures, novel 17β-HSD2 inhibitors of natural origin were searched using in silico methods. Previously, a procedure to discover new synthetic chemicals that inhibit 17β-HSD2 was established.19 In this previous study, pharmacophore models representing the chemical functionalities and steric requirements essential for the activity of small molecules toward 17β-HSD2 were constructed and employed for virtual screening of a commercial synthetic chemical database. From this previous experimental validation, the two pharmacophore models 1 and 2 (Figure 2) showed good predictive power, with positive hit rates of 50% and 10%, respectively. Although the models are very similar in feature types and distribution, they differ slightly in feature location, which is why they may lead to somewhat different virtual hits. Thus, both of these models were selected for virtual screening of selected natural product databases.
Figure 2.

Pharmacophore models for 17β-HSD2 inhibitors. (A) Chemical features of models 1 and 2 describing the types, locations, and tolerance spheres of inhibitory chemical functionalities. Pharmacophore features are colored as follows: red, hydrogen-bond acceptor; green, hydrogen-bond donor; yellow, hydrophobic; and blue, aromatic ring. Optional features are depicted in scattered style. (B) Full versions of models 1 and 2 with gray exclusion volumes as steric restraints for inhibitor size (forbidden areas). A 3D video view of model 1 is available as Supporting Information.
Results and Discussion
In-house natural product databases based on input from several academic institutions (total of 439 entries) and the Sigma-Aldrich catalogue (Sigma-Aldrich, St. Louis, MO, USA), containing natural products and synthetic compounds, were screened virtually using the two pharmacophore models. The virtual screening procedure and its results are described in detail in the Supporting Information (text and Table S1). As the full models were quite restrictive, most databases were also screened in models where one omitted feature (omf) was applied during the pharmacophore mapping.
The 36 selected virtual hits were evaluated in an in vitro assay using lysates of cells expressing the recombinant human enzyme 17β-HSD2. Initially, all compounds were tested at a final concentration of 20 μM. Compounds showing more than 50% inhibition at that concentration are shown in Tables 1 and 2 as well as Figures 3 and 4. For all compounds inhibiting 17β-HSD2 activity by at least 70% (remaining activity ≤30% of vehicle control), IC50 values were determined. The complete list of the compounds tested is provided in Table S2, Supporting Information.
Table 1. Active Hit Compounds of Natural Origin, Databases, Mapping Pharmacophore Models, and Activities against 17β-HSD2.
| compound | database | pharmacophore models | remaining activity at 20 μM (% of control) or IC50 |
|---|---|---|---|
| nordihydroguaiaretic acid (1) | Atanasov | models 1 and 2 | 0.38 ± 0.04 μM |
| oleanolic acid (2) | Atanosov | model 1 omfa | 49 ± 6% |
| curcumin (3) | Atanosov | models 1 and 2 omf | 1.73 ± 0.2 μM |
| (−)-dihydroguaiaretic acid (4) | Davis | models 1 and 2 | 0.94 ± 0.02 μM |
| jaceosidin (5) | Davis | models 1 and 2 omf | 9.3 ± 2.3 μM |
| isoliquiritigenin (6) | Davis | models 1 and 2 | 0.36 ± 0.08 μM |
| pinoresinol (7) | Waltenberger | models 1 and 2 | 42 ± 5% |
| lupinalbin A (8) | Krenn | model 2 omf | 1.52 ± 0.15 μM |
| 2′-hydroxygenistein (9) | Krenn | model 2 omf | 2.03 ± 0.37 μM |
| butein (10) | Sigma | model 1 | 7.3 ± 2.7 μM |
| rosmarinic acid (11) | Sigma | model 1 | 3.72 ± 0.17 μM |
| ethyl vanillate (12) | Sigma | model 1 | 1.28 ± 0.26 μM |
omf, screening by allowing one omitted feature.
Table 2. Active Semisynthetic Fungal Natural Products, Origin, Mapping Pharmacophore Models, and Activities against 17β-HSD2.
| compound | database | pharmacophore models | remaining activity at 20 μM (% of control) or IC50 |
|---|---|---|---|
| 2-(3-chloro-4-hydroxyphenyl)-N-phenethylacetamide (13) | Davis | model 1 | 1.57 ± 0.16 μM |
| 2-(3-chloro-4-hydroxyphenyl)-N-(2-methoxyethyl)acetamide (14) | Davis | model 1 omfa | 37 ± 3% |
| N-butyl-2-(3-chloro-4-hydroxyphenyl)acetamide (15) | Davis | model 1 | 33 ± 6% |
| N-benzyl-2-(3-chloro-4-hydroxyphenyl)acetamide (16) | Davis | model 1 | 3.42 ± 0.74 μM |
| N-(2-(1H-indol-3-yl)ethyl)-2-(3-chloro-4-hydroxyphenyl)acetamide (17) | Davis | model 1 | 0.98 ± 0.24 μM |
| 2-(3-chloro-4-hydroxyphenyl)-N-(2-chlorobenzyl)acetamide (18) | Davis | model 1 | 0.78 ± 0.16 μM |
omf, screening by allowing one omitted feature.
Figure 3.

Structures of natural products identified in this study that inhibit 17β-HSD2.
Figure 4.

Semisynthetic fungal natural products that inhibit 17β-HSD2.
From the selected 36 tested compounds, 12 were active with IC50 values of <5 μM, six were moderately active showing at least 50% inhibition at a compound concentration of 20 μM, and the remaining compounds were considered inactive. Altogether, this corresponds to a 50% hit rate, indicating that the pharmacophore models performed explicitly well, not only for synthetic molecules but also for natural compounds. This is an important aspect, because natural products often differ from synthetic drug-like structures. From the 33 in-house database-derived test compounds, 10 fit into model 1 and four into model 2, respectively, without omitted features during the screening (Tables 1 and 2). Remarkably, all these hits were active in vitro. Additionally, the strategy of allowing one pharmacophore feature to be left out during the natural product database screening proved successful: The hits obtained by allowing one omitted feature additionally included the active compounds oleanolic acid (2), curcumin (3), jaceosidin (5), lupinalbin A (8), 2′-hydroxygenistein (9), and the semisynthetic derivative 2-(3-chloro-4-hydroxyphenyl)-N-(2-methoxyethyl)acetamide (14). Although, admittedly, all inactive compounds from this study have also been identified in the screenings with one omitted feature, these additional active hits encourage this screening mode, when a wider range of chemically diverse 17β-HSD2 inhibitors is sought and a higher number of false positive virtual hits is acceptable.
For a possible therapeutic use of a 17β-HSD2 inhibitor, a compound must be selective over 17β-HSD1, which catalyzes the reverse reaction. Therefore, the most active newly identified 17β-HSD2 inhibitors were screened at a final concentration of 20 μM in vitro using lysates of cells expressing the recombinant human 17β-HSD1 enzyme. For all compounds inhibiting 17β-HSD1 by 70% or more, IC50 values and corresponding selectivity factors were determined. The results are shown in Table 3. Follow-up experiments should include additional SDR enzymes such as 11β-HSDs, 3α/β-HSDs, and retinol dehydrogenases as well as a careful assessment of the cytotoxic potential of the identified compounds.
Table 3. Selectivity of the Most Active 17β-HSD2 Inhibitors toward 17β-HSD1.
| compound | 17β-HSD2 activity (IC50) | 17β-HSD1 activity (IC50 or remaining activity at 20 μM) | selectivity factor |
|---|---|---|---|
| nordihydroguaiaretic acid (1) | 0.38 ± 0.04 μM | 5.5 ± 1.3 μM | 15 |
| curcumin (3) | 1.73 ± 0.20 μM | 52.2 ± 7.1% | ∼12 |
| (−)-dihydroguaiaretic acid (4) | 0.94 ± 0.02 μM | 7.7 ± 2.2 μM | 8 |
| isoliquiritigenin (6) | 0.36 ± 0.08 μM | 2.83 ± 0.80 μM | 8 |
| lupinalbin A (8) | 1.52 ± 0.15 μM | 0.049 ± 0.019 μM | 0.03 |
| 2′-hydroxygenistein (9) | 2.03 ± 0.37 μM | 1.09 ± 0.06 μM | 0.5 |
| rosmarinic acid (11) | 3.72 ± 0.17 μM | n.i.a | >5 |
| ethyl vanillate (12) | 1.28 ± 0.26 μM | n.i. | >15 |
| 2-(3-chloro-4-hydroxyphenyl)-N-phenethylacetamide (13) | 1.57 ± 0.16 μM | n.i. | >12 |
| N-benzyl-2-(3-chloro-4-hydroxyphenyl)acetamide (16) | 3.42 ± 0.74 μM | n.i. | >5 |
| N-(2-(1H-indol-3-yl)ethyl)-2-(3-chloro-4-hydroxyphenyl)acetamide (17) | 0.98 ± 0.24 μM | n.i. | >20 |
| 2-(3-chloro-4-hydroxyphenyl)-N-(2-chlorobenzyl)acetamide (18) | 0.78 ± 0.16 μM | 54.8 ± 5.8% | ∼25 |
n.i., no inhibition.
Most of the active hits found in this study belong to compound classes associated with steroidogenic activities. This includes the triterpene oleanolic acid (2), which belongs to a compound class containing several 11β-HSD inhibitors.30−33 Compounds 5, 8, and 9 are flavonoids, a class known to have estrogenic activity. Nordihydroguaiaretic acid (1) is a lignan found at high concentrations in the leaves of Larrea tridentata (Sessé & Moc. ex DC.) Coville, a common shrub in the United States and in Mexico.34 The leaves have been used in the preparation of a tea for the treatment of cancer, arthritis, and tuberculosis. Compound 1 is an antioxidant that also inhibits lipoxygenase, thus influencing the leukotriene cascade and suppressing ovulation in rats.35 Thereby, it may pose a potential risk for reproductive toxicity if ingested in large amounts. Compound 1 was proposed to be converted into a phytoestrogen by gut flora.36 In addition, it was shown to have estrogenic effects, being an ERα-agonist, with a tendency to be selective over ERβ.37 Additionally, compound 1 was shown to inhibit the formation of β-amyloid fibrils in the central nervous system and the accumulation of β-peptides. These properties suggest that 1 is an interesting compound for the development of potential anti-Alzheimer disease (AD) pharmaceuticals.38 Similar anti-amyloidogenic effects were also reported in studies with mice for 1, 3, and 11, supporting the potential preventive properties of these natural compounds against AD.39
Curcumin (3) is a tautomeric diarylheptanoid compound that is found in the roots of Curcuma longa L. and has a great variety of potential therapeutic activities.40,41 It is one of the main ingredients of curry spice mixtures and is responsible for the yellow color.42 Many papers have been published in the past few decades describing anti-inflammatory,43 anticancer,44,45 and antioxidant properties of 3.40 In Asian medicine, 3 was used for topical or oral application to treat a variety of diseases for thousands of years. Despite the low bioavailability and rapid hepatic metabolism, 3 was shown to be therapeutically active against several diseases.46 There is debate as to whether 3 may be an invalid bioactive compound because of its PAINS properties47−49 or may still have some potential as a lead structure candidate for certain conditions.50 According to the experiments and observations from this study, 3 directly and specifically inhibits 17β-HSD2 and 17β-HSD1. A detailed discussion on this issue is provided in the Supporting Information (p S9). Although 3 may not be a suitable lead compound for various reasons, it still reflects the ability of the virtual screening workflow to detect structurally diverse 17β-HSD2 inhibitors.
Dihydroguaiaretic acid (4) is another lignan that is present in various plant extracts, such as those derived from the bark of Machilus thunbergii(51) Siebold & Zucc. and the seeds of Myristica fragrans Houtt.52 These plants are found predominantly in tropical and subtropical Asian countries. Compound 4 was reported to possess antibacterial,53 antioxidative,54 and potential anticancer properties.55 Little is known about the potential interference of 4 with estrogen-metabolizing hormones. In 2001, Filleur et al. reported that 4 showed no effects on 17β-HSD activity in placenta microsomes.56 This is in contrast with the potent inhibition (IC50, 940 ± 20 nM) of 17β-HSD2 by 4 found in the present study. The reason for this discrepancy is unclear but may be due to experimental differences, as in the present study recombinant human enzyme was used. In contrast, in the study by Filleur et al. placenta microsomes that also express other steroid-metabolizing enzymes were applied.
Isoliquiritigenin (6) is a hydroxylated chalcone found in Glycyrrhiza uralensis Fisch. ex. DC.57 and other various plant preparations. Many pharmacological effects of 6 have been described in the literature such as antitumor, antioxidative, and antibacterial properties.58 Using a recombinant protein, it was reported that 6 inhibits aromatase activity with an IC50 value of 3.8 μM.59 This would lower the amount of estrogens produced from androgens, which may aggravate osteoporosis. Nevertheless, 6 is a moderately potent inhibitor of aromatase, and efficient inhibition of 17β-HSD2 was achieved at concentrations 10 times lower. Importantly, 6 did not inhibit 17β-HSD1. Using yeast strains expressing human receptors, 6 was shown to bind to ERα (IC50 to displace estradiol of 1.87 μM) and ERβ (IC50 of 269 nM), however, with much lower affinity than estradiol.60
Compounds 8 and 9 are major constituents contained in a methanolic extract of the aerial parts of Eriosema laurentii De Wild, which was shown to have protective effects against femur mass loss and significantly increased calcium and inorganic phosphorus content in the femur in ovariectomized rats.61,62 Inhibition of 17β-HSD2 by these compounds may enhance local levels of estradiol, thereby potentiating estrogen receptor α (ERα)-mediated signaling. However, some of these effects may be explained by direct effects of the compounds on steroid receptors and/or helix–loop–helix transcription factors. In yeast systems expressing the human ERα and the human aryl hydrocarbon receptor, 8 showed agonistic effects with EC50 values of 21.4 nM and 1.34 μM, respectively.63 Additionally, 9 was reported to activate ERα with an EC50 value of 6.1 μM. Regarding 8 and 9, it needs to be noted that these compounds exert more potent inhibitory effects against 17β-HSD1 than 17β-HSD2. In fact, 8 potently inhibited 17β-HSD1 with an IC50 of 49 ± 19 nM and an approximately 30-fold selectivity over 17β-HSD2. This in vitro information suggests that 8 most potently activates ERα and potently inhibits estrone to estradiol conversion by 17β-HSD1 but shows weaker effects on 17β-HSD2-mediated estradiol inactivation. Depending on the tissue and cell type, ERα is expressed together with either 17β-HSD1 or 17β-HSD2, which may result in cell-specific estrogenic effects of 8.
Rosmarinic acid (11) was first isolated from an extract of Rosmarinus officinalis L.64 This compound was studied for many years and showed antinociceptive and anti-inflammatory effects in animal studies.65 In addition, several clinical trials showed positive effects of comfrey roots containing 11 as a topical treatment against pain.66 Antinociceptive effects would clearly be beneficial in the treatment of osteoporosis because of increasing pain with progression of the disease. Compound 11 selectively inhibited 17β-HSD2 over 17β-HSD1, although with rather moderate activity. It therefore remains to be seen whether such concentrations can be reached in bone cells. Alternatively, paracrine effects from neighboring cells may affect estrogen availability and therefore bone metabolism.
Ethyl vanillate (12) is an antioxidative67 compound that has been found in hedge mustard [Sisymbrium officinale (L.) Scop.] and also in Pinot noir wine.68 Although 12 has been known for quite some time, due to its intense vanilla taste and its use as a flavoring additive, its biological properties remain poorly investigated.
Most of the newly discovered 17β-HSD2 inhibitors were already known as phytoestrogens or compounds that are converted into phytoestrogen by gut flora (e.g., pinoresinol (7) and 1).36 The rationale why the pharmacophore model found these ER-active compounds was that the substrate (estradiol) of 17β-HSD2 is the endogenous ER agonist, and thus the binding pockets of ER and 17β-HSD2 are obviously able to accommodate similar compounds that may be considered as steroid mimetics. This was reflected by the pharmacophore model that is based on the properties of compounds binding to 17β-HSD2: the compounds that share features needed for binding to 17β-HSD2 are likely to bind to ERα and ERβ as well.
Many of the active hits share considerable structural similarity. Interestingly, the most active substance, 6, has one phenolic hydroxy group less than 10. This difference led to a drastic effect on the activity of these compounds: 6 gave an IC50 value of 0.36 ± 0.08 μM, whereas 10 was 20-fold less active, with an IC50 of 7.3 ± 2.7 μM. However, the difference in the overall lipophilicity of these compounds may also play a role in their different activities.
The semisynthetic fungal natural products (13–1869) followed a clear structure–activity relationship (SAR), with the activity shown to increase when a second aromatic ring was present. The parent compound (i.e., natural product) for this semisynthetic series, 2-(3-chloro-4-hydroxyphenyl)acetamide (S11), and the related natural products 2-(3-chloro-4-hydroxyphenyl)acetic acid (S15) and 2-(4-hydroxyphenyl)acetamide (S16) (see Table S1, Supporting Information, for their chemical structures), did not inhibit 17β-HSD2, whereas compounds 2-(3-chloro-4-hydroxyphenyl)-N-(2-methoxyethyl)acetamide (14) and N-butyl-2-(3-chloro-4-hydroxyphenyl)acetamide (15) were moderately active. The most active compounds from this series were 2-(3-chloro-4-hydroxyphenyl)-N-phenethylacetamide (13) and 16–18, which all shared a similar interaction pattern (Figure 5A). However, if the acetamide fragment is extended with, for example, an N-butyl chain, the compound can form additional hydrophobic interactions with the enzyme, resulting in an increased activity (Figure 5B). In addition to the alkyl chain, the most active compounds have a second aryl ring that can form aromatic interactions with the enzyme (Figure 5C). On the basis of the activities of these compounds, it can be proposed that 17β-HSD2 has a hydrophobic ligand binding pocket and aromatic amino acid residues in the active site that may contribute to the affinities of these ligands.
Figure 5.
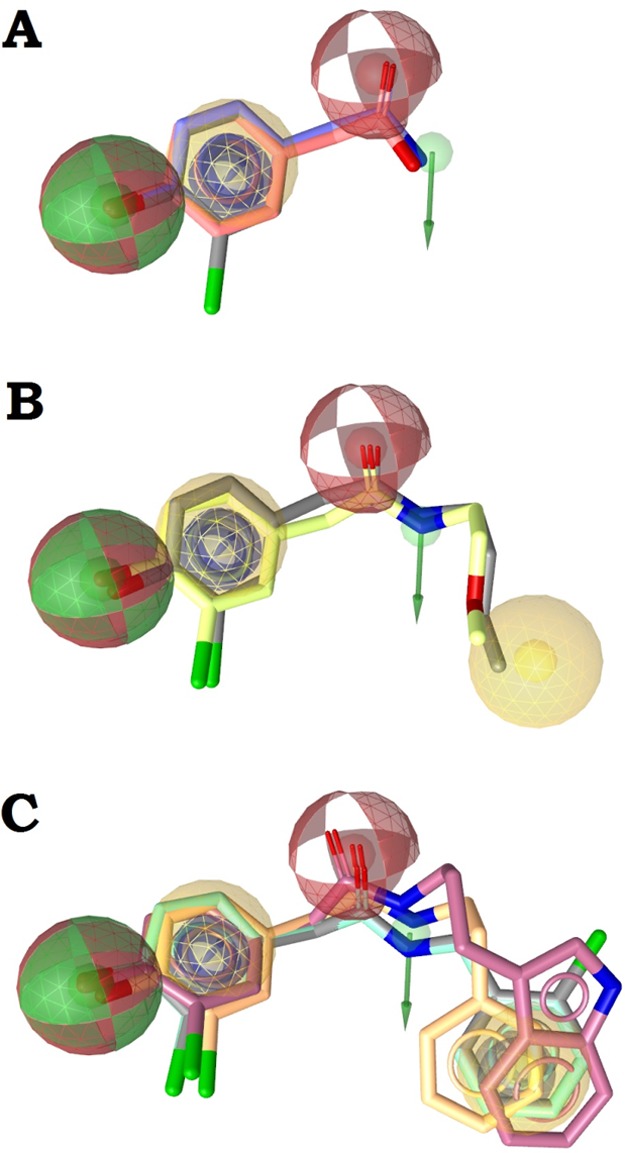
Illustration of the SAR of semisynthetic natural product derivatives (Table 3). (A) The core structure with compounds S12 (gray), S15 (red), and S11 (blue) with a pharmacophore model illustrating the interaction pattern. (B) The moderately active compounds 14 (yellow) and 15 (gray) with the additional hydrophobic feature. (C) The most active compounds 13 (orange), N-benzyl-2-(3-chloro-4-hydroxyphenyl)acetamide (16, green), N-(2-(1H-indol-3-yl)ethyl)-2-(3-chloro-4-hydroxyphenyl) (17, purple), and 18 (gray) with the additional aromatic ring feature.
Most of the tested compounds inhibited selectively 17β-HSD2 over 17β-HSD1, except for compounds 8 and 9. The semisynthetic compounds 13 and 16–18 also showed good selectivity in terms of the inhibition of 17β-HSD2. The two most potent compounds, 1 and 6, were 15 and 8 times more active toward 17β-HSD2 than 17β-HSD1. Both compounds are potential natural lead structures that could be used for the development of 17β-HSD2 drug candidates. Unlike many other related compounds that are possibly rapidly metabolized due to the presence of several hydroxy groups, 2-(3-chloro-4-hydroxyphenyl)-N-(2-chlorobenzyl)acetamide (18) has only a single hydroxy group and might therefore be less prone to rapid biotransformation. Compound 18 still potently and selectively inhibited 17β-HSD2 with an IC50 of 0.78 ± 0.16 μM.
Among the most active compounds identified during these studies were also the flavonoids 5 and 9. Schuster et al. earlier reported several flavonoids inhibiting 17β-HSD2. Taking the data together (Table 4),24 a SAR model for the flavonoids that inhibit this enzyme could be established (Figure 6).
Table 4. Flavonoid Structures and Activities Used for Deriving a Flavonoid SAR Model of 17β-HSD2 Inhibitors.

Figure 6.
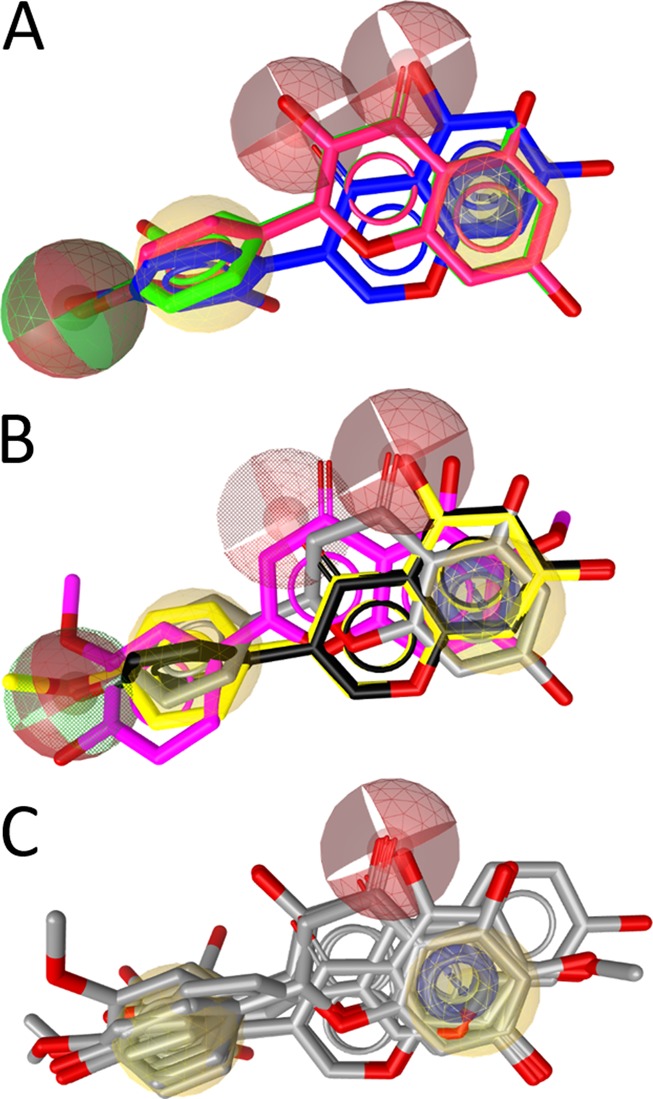
SAR of the flavonoids inhibiting 17β-HSD2. (A) The three most active compounds, 9 (2′-hydroxygenistein, blue), kaempferol (30, red), and quercetin (31, green), share a combined hydrogen bond acceptor/donor at position C-4′, a hydrophobic (aromatic) ring (ring B), two neighboring hydrogen bond acceptors on rings A and C, and the aromatic ring A. (B) The moderately active inhibitors 5 (magenta), naringenin (23, gray), genistein (32, black), and biochanin A (33, yellow) fit into the SAR-pharmacophore illustrating the importance of the hydrogen bond acceptor features on the B and C rings, respectively. (C) For comparison, the general flavonoid model not distinguishing active from inactive compounds is shown with all 17 flavonoids from Table 4.
In general, the active flavonoids share a typical pharmacophore model containing hydrogen bond acceptors and donors and hydrophobic and aromatic features (Figure 6A). The hydrogen bond acceptor in position C-3 (scaffold A) was found to be beneficial for activity, as the most active flavonoids, 30 and 31, contain a hydroxy group at this position (Figure 6B). If this feature was absent, the activity decreased or the compound was inactive. Furthermore, the hydrogen bond acceptor unit at the C-4′-position is important and shared by all active compounds. If the hydrogen-bonding feature at this position was deleted, active and inactive compounds were no longer distinguished (Figure 6C).
To learn more about the general properties of 17β-HSD2 inhibitors, model 1 and the flavonoid model were aligned (Figure 7). Every model contains an aromatic ring feature next to a hydrogen bond donor/acceptor feature. Among the compounds mapped, this combination was often represented by a phenolic hydroxy group. Another common feature was the hydrophobic/aromatic group in a certain distance from the first feature group. Interestingly, in between these aligned hydrophobic/aromatic features, there were hydrogen bond acceptor features. These indicate that in the binding pocket there may be two hydrophobic regions that tolerate aromatic interactions, and in between these pockets, there was most likely a hydrogen-bonding partner. This feature arrangement is in line with the architecture of already crystallized 11β-HSD1 and 17β-HSD1, where inhibitors are anchored to the catalytically active amino acids by central hydrogen bonds and form further, adjacent hydrophobic contacts (e.g., the PDB structures 4c7j70 and 3hb571).
Figure 7.
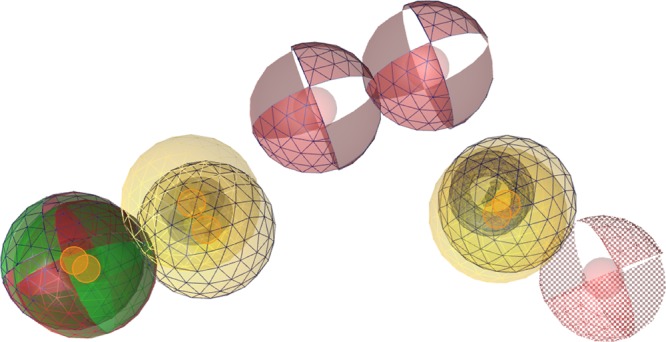
Alignment of the 17β-HSD2 inhibitor model 1 from Vuorinen et al.19 and the SAR model (features highlighted by grid) for highly active flavonoids. The pharmacophore features are color-coded: hydrogen bond acceptor, red; hydrogen bond donor, green; hydrophobic, yellow; aromatic ring, blue. The alignment centers are indicated with orange spheres.
The present virtual screening approach for the identification of natural products-derived 17β-HSD2 inhibitors was productive. Thus, only 38 compounds had to be tested to yield 17 active hits with sub- and low-micromolar IC50 values. The most potent bioactive compound, 6, exhibited an IC50 value of 360 ± 80 nM. Thus, the present approach had a success rate of 47% within the virtual hit lists. The fact that so many interesting 17β-HSD2 inhibitors were obtained within this relatively small natural product collection points toward the probable presence of more potent active compounds among other natural products.
Furthermore, SAR information was derived for two compound classes, providing more detailed insight into the binding pocket of the enzyme. Only 8 and 9, which were identified by model 2 with one omitted feature, were not selective and even preferentially inhibited 17β-HSD1. Consequently, both compounds seem not to be suitable lead structures for further development as antiosteoporosis leads. All other newly discovered 17β-HSD2 inhibitors were preferentially selective over 17β-HSD1, and therefore they could serve as lead structures for further optimization. It needs to be noted that the activities of these compounds toward 17β-HSD2 are at least an order of magnitude lower than that of reported synthetic, chemically optimized compounds.18,20,21 To further develop potential lead candidates, additional investigations into the bioavailability, metabolism, and tissue distribution of the identified natural compounds are needed. Inhibition of 17β-HSD2 is expected to result in tissue-specific elevated levels of estradiol, and potential adverse effects include endometrial hyperplasia and impaired growth control of the glandular epithelium of the breast.72−74 Thus, compounds that are primarily active in the bone would be preferred for future drug development.
Experimental Section
Databases
The Davis Compound Library (Griffith Institute for Drug Discovery, Griffith University) consisted of 352 compounds, of which the majority were obtained from Australian natural sources, such as endophytic fungi,75 macrofungi,76 plants,77 and marine invertebrates.78,79 Approximately 15% of the entries of this library were semisynthetic natural product analogues,80,76 while a small percentage (∼5%) are known commercial drugs or synthetic compounds inspired by natural products. The Atanasov and Krenn databases consisted of 51 and 13 in-house available natural products, respectively, from the Department of Pharmacognosy at the University of Vienna, Austria. From the University of Innsbruck, 23 selected plant- and lichen-derived compounds81−84,62 available in-house at the Institute of Pharmacy/Pharmacognosy were collected in the Waltenberger database. Finally, the Sigma-Aldrich catalogue was also screened, as it includes some commercially available natural products.
Virtual Screening
The databases were prepared for virtual screening by deleting counterions and generating multiconformational databases using OMEGA implemented in LigandScout 3.03b. For the relatively small in-house databases used, BEST settings were employed with a maximum of 500 conformers per molecule. For the larger Sigma-Aldrich database, FAST settings were used, which allowed for a maximum of 50 conformations per compound.
Origin, Isolation, and Purification of the Natural Compounds
All natural products from the Davis Compound Library were isolated from plants, marine invertebrates, or endophytic fungi archived at the Griffith Institute for Drug Discovery, Griffith University, Australia, or purchased from Sigma-Aldrich. The extraction and isolation of the natural products featured in this paper have been previously reported by Davis et al.69,85−88 The synthesis and characterization of the semisynthetic fungal analogues 13–18 have also been previously reported in the literature.69 All compounds from the Davis collection were analyzed for purity prior to screening and were shown by LC-MS or 1H NMR analysis to have purities of >95%. The compounds from the Atanasov library were obtained from Sigma-Aldrich, except for 2, 3, and butyl gallate (S3), which were purchased from Fisher, Molekula, and ABCR GmbH & Co. KG, respectively. All compounds were purchased at a purity of ≥90%. Compounds 8 and 9 were isolated in an activity-guided approach from a MeOH extract from Eriosema laurentii de Wild and unambiguously identified by following MS and NMR analysis. HPLC was applied to determine purity and resulted in 98.7% purity for 8 and 92.1% purity for 9. The compounds from the Waltenberger library were isolated from different plant and lichen species in the course of the project “Drugs from Nature Targeting Inflammation” (DNTI).89 Compound 7 was isolated from a MeOH extract of the bark material of Himatanthus sucuuba (Spruce) Woodson as described elsewhere.62 The purity of this compound was determined by HPLC and NMR experiments as >95%.
Activity Assays for 17β-HSD1 and 17β-HSD2 Using Cell Lysates
The 17β-HSD1 and 17β-HSD2 activity assays were performed as described previously.19 Briefly, lysates of human embryonic kidney cells (HEK-293, ATCC, Manassas, VA, USA) expressing either human 17β-HSD1 or human 17β-HSD2 were incubated for 10 min at 37 °C in TS2 buffer (100 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1 mM MgCl2, 250 mM sucrose, 20 mM Tris-HCl, pH 7.4) in a final volume of 22 μL containing either solvent (0.1% DMSO) or the inhibitor at the respective concentration. 17β-HSD1 activity was measured in the presence of 200 nM estrone, containing 50 nCi of [2,4,6,7-3H]-estrone, and 500 μM NADPH. In contrast, 17β-HSD2 activity was determined in the presence of 200 nM estradiol, containing 50 nCi of [2,4,6,7-3H]-estradiol, and 500 μM NAD+. Reactions were stopped after 10 min by adding an excess of unlabeled estradiol and estrone (2 mM of each in methanol). Unlabeled steroids and cofactors were purchased from Sigma-Aldrich and radiolabeled compounds from PerkinElmer (Boston, MA, USA). The steroids were separated by TLC, followed by scintillation counting and calculation of substrate conversion. Data were collected from at least three independent measurements. Compound 29(24) was used as a positive control for 17β-HSD1 assays and compound 22 from Vuorinen et al.19 as a positive control for 17β-HSD2 tests.
Structure–Activity-Relationship Modeling
The SAR models were generated using LigandScout 4.09 with default settings (Wolber 2005 JCIM;90 LigandScout 4.09, 2005–2016, Inte:Ligand GmbH, Vienna, Austria, www.inteligand.com). For all compounds, BEST conformational models using iCon (max 500 conformers per entry) were calculated and overlaid by chemical features using the pharmacophore-based alignment algorithm of the program.
Acknowledgments
This study was supported by the Swiss National Science Foundation (31003A-159454 to A.O.), the Novartis Research Foundation (A.O.), the Austrian Science Fund (P26782 to D.S. and P25971-B23 to A.G.A.), the Hochschuljubiläumsfond (H-297322/2014 to A.G.A.), the Ernst Mach Stipendium (to S.B.A.), the National Health and Medical Research Council (NHMRC) (APP1024314 to R.A.D.), and the Australian Research Council (ARC) (LE0668477, LE0237908, LP120200339 to R.A.D.). D.S. is an Ingeborg Hochmair professor of the University of Innsbruck. We thank P. Schuster and G. Begg for help in preparing the manuscript and Inte:Ligand GmbH for providing LigandScout software free of charge.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.6b00950.
Author Contributions
# A. Vuorinen and R. T. Engeli contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Persson B.; Kallberg Y.; Bray J. E.; Bruford E.; Dellaporta S. L.; Favia A. D.; Duarte R. G.; Jornvall H.; Kavanagh K. L.; Kedishvili N.; Kisiela M.; Maser E.; Mindnich R.; Orchard S.; Penning T. M.; Thornton J. M.; Adamski J.; Oppermann U. Chem.-Biol. Interact. 2009, 178, 94–98. 10.1016/j.cbi.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y.; Qiu Q. Q.; Debear J.; Lathrop W. F.; Bertolini D. R.; Tamburini P. P. J. Bone Miner. Res. 1998, 13, 1539–1546. 10.1359/jbmr.1998.13.10.1539. [DOI] [PubMed] [Google Scholar]
- Mustonen M.; Poutanen M.; Kellokumpu S.; de Launoit Y.; Isomaa V.; Vihko R.; Vihko P. J. Mol. Endocrinol. 1998, 20, 67–74. 10.1677/jme.0.0200067. [DOI] [PubMed] [Google Scholar]
- Mustonen M. V.; Isomaa V. V.; Vaskivuo T.; Tapanainen J.; Poutanen M. H.; Stenback F.; Vihko R. K.; Vihko P. T. J. Clin. Endocrinol. Metab. 1998, 83, 1319–1324. 10.1210/jcem.83.4.4709. [DOI] [PubMed] [Google Scholar]
- Takeyama J.; Sasano H.; Suzuki T.; Iinuma K.; Nagura H.; Andersson S. J. Clin. Endocrinol. Metab. 1998, 83, 3710–3715. 10.1210/jcem.83.10.5212. [DOI] [PubMed] [Google Scholar]
- Puranen T. J.; Kurkela R. M.; Lakkakorpi J. T.; Poutanen M. H.; Itaranta P. V.; Melis J. P.; Ghosh D.; Vihko R. K.; Vihko P. T. Endocrinology 1999, 140, 3334–3341. 10.1210/endo.140.7.6861. [DOI] [PubMed] [Google Scholar]
- Wu L.; Einstein M.; Geissler W. M.; Chan H. K.; Elliston K. O.; Andersson S. J. Biol. Chem. 1993, 268, 12964–12969. [PubMed] [Google Scholar]
- Lukacik P.; Kavanagh K. L.; Oppermann U. Mol. Cell. Endocrinol. 2006, 248, 61–71. 10.1016/j.mce.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Dufort I.; Rheault P.; Huang X. F.; Soucy P.; Luu-The V. Endocrinology 1999, 140, 568–574. 10.1210/endo.140.2.6531. [DOI] [PubMed] [Google Scholar]
- Geissler W. M.; Davis D. L.; Wu L.; Bradshaw K. D.; Patel S.; Mendonca B. B.; Elliston K. O.; Wilson J. D.; Russell D. W.; Andersson S. Nat. Genet. 1994, 7, 34–39. 10.1038/ng0594-34. [DOI] [PubMed] [Google Scholar]
- Ghosh D.; Vihko P. Chem.-Biol. Interact. 2001, 130–132, 637–650. 10.1016/S0009-2797(00)00255-6. [DOI] [PubMed] [Google Scholar]
- Soubhye J.; Alard I. C.; van Antwerpen P.; Dufrasne F. Future Med. Chem. 2015, 7, 1431–1456. 10.4155/fmc.15.74. [DOI] [PubMed] [Google Scholar]
- Compston J. E. Physiol. Rev. 2001, 81, 419–447. [DOI] [PubMed] [Google Scholar]
- Riggs B. L.; Khosla S.; Melton L. J. J. Bone Miner. Res. 1998, 13, 763–773. 10.1359/jbmr.1998.13.5.763. [DOI] [PubMed] [Google Scholar]
- Chin K.-Y.; Ima-Nirwana S. Int. J. Endocrinol. 2012, 2012, 208719. 10.1155/2012/208719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael H.; Härkönen P. L.; Väänänen H. K.; Hentunen T. A. J. Bone Miner. Res. 2005, 20, 2224–2232. 10.1359/JBMR.050803. [DOI] [PubMed] [Google Scholar]
- Bagi C. M.; Wood J.; Wilkie D.; Dixon B. J. Musculoskelet. Neuronal. Interact. 2008, 8, 267–280. [PubMed] [Google Scholar]
- Perspicace E.; Cozzoli L.; Gargano E. M.; Hanke N.; Carotti A.; Hartmann R. W.; Marchais-Oberwinkler S. Eur. J. Med. Chem. 2014, 83, 317–337. 10.1016/j.ejmech.2014.06.036. [DOI] [PubMed] [Google Scholar]
- Vuorinen A.; Engeli R.; Meyer A.; Bachmann F.; Griesser U. J.; Schuster D.; Odermatt A. J. Med. Chem. 2014, 57, 5995–6007. 10.1021/jm5004914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel M.; Marchais-Oberwinkler S.; Perspicace E.; Möller G.; Adamski J.; Hartmann R. W. J. Med. Chem. 2011, 54, 7547–7557. 10.1021/jm2008453. [DOI] [PubMed] [Google Scholar]
- Xu K.; Al-Soud Y. A.; Wetzel M.; Hartmann R. W.; Marchais-Oberwinkler S. Eur. J. Med. Chem. 2011, 46, 5978–5990. 10.1016/j.ejmech.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Deluca D.; Krazeisen A.; Breitling R.; Prehn C.; Möller G.; Adamski J. J. Steroid Biochem. Mol. Biol. 2005, 93, 285–292. 10.1016/j.jsbmb.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Le Bail J. C.; Laroche T.; Marre-Fournier F.; Habrioux G. Cancer Lett. 1998, 133, 101–106. 10.1016/S0304-3835(98)00211-0. [DOI] [PubMed] [Google Scholar]
- Schuster D.; Nashev L. G.; Kirchmair J.; Laggner C.; Wolber G.; Langer T.; Odermatt A. J. Med. Chem. 2008, 51, 4188–4199. 10.1021/jm800054h. [DOI] [PubMed] [Google Scholar]
- Atanasov A. G.; Waltenberger B.; Pferschy-Wenzig E. M.; Linder T.; Wawrosch C.; Uhrin P.; Temml V.; Wang L.; Schwaiger S.; Heiss E. H.; Rollinger J. M.; Schuster D.; Breuss J. M.; Bochkov V.; Mihovilovic M. D.; Kopp B.; Bauer R.; Dirsch V. M.; Stuppner H. Biotechnol. Adv. 2015, 33, 1582–614. 10.1016/j.biotechadv.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2012, 75, 311–335. 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder J.; Sedrani R.; Wiesmann C. Nat. Rev. Drug Discovery 2014, 13, 577–587. 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- Williamson E. M.; Heinrich M.; Jäger A. K., Eds. Ethnopharmacology; John Wiley & Sons Ltd: Chichester, West Sussex, UK, 2015; pp 213–226. [Google Scholar]
- Sharma C.; Kumari T.; Arya K. R. Int. J. Pharm. Res. Health Sci. 2014, 2, 185–190. [Google Scholar]
- Blum A.; Favia A. D.; Maser E. Mol. Cell. Endocrinol. 2009, 301, 132–136. 10.1016/j.mce.2008.08.028. [DOI] [PubMed] [Google Scholar]
- Kratschmar D. V.; Vuorinen A.; Da Cunha T.; Wolber G.; Classen-Houben D.; Doblhoff O.; Schuster D.; Odermatt A. J. Steroid Biochem. Mol. Biol. 2011, 125, 129–142. 10.1016/j.jsbmb.2010.12.019. [DOI] [PubMed] [Google Scholar]
- Rollinger J. M.; Kratschmar D. V.; Schuster D.; Pfisterer P. H.; Gumy C.; Aubry E. M.; Brandstötter S.; Stuppner H.; Wolber G.; Odermatt A. Bioorg. Med. Chem. 2010, 18, 1507–1515. 10.1016/j.bmc.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Vuorinen A.; Seibert J.; Papageorgiou V. P.; Rollinger J. M.; Odermatt A.; Schuster D.; Assimopoulou A. N. Planta Med. 2015, 81, 525–532. 10.1055/s-0035-1545720. [DOI] [PubMed] [Google Scholar]
- Lambert J. D.; Zhao D.; Meyers R. O.; Kuester R. K.; Timmermann B. N.; Dorr R. T. Toxicon 2002, 40, 1701–1708. 10.1016/S0041-0101(02)00203-9. [DOI] [PubMed] [Google Scholar]
- Mikuni M.; Yoshida M.; Hellberg P.; Peterson C. A.; Edwin S. S.; Brännström M.; Peterson C. M. Biol. Reprod. 1998, 58, 1211–1216. 10.1095/biolreprod58.5.1211. [DOI] [PubMed] [Google Scholar]
- Benassayag C.; Perrot-Applanat M.; Ferre F. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2002, 777, 233–248. 10.1016/S1570-0232(02)00340-9. [DOI] [PubMed] [Google Scholar]
- Fujimoto N.; Kohta R.; Kitamura S.; Honda H. Life Sci. 2004, 74, 1417–1425. 10.1016/j.lfs.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Ono K.; Hasegawa K.; Yoshiike Y.; Takashima A.; Yamada M.; Naiki H. J. Neurochem. 2002, 81, 434–40. 10.1046/j.1471-4159.2002.00904.x. [DOI] [PubMed] [Google Scholar]
- Yamada M.; Ono K.; Hamaguchi T.; Noguchi-Shinohara M. Adv. Exp. Med. Biol. 2015, 863, 79–94. 10.1007/978-3-319-18365-7_4. [DOI] [PubMed] [Google Scholar]
- Gupta S. C.; Patchva S.; Koh W.; Aggarwal B. B. Clin. Exp. Pharmacol. Physiol. 2012, 39, 283–299. 10.1111/j.1440-1681.2011.05648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolova Y.; Deneva V.; Antonov L.; Drakalska E.; Momekova D.; Lambov N. Spectrochim. Acta, Part A 2014, 132, 815–820. 10.1016/j.saa.2014.05.096. [DOI] [PubMed] [Google Scholar]
- Eigner D.; Scholz D. J. Ethnopharmacol. 1999, 67, 1–6. 10.1016/S0378-8741(98)00234-7. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Banerjee S.; Sil P. C. Food Chem. Toxicol. 2015, 83, 111–24. 10.1016/j.fct.2015.05.022. [DOI] [PubMed] [Google Scholar]
- Epstein J.; Sanderson I. R.; Macdonald T. T. Br. J. Nutr. 2010, 103, 1545–1557. 10.1017/S0007114509993667. [DOI] [PubMed] [Google Scholar]
- Ko E. Y.; Moon A. J. Cancer Prev. 2015, 20, 223–231. 10.15430/JCP.2015.20.4.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P.; Kunnumakkara A. B.; Newman R. A.; Aggarwal B. B. Mol. Pharmaceutics 2007, 4, 807–818. 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- Baell J. B. J. Nat. Prod. 2016, 79, 616–28. 10.1021/acs.jnatprod.5b00947. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. J. Med. Chem. 2010, 53, 2719–40. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Bisson J.; McAlpine J. B.; Friesen J. B.; Chen S. N.; Graham J.; Pauli G. F. J. Med. Chem. 2016, 59, 1671–90. 10.1021/acs.jmedchem.5b01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K. M.; Dahlin J. L.; Bisson J.; Graham J.; Pauli G. F.; Walters M. A. J. Med. Chem. 2017, 60, 1620. 10.1021/acs.jmedchem.6b00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C. J.; Sung S. H.; Kim Y. C. Planta Med. 2004, 70, 79–80. 10.1055/s-2004-815463. [DOI] [PubMed] [Google Scholar]
- Kwon H. S.; Kim M. J.; Jeong H. J.; Yang M. S.; Park K. H.; Jeong T. S.; Lee W. S. Bioorg. Med. Chem. Lett. 2008, 18, 194–198. 10.1016/j.bmcl.2007.10.098. [DOI] [PubMed] [Google Scholar]
- Favela-Hernandez J. M.; Garcia A.; Garza-Gonzalez E.; Rivas-Galindo V. M.; Camacho-Corona M. R. Phytother. Res. 2012, 26, 1957–1960. 10.1002/ptr.4660. [DOI] [PubMed] [Google Scholar]
- Yamauchi S.; Masuda T.; Sugahara T.; Kawaguchi Y.; Ohuchi M.; Someya T.; Akiyama J.; Tominaga S.; Yamawaki M.; Kishida T.; Akiyama K.; Maruyama M. Biosci., Biotechnol., Biochem. 2008, 72, 2981–2986. 10.1271/bbb.80461. [DOI] [PubMed] [Google Scholar]
- Choi M. S.; Jeong H. J.; Kang T. H.; Shin H. M.; Oh S. T.; Choi Y.; Jeon S. Life Sci. 2015, 141, 81–89. 10.1016/j.lfs.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Filleur F.; Le Bail J. C.; Duroux J. L.; Simon A.; Chulia A. J. Planta Med. 2001, 67, 700–704. 10.1055/s-2001-18349. [DOI] [PubMed] [Google Scholar]
- Li S.; Li W.; Wang Y.; Asada Y.; Koike K. Bioorg. Med. Chem. Lett. 2010, 20, 5398–401. 10.1016/j.bmcl.2010.07.110. [DOI] [PubMed] [Google Scholar]
- Peng F.; Du Q.; Peng C.; Wang N.; Tang H.; Xie X.; Shen J.; Chen J. Phytother. Res. 2015, 29, 969–977. 10.1002/ptr.5348. [DOI] [PubMed] [Google Scholar]
- Ye L.; Gho W. M.; Chan F. L.; Chen S.; Leung L. K. Int. J. Cancer 2009, 124, 1028–36. 10.1002/ijc.24046. [DOI] [PubMed] [Google Scholar]
- Choi S. Y.; Ha T. Y.; Ahn J. Y.; Kim S. R.; Kang K. S.; Hwang I. K.; Kim S. Planta Med. 2008, 74, 25–32. 10.1055/s-2007-993760. [DOI] [PubMed] [Google Scholar]
- Ateba S. B.; Njamen D.; Medjakovic S.; Hobiger S.; Mbanya J. C.; Jungbauer A.; Krenn L. J. Ethnopharmacol. 2013, 150, 298–307. 10.1016/j.jep.2013.08.050. [DOI] [PubMed] [Google Scholar]
- Waltenberger B.; Rollinger J. M.; Griesser U. J.; Stuppner H.; Gelbrich T. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2011, 67, o409–12. 10.1107/S0108270111035761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ateba S. B.; Njamen D.; Medjakovic S.; Zehl M.; Kaehlig H.; Jungbauer A.; Krenn L. BMC Complementary Altern. Med. 2014, 14, 294. 10.1186/1472-6882-14-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M.; Simmonds M. S. Phytochemistry 2003, 62, 121–125. 10.1016/S0031-9422(02)00513-7. [DOI] [PubMed] [Google Scholar]
- Boonyarikpunchai W.; Sukrong S.; Towiwat P. Pharmacol., Biochem. Behav. 2014, 124, 67–73. 10.1016/j.pbb.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Staiger C. Phytother. Res. 2012, 26, 1441–1448. 10.1002/ptr.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai A.; Sawano T.; Ito H. Biosci., Biotechnol., Biochem. 2012, 76, 314–318. 10.1271/bbb.110700. [DOI] [PubMed] [Google Scholar]
- Blazevic I.; Radonic A.; Mastelic J.; Zekic M.; Skocibusic M.; Maravic A. Chem. Biodiversity 2010, 7, 2023–2034. 10.1002/cbdv.200900234. [DOI] [PubMed] [Google Scholar]
- Davis R. A.; Pierens G. K.; Parsons P. G. Magn. Reson. Chem. 2007, 45, 442–445. 10.1002/mrc.1984. [DOI] [PubMed] [Google Scholar]
- Goldberg F. W.; Dossetter A. G.; Scott J. S.; Robb G. R.; Boyd S.; Groombridge S. D.; Kemmitt P. D.; Sjögren T.; Gutierrez P. M.; deSchoolmeester J.; Swales J. G.; Turnbull A. V.; Wild M. J. J. Med. Chem. 2014, 57, 970–986. 10.1021/jm4016729. [DOI] [PubMed] [Google Scholar]
- Mazumdar M.; Fournier D.; Zhu D. W.; Cadot C.; Poirier D.; Lin S. X. Biochem. J. 2009, 424, 357–366. 10.1042/BJ20091020. [DOI] [PubMed] [Google Scholar]
- Gunnarsson C.; Hellqvist E.; Stal O. Br. J. Cancer 2005, 92, 547–52. 10.1038/sj.bjc.6602375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnarsson C.; Olsson B. M.; Stal O. Southeast Sweden Breast Cancer Group. Cancer Res. 2001, 61, 8448–51. [PubMed] [Google Scholar]
- Kitawaki J.; Koshiba H.; Ishihara H.; Kusuki I.; Tsukamoto K.; Honjo H. J. Clin. Endocrinol. Metab. 2000, 85, 3292–6. 10.1210/jcem.85.9.6829. [DOI] [PubMed] [Google Scholar]
- Davis R. A.; Carroll A. R.; Andrews K. T.; Boyle G. M.; Tran T. L.; Healy P. C.; Kalaitzis J. A.; Shivas R. G. Org. Biomol. Chem. 2010, 8, 1785–1790. 10.1039/b924169h. [DOI] [PubMed] [Google Scholar]
- Choomuenwai V.; Andrews K. T.; Davis R. A. Bioorg. Med. Chem. 2012, 20, 7167–7174. 10.1016/j.bmc.2012.09.052. [DOI] [PubMed] [Google Scholar]
- Levrier C.; Balastrier M.; Beattie K. D.; Carroll A. R.; Martin F.; Choomuenwai V.; Davis R. A. Phytochemistry 2013, 86, 121–126. 10.1016/j.phytochem.2012.09.019. [DOI] [PubMed] [Google Scholar]
- Barnes E. C.; Said N. A. B. M.; Williams E. D.; Hooper J. N. A.; Davis R. A. Tetrahedron 2010, 66, 283–287. 10.1016/j.tet.2009.10.109. [DOI] [Google Scholar]
- Liberio M. S.; Sooraj D.; Williams E. D.; Feng Y.; Davis R. A. Tetrahedron Lett. 2011, 52, 6729–6731. 10.1016/j.tetlet.2011.09.151. [DOI] [Google Scholar]
- Barnes E. C.; Choomuenwai V.; Andrews K. T.; Quinn R. J.; Davis R. A. Org. Biomol. Chem. 2012, 10, 4015–4023. 10.1039/c2ob00029f. [DOI] [PubMed] [Google Scholar]
- Atanasov A. G.; Wang J. N.; Gu S. P.; Bu J.; Kramer M. P.; Baumgartner L.; Fakhrudin N.; Ladurner A.; Malainer C.; Vuorinen A.; Noha S. M.; Schwaiger S.; Rollinger J. M.; Schuster D.; Stuppner H.; Dirsch V. M.; Heiss E. H. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 4813–9. 10.1016/j.bbagen.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J.; Waltenberger B.; Noha S. M.; Schuster D.; Rollinger J. M.; Boustie J.; Chollet M.; Stuppner H.; Werz O. ChemMedChem 2012, 7, 2077–2081. 10.1002/cmdc.201200345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrudin N.; Ladurner A.; Atanasov A. G.; Heiss E. H.; Baumgartner L.; Markt P.; Schuster D.; Ellmerer E. P.; Wolber G.; Rollinger J. M.; Stuppner H.; Dirsch V. M. Mol. Pharmacol. 2010, 77, 559–566. 10.1124/mol.109.062141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oettl S. K.; Gerstmeier J.; Khan S. Y.; Wiechmann K.; Bauer J.; Atanasov A. G.; Malainer C.; Awad E. M.; Uhrin P.; Heiss E. H.; Waltenberger B.; Remias D.; Breuss J. M.; Boustie J.; Dirsch V. M.; Stuppner H.; Werz O.; Rollinger J. M. PLoS One 2013, 8, e76929. 10.1371/journal.pone.0076929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes E. C.; Kavanagh A. M.; Ramu S.; Blaskovich M. A.; Cooper M. A.; Davis R. A. Phytochemistry 2013, 93, 162–166. 10.1016/j.phytochem.2013.02.021. [DOI] [PubMed] [Google Scholar]
- Baron P. S.; Neve J. E.; Camp D.; Suraweera L.; Lam A.; Lai J.; Jovanovic L.; Nelson C.; Davis R. A. Magn. Reson. Chem. 2013, 51, 358–363. 10.1002/mrc.3958. [DOI] [PubMed] [Google Scholar]
- Davis R. A.; Barnes E. C.; Longden J.; Avery V. M.; Healy P. C. Bioorg. Med. Chem. 2009, 17, 1387–1392. 10.1016/j.bmc.2008.12.030. [DOI] [PubMed] [Google Scholar]
- Healy P. C.; Hocking A.; Tran-Dinh N.; Pitt J. I.; Shivas R. G.; Mitchell J. K.; Kotiw M.; Davis R. A. Phytochemistry 2004, 65, 2373–2378. 10.1016/j.phytochem.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Waltenberger B.; Atanasov A. G.; Heiss E. H.; Bernhard D.; Rollinger J. M.; Breuss J. M.; Schuster D.; Bauer R.; Kopp B.; Franz C.; Bochkov V.; Mihovilovic M. D.; Dirsch V. M.; Stuppner H. Monatsh. Chem. 2016, 147, 479–491. 10.1007/s00706-015-1653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber G.; Langer T. J. Chem. Inf. Model. 2005, 45, 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.