

Abstract
Aims
To resolve the controversy as to whether periostin plays a role in myocardial regeneration after myocardial infarction (MI), we created a neonatal mouse model of MI to investigate the influence of periostin ablation on myocardial regeneration and clarify the underlying mechanisms.
Methods and results
Neonatal periostin-knockout mice and their wildtype littermates were subjected to MI or sham surgery. In the wildtype mice after MI, fibrosis was detectable at 3 days and fibrotic tissue was completely replaced by regenerated myocardium at 21 days. In contrast, in the knockout mice, significant fibrosis in the infarcted area was present at even 3 weeks after MI. Levels of phosphorylated-histone 3 and aurora B in the myocardium, detected by immunofluorescence and western blotting, were significantly lower in knockout than in wildtype mice at 7 days after MI. Similarly, angiogenesis was decreased in the knockout mice after MI. Expression of both the endothelial marker CD-31 and α-smooth muscle actin was markedly lower in the knockout than in wildtype mice at 7 days after MI. The knockout MI group had elevated levels of glycogen synthase kinase (GSK) 3β and decreased phosphatidylinositol 3-kinase (PI3K), phosphorylated serine/threonine protein kinase B (p-Akt), and cyclin D1, compared with the wildtype MI group. Similar effects were observed in experiments using cultured cardiomyocytes from neonatal wildtype or periostin knockout mice. Administration of SB216763, a GSK3β inhibitor, to knockout neonatal mice decreased myocardial fibrosis and increased angiogenesis in the infarcted area after MI.
Conclusion
Ablation of periostin suppresses post-infarction myocardial regeneration by inhibiting the PI3K/GSK3β/cyclin D1 signalling pathway, indicating that periostin is essential for myocardial regeneration.
Keywords: Periostin, Myocardial regeneration, Glycogen synthase kinase 3β, Cyclin D1, Myocardial infarction
1. Introduction
While it remains under debate whether the myocardium can regenerate after ischaemic injury in the adult heart,1 neonatal mice show a capacity for myocardial regeneration after injuries such as myocardial infarction (MI) or resection of the left ventricular apex, but they lose this capacity within 7 days after birth.2–4 Recent studies have revealed that certain relevant cytokines, proteins, physical and chemical factors, and genes may be involved in myocardial regeneration.3,5–7
Periostin plays important roles during cardiac development and in the epithelial-mesenchymal transition.8 It was also linked to cardiovascular diseases such as dilated cardiomyopathy and MI.9,10 Several studies demonstrated function for periostin in regeneration of tissues including the myocardium.4,11,12 However, it remains controversial whether periostin can promote myocardial regeneration.4,13 A previous study using an adult MI model reported no significant differences in myocardial regeneration in periostin deficient or periostin transgenic mice, as compared with their corresponding wildtype strains.13 Intriguingly, using a neonatal heart resection model, a recent study demonstrated that signal transducers and activators of transcription 3 (STAT3)/periostin signalling is a critical mediator of interleukin 13 signalling in the regenerating mouse heart.4 Therefore, it may be a good way to resolve this controversy using an MI model in neonatal periostin knockout mice. We therefore used this approach to test our hypothesis that periostin is necessary for post-infarction myocardial regeneration in the neonatal heart.
In our study, we compared the myocardial regenerative capacity of neonatal periostin knockout and wildtype mice and further clarified the influence of periostin on the phosphatidylinositol 3-kinase (PI3K)/glycogen synthase kinase 3β (GSK 3β)/cyclin D1 signalling pathway.
2. Methods
All procedures were performed in accordance with our institution’s guidelines for animal research that conform to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication, 8th Edition, 2011). Approval for this study was granted by our university’s ethics review board.
2.1 Neonatal mouse MI model
Periostin knockout mice (B6; 129-Postntm1Jmol/J, Targeted: Null/Knockout, Stock No: 009067. Donated by Jeffery D. Molkentin, Cincinnati Children's Hospital) were from the Jackson Laboratory (Bar Harbor, ME, USA). The corresponding heterozygous mice were used for breeding. MI surgeries were performed on neonatal mice as described previously.3 Briefly, neonatal knockout mice and wildtype littermates, on the second day after birth, were anesthetized on ice for 3–4 min and maintained on ice during the surgical procedure. Anesthesia effectiveness was assessed based on reduced respiration. After disinfection of the incision area, the chest was opened with a horizontal incision through the muscle between the third and fourth intercostal spaces. The left coronary artery was permanently ligated with an 8-0 silk suture and the thoracic wall and skin were closed, also with an 8-0 silk suture. Sham-operated animals underwent an analogous surgical operation, but without occlusion of the coronary artery. After surgery, the skin was disinfected and the animals revived, while maintained on a thermal insulation blanket.14 Myocardial ischaemia was confirmed by an electrocardiogram ST-segment elevation after the animal was revived. Some mice were sacrificed at 1, 3, 7, or 10 days after surgery by putting them on ice for 5 min until respiration ceased. At 14 or 21 days after surgery, other mice were sacrificed by an overdose of pentobarbital sodium anesthesia (150 mg/kg intraperitoneal injection) or cervical dislocation.
In some periostin knockout mice, SB216763 (Sigma Aldrich, St. Louis, MO, USA), a GSK3β inhibitor, was administrated intraperitoneally (10 mg/kg/day, in dimethyl sulfoxide vehicle) beginning on the first day after surgery and continuing for 7 days.
2.2 Cell culture
Ventricular myocytes were isolated from neonatal periostin knockout and wildtype mice as described previously.15 The neonatal mice were killed by 2% isoflurane inhalation and cervical dislocation. Cardiomyocytes were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Sigma Aldrich) supplemented with 10% fetal bovine serum (Equitech-Bio, Kerrville, TX, USA) for 72 h and then in serum-free DMEM for 48 h prior to use in experiments. The cells were exposed to anoxia for 3 h and reoxygenation for 24 h (AR) in the presence or absence of SB216763 (a GSK3β inhibitor, 3 μM, in dimethyl sulfoxide, added 2 h prior to AR).
2.3 Triphenyl tetrazolium chloride (TTC) staining
One day after surgery, some mice were killed, their hearts harvested and each heart cut into three pieces. MI was confirmed by staining with 1% TTC (Sigma Aldrich) at 37 °C for 20 min. Myocardial infarct size was measured using Image J Analysis software (National Institutes of Health, Bethesda, MD, USA).
2.4 Western blotting
Proteins were obtained from whole-heart homogenates, with tissue samples from three animals pooled for each biological replicate. Immunoblotting was performed with primary antibodies against GAPDH, PI3K, phosphorylated serine/threonine protein kinase B (p-Akt), Akt, p-GSK3β (1:1000, Cell Signaling Technology, Boston, MA, USA), phospho-histone3 (p-H3) (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), periostin (1:2000, Abcam, Cambrige, UK), GSK3β (1:1000, Santa Cruz Biotechnology), cyclin D1 (1:10 000, Abcam) and atrial natriuretic peptide (ANP) (1:1000, Santa Cruz Biotechnology). Samples containing equal amounts of protein per lane were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinyl difluoride membranes. The membranes were blocked with 5% bovine serum albumin (BSA) at room temperature for 2 h and then incubated overnight at 4 °C with the appropriate primary antibody. The blots were detected using a Super Signal ECL kit (Invitrogen, Carlsbad, CA, USA) in a western blotting detection system (Kodak Digital Science, Rochester, NY, USA) and quantified by densitometry using Image J Analysis software. For detection of p-Akt and p-GSK3β proteins, a stripping buffer (Thermo Fisher, Waltham, MA, USA) was used to remove conjugated antibody and the blot was then incubated with the second primary antibody. Electrophoresis of each target protein and its loading control, GAPDH, was performed on the same gel, and then the gel was cut into two parts, in accordance with the molecular weight of each protein. Each gel protein was then processed separately for western blotting.
2.5 Histological examinations
Heart tissue from different groups was excised, rinsed with phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde, embedded in paraffin and cut into 4–6 μm sections. Masson’s trichrome (Azan) staining was used to evaluate myocardial fibrosis. For immunohistochemistry (IHC), after antigen retrieval by incubation in citrate buffer (pH 6.0), sections were incubated with rabbit anti-mouse periostin or rabbit anti-mouse CD31 (Abcam) antibody overnight at 4 °C. The extent of new blood vessels was evaluated based on the density of endothelial cells using the ratio of positively stained area to gross area. To assess CD31 and periostin staining, each tissue section was scanned entirely and its staining intensity would be scored as 0 (negative), 1 (weak), 2 (medium), or 3 (strong). The extent of staining was scored as 0 (0%), 1 (1–25%), 2 (26–50%), 3 (51–75%), or 4 (76–100%), according to the percentages of positively stained areas in relation to those of the whole view field. For each sample, the sum of these two scores was reported as the final staining score (0–7).
For phospho-histone H3 (p-H3)/troponin T and aurora B/troponin T co-staining, paraffin sections were washed three times in PBS, blocked in buffer (10% BSA for 20 min) then washed three times in PBS. Antigen retrieval was achieved by boiling sections in sodium citrate solution for 20 min. Sections were then incubated overnight at 4 °C with primary antibodies against p-H3 (Ser10) (1:100, rabbit polyclonal, Santa Cruz Biotechnology) and cardiac troponin T (1:100, mouse monoclonal, Santa Cruz Biotechnology) or aurora B (5 μg/mL, Abcam) and cardiac troponin T (1:100, mouse monoclonal, Santa Cruz Biotechnology). The next day, slides were washed in PBS 3 times then incubated for 1 h at room temperature with secondary antibodies conjugated to Alexa Fluor 488 or 555 (1:100 dilution, Santa Cruz Biotechnology) to stain for p-H3 or aurora B, respectively. Slides were washed 3 times in PBS and stained with 4,6-diamidino-2-phenylindole (DAPI) for 30 min to label nuclei.
Apoptosis in the myocardium was determined using a terminal deoxynucleotidyltransferase mediated (dUTP) nick-end labelling (TUNEL) assay. Briefly, apoptotic cells were detected with an In Situ Cell Death Detection Kit, TMR red (Roche, Basel, Switzerland). Heart tissues, embedded in paraffin and sectioned, were used. After deparaffinization, sections were treated with proteinase K for 20 min, incubated with the TUNEL reaction mixture or a negative control solution for 60 min at 37 °C and then stained with DAPI solution for 10 min. Slides were washed twice with PBS after each step. The ratio of positive TUNEL-labelled nuclei was calculated from four different randomly selected areas, visualized by confocal microscopy.
2.6 Quantitative real-time polymerase chain reaction
Total RNA was extracted from heart tissues using Trizol reagent (Invitrogen). Real-time quantitative real-time polymerase chain reaction (qPCR) to detect mRNA for periostin, ANP and brain natriuretic peptide (BNP) and STAT3 in heart tissues was performed using a Quantitect SYBR RT-PCR kit (DRR420A, Takara, Japan). The primer sequences are shown in Supplementary material online, Table S1.
2.7 Echocardiography
Heart dimensions and cardiac function of mice at 21 days after surgery were evaluated by echocardiography using the Vevo 770 Ultrasound machine (VisualSonics, Toronto, Ontario, Canada) with a 30 M-Hz probe. After mice were anesthetized with 1.5% inhalational isoflurane, two-dimensional parasternal short-axis images of the left ventricle were obtained at the level of the papillary muscles. M-mode echocardiography was performed to evaluate internal diastolic and systolic left ventricular diameter (LVEDd, LVESd), and fractional shortening (LVFS).
2.8 Statistical analysis
Data were expressed as means ± standard error of the mean (SEM), and P < 0.05 was considered to be statistically significant. Statistical significance between two experimental groups was determined using Student’s two-tailed t-test, while comparisons of parameters among ≥3 groups with two factor levels were analyzed by two-way ANOVA, followed by Bonferroni’s correction for post hoc multiple comparisons.
3. Results
3.1 Periostin in the infarcted area was upregulated in response to MI in adult and neonatal mice
In neonatal mice, periostin mRNA was significantly upregulated at 1, 3, and 7 days after MI (Figure 1A). IHC staining revealed that periostin expression in the infarcted area peaked at 7 days and returned to baseline levels by 21 days after MI (Figure 1B and C). Periostin expression was detectable in the infarcted, border, and remote areas, but its abundance was greater in the border than in other areas (Figure 1C). In the adult mouse heart subjected to MI, periostin expression was also significantly increased (Supplementary material online, Figure S1A and B). Real-time qPCR and western blotting showed that periostin was significantly increased at 7 days after MI, compared with in the sham group (Figure 1D and E). The PCR results showed that periostin was expressed in both cardiomyocytes and fibroblasts derived from the neonatal mouse heart (Figure 1F).
Figure 1.

Periostin expression was increased in neonatal mice with myocardial infarction (MI). (A) Time course of periostin mRNA expression in hearts from mice subjected to MI. *P < 0.01 vs. the corresponding sham (S) group, n = 5 per group (two-way ANOVA analysis). (B) Time course of periostin immunostaining in mouse hearts at 3, 7, 14, or 21 days after MI. Scale bar = 50 μm. (C) Semi-quantitative analysis of periostin expression in four groups, with the insert picture showing the location of periostin staining in infarcted, border and remote areas. *P < 0.01 vs. sham group, n = 8 per group (two-way ANOVA analysis). (D) mRNA expression of periostin was determined by real-time qPCR in neonatal mouse heart samples, obtained 7 days after MI. *P < 0.01 vs. sham group, n = 5 per group (t test). The insert picture is an amplification curve. (E) Western blot for periostin in neonatal mouse heart samples, obtained 7 days after MI. *P < 0.05 vs. sham group, n = 8 per group (t test). (F) mRNA expression of periostin in neonatal mouse cardiomyocytes and fibroblasts was determined by PCR.
3.2 Wildtype neonatal mice had the capacity to regenerate myocardium after MI
To investigate whether wildtype mice would have the capacity to regenerate myocardium after cardiac injury, we created an MI model in neonatal mice. The left coronary artery was permanently ligated, as shown in Figure 2A, and elevation of the ST segment after coronary ligation was confirmed (Figure 2A). We further verified this model with TTC staining. At 24 h after MI, the infarct size was about 60% of the left ventricular area in both the periostin knockout and wildtype groups (Figure 2B). At 7 days after MI, immunofluorescence staining for p-H3 and aurora B, as well as for cell cycle entry and cytokinesis markers, was detectable in the infarcted, border and remote areas (Figure 2C and D). These findings indicated myocardial regeneration. A time-course of Masson’s trichrome staining patterns showed significant fibrosis in the infarcted area at 1, 3, 7, 10, 14, and 17 days after MI (Figure 2E). The fibrosis had almost completely disappeared by 21 days after MI, indicating that myocardial regeneration was complete (Figure 2E).
Figure 2.

Capacity for myocardial regeneration existed in neonatal mice subjected to myocardial infarction (MI). (A) Schematic diagram showing location of left coronary ligation and ST segment elevation in electrocardiogram monitoring. Scale bar = 1 mm. (B) Confirmation of MI by autopsy and TTC staining in mice 24 h after MI. Scale bar = 1 mm. The infarct size (IS) was about 57% in wildtype (WT) and periostin knockout (KO) groups (t test). LV: left ventricle. (C, D) Representative images showing immunofluorescence staining for p-H3 and aurora B (arrow), in hearts from sham and MI mice obtained at 7 days after surgery and quantitative analysis. Inserts show high magnification images of p-H3 and aurora B- positive cardiomyocytes. *P < 0.01 vs. sham group, n = 8 per group (two-way ANOVA analysis). Scale bar = 50 μm (in the insert images, scale bar = 10 μm). (E) Masson’s trichrome staining of hearts obtained at 1, 3, 7, 10, 14, 17, or 21 days after MI. The lower panels show magnifications of the regions indicated by black boxes in the upper panels. Scale bar = 1 mm (upper) and 100 μm (lower).
3.3 Periostin deficiency hindered recovery of post-MI remodelling
At 7 days after surgery, there were no significant differences in the ratios of heart to body weight (HW/BW) between knockout and wildtype mice in either the sham or MI groups (Figure 3A). However, at 21 days after surgery, HW/BW in knockout MI- and sham-treated mice was significantly greater than in the corresponding wildtype groups (Figure 3A). HW/BW in both wildtype and knockout mice was not increased in response to MI (Figure 3A). At 7 days after MI, fibrosis, as detected by Masson’s trichrome staining, was similar in knockout mice and their wildtype littermates. However, at 21 days after MI, fibrosis was greater in the infarcted area of the knockout mice, while it had almost completely disappeared in the wildtype mice (Figure 3B and C). This suggested that periostin deficiency suppressed myocardial regeneration after MI. TUNEL assay results showed no significant difference in apoptotic cell death between the wildtype and knockout groups at 7 days after MI (Figure 3D). Echocardiographic LVFS at 21 days after MI was significantly lower in the periostin knockout than in the wildtype group, while, in wildtype mice, there was no significant difference between sham and MI groups (Figure 3F). Echocardiographic data for left ventricular diameters are shown in Supplementary material online, Table S2.
Figure 3.

Periostin deficiency resulted in post-MI cardiac fibrosis and dysfunction caused by attenuated myocardial regeneration. (A) Heart weight to body weight ratios (HW/BW), at 7 and 21 days after MI or sham surgeries in neonatal mice. *P < 0.05 compared with corresponding wildtype group, n = 5 per group (two-way ANOVA analysis). (B) Masson’s trichromatic (Azan) staining of the myocardium from wildtype (WT) and periostin knockout (KO) groups, at 7 and 21 days after MI. The lower panels show magnifications of the regions indicated by black boxes in the upper panels. Scale bar = 1 mm (upper) and 100 μm (lower). (C) Semi-quantitative analysis of myocardial fibrosis in panel B. The insert images are multiple cross sections of Azan staining in hearts from mice, obtained at 7 days after MI surgery. *P < 0.01 vs. the corresponding sham group, #P < 0.05 vs. WT MI (at 21 d) group, n = 8 per group (two-way ANOVA analysis). (D) Apoptotic myocardial cell death, visualized by TUNEL staining, in knockout and wildtype mice subjected to sham and MI surgeries, after 7 d. (E) Quantitative analysis showed no differences between wildtype and knockout groups. *P < 0.01 vs. the corresponding sham group, n = 8 per group (two-way ANOVA analysis). Scale bar = 100 μm. (F) Left ventricular systolic function, quantified by left ventricle fractional shortening (LVFS) at 21 days after MI in four groups. *P < 0.05 vs. the corresponding sham group, #P < 0.01 vs. WT MI group, n = 8 per group (two-way ANOVA analysis).
3.4 Periostin deficiency suppressed myocardial regeneration
Western blotting results showed significantly lower levels of p-H3 in periostin knockout mice than in wildtypes at 7 days after MI (Figure 4A). Immunofluorescence staining showed that p-H3 and aurora B expression at 7 days after MI was significantly lower in knockout than in wildtype mice (Figure 4B and C), indicating involvement of periostin in myocardial regeneration after MI. In wildtype mice, qPCR and western blotting results showed, respectively, that myocardial mRNA of ANP and BNP, and ANP protein were significantly downregulated in response to MI at 7 days and ANP expression was, then, significantly upregulated at 21 days (Figure 4D and E). In periostin knockout mice, the only notable change was that ANP mRNA and protein expression were markedly upregulated at 21 days after MI, though their levels were significantly lower than in the corresponding wildtype MI group (Figure 4D and E). At 21 days after MI, ANP mRNA and protein expression were increased in hearts of both wildtype and knockout mice (Figure 4D and E). Expression of STAT3 mRNA was significantly lower in the knockout MI than in the wildtype MI group (Figure 4F).
Figure 4.

Periostin knockout (KO) suppressed cardiomyocyte regeneration. (A) Western blots for phospho-histone 3 (p-H3) and semi-quantitative analysis of western blotting results. *P < 0.05 vs. the corresponding sham group, #P < 0.01 vs. WT MI group (n = 8 per group). Immunofluorescence for p-H3 (B) and aurora B (C) in cardiomyocytes counterstained for troponin T and the results of quantitative analysis for p-H3 and aurora B stained cells (indicated with arrows). Scale bar = 50 μm. *P < 0.05 vs. the corresponding sham groups, #P < 0.01 vs. WT MI group, n = 8 per group. (D) mRNA expression of myocardial ANP and BNP was determined by real-time qPCR in neonatal mice at 7 or 21 days after MI or sham surgeries. *P < 0.05 vs. corresponding sham group, #P < 0.01 vs. WT MI group (n = 8 per group). (E) Western blot for ANP in neonatal hearts from four groups, obtained 7 or 21 days after MI. *P < 0.05 vs. corresponding sham group, #P < 0.01 vs. WT MI group (n = 8 per group). (F) mRNA expression of STAT3 determined by real-time qPCR in hearts from four groups, obtained at 7 days after MI. *P < 0.05 vs. corresponding sham group, #P < 0.01 vs. WT MI group (n = 8 per group). Two-way ANOVA analysis was used for (A–F). WT, wildtype; MI, myocardial infarction.
3.5 Periostin affected the PI3K/Akt/GSK3β/cyclin D1 signalling pathway
At 7 days after MI, levels of PI3K, p-Akt, Akt and cyclin D1 in wildtype mice were significantly higher than in periostin knockout mice. In contrast, levels of phosphorylated GSK3β were lower in the periostin knockout than in wildtype mice subjected to MI (Figure 5A–C). We prepared cardiomyocytes from newborn wildtype and periostin knockout mice. When these cultured newborn mouse cardiomyocytes were treated with AR, we obtained similar results to those from the in vivo experiments (Figure 5D and E).
Figure 5.

Periostin deficiency inhibited the PI3K/GSK3β/cyclin D1 signalling pathway. (A) Representative western blots for PI3K, p-Akt, Akt, and p-GSK3β in hearts obtained from the in vivo experimental groups. (B) Representative western blots for GSK3β and cyclin D1 in hearts obtained from the in vivo experimental groups. (C) Quantitative analysis of protein expression levels for panels A and B. *P < 0.05 vs. the corresponding sham group, #P < 0.05 vs. WT (wildtype) MI (myocardial infarction) group (n = 8 per group). Heart samples obtained from neonatal mice at 7 days after MI or sham operations. KO, periostin knockout mice. (D) Western blots for PI3K, p-Akt, Akt, p-GSK3β, GSK3β, and cyclin D1 in cultured cardiomyocytes exposed to 3 h anoxia and 24 h reoxygenation (AR) or normoxia (con: control). (E) Quantitative analysis of protein expression levels for panel D. *P < 0.05 vs. the corresponding control group, #P < 0.05 vs. WT AR group (n = 8 per group). Cardiomyocytes were derived from hearts of neonatal mice. For panels C and E, two-way ANOVA analysis was used.
3.6 Periostin deficiency inhibited post-MI angiogenesis
We next investigated whether myocardial angiogenesis was affected by periostin ablation. CD31 was used to label endothelial cells, thus staining the blood vessels. At 7 days after MI, periostin knockout mice had less CD31 staining than wildtype mice (Figure 6A). To further confirm this result, we stained for a smooth muscle cell marker, smooth muscle actin (α-SMA) (Figure 6B). α-SMA staining confirmed the marked decrease in vascular area in knockout, compared with wildtype mice after MI (Figure 6B). Because CD31 and α-SMA stains can label both pre-existing and newly formed capillaries and arterioles, respectively, vascular density was compared with that in the control mice, not receiving MI, to ascertain which vessels had resulted from angiogenesis. Our findings suggested a role for periostin in post-MI angiogenesis in neonatal mice.
Figure 6.
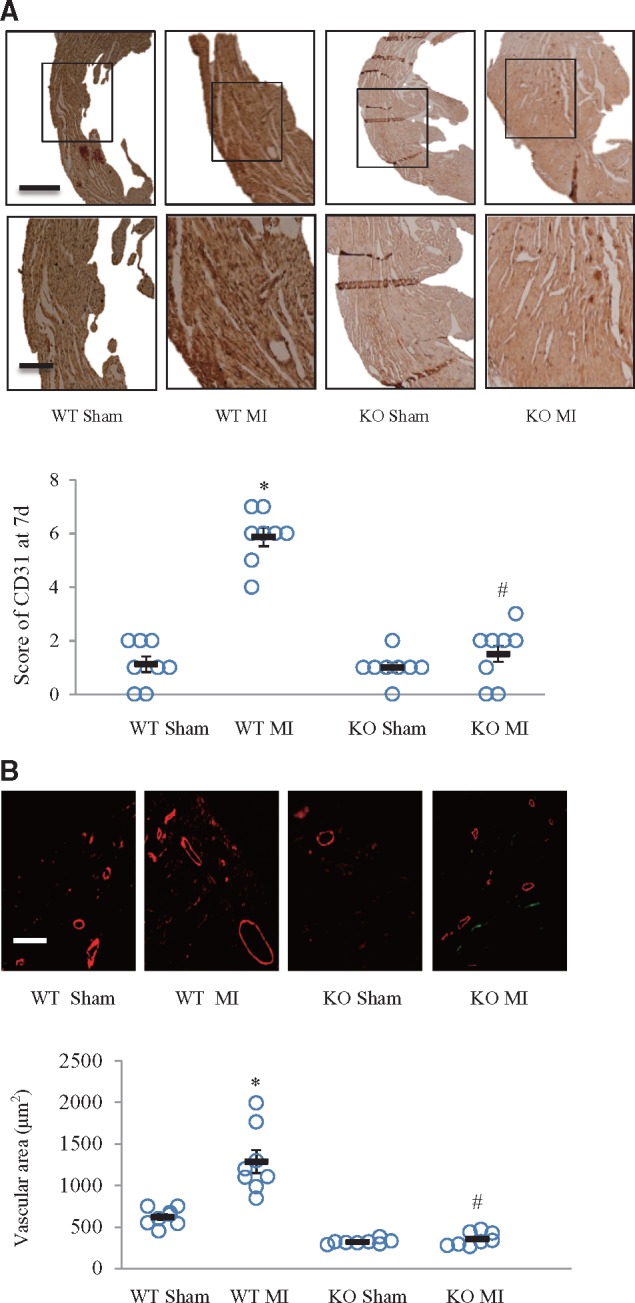
Periostin deficiency suppressed post-MI angiogenesis. In hearts from neonatal mice, obtained at 7 days after MI or sham operations, angiogenesis was examined by immunohistochemistry. (A) CD31 was used to label the endothelial cells in capillaries. The lower panels show magnifications of the regions in the upper panels. Semi-quantitative analysis of CD-31 staining in each group is shown in the upper panel. *P < 0.01 vs. WT sham group, #P < 0.01 vs. WT MI group. Scale bar = 1 mm (upper) and 50 μm (lower) (n = 8 per group). (B) α-SMA (smooth muscle actin) immunofluorescence staining was used to label smooth muscle cells in arterioles. Scale bar = 50 μm. *P < 0.01 vs. WT sham group, #P < 0.01 vs. WT MI group (n = 8 per group). For (A) and (B), two-way ANOVA analysis was used.
3.7 GSK3β inhibition in periostin knockout mice promoted myocyte regeneration and angiogenesis
We further performed rescue experiments to test whether the GSK-3β inhibitor SB216763 would improve cardiomyocyte regeneration and angiogenesis in the periostin knockout mice. SB216763 (10 mg/kg/d) was intraperitoneally injected for 7 d. Myocardial GSK-3β expression was decreased and that of cyclin D1 was increased at 7 days after MI in the SB216763 treated mice (Figure 7A). Histological immunofluorescence staining showed significantly higher levels of p-H3 and aurora B in SB216763 treated than in untreated mice at 7 days after MI (Figure 7B and C). At 21 days after MI, myocardial fibrosis was significantly lower in SB216763 treated than in untreated mice (Figure 7D). At 7 days after MI, there was a larger area of α-SMA stained vessels in SB216763 treated than in untreated mice (Figure 7E). SB216763 treatment significantly improved echocardiographic LVFS at 21 days after MI (Figure 7F; Supplementary material online, Table S3).
Figure 7.

GSK3β inhibitor SB216763 improved cardiomyocyte regeneration and angiogenesis in periostin knockout mice. (A) SB216763 (SB) downregulated GSK3β and upregulated cyclin D1 in periostin knockout (KO) mice at 7 days after myocardial infarction (MI). (B) Phosphorylated histone 3 (p-H3) stained cells. (C) Aurora B stained cells. (D) SB216763 attenuated myocardial fibrosis in periostin knockout neonatal mice at 21 days after MI or sham operations. Fibrotic areas were detected by Masson’s trichrome staining. Scale bar = 1 mm (upper) and 100 μm (lower). (E) α-SMA (smooth muscle actin) immunofluorescence staining was used to determine arteriolar density in periostin knockout neonatal mice at 21 days after MI or sham operations. Scale bar = 50 μm. (F) SB216763 treatment improved left ventricular fractional shortening, as determined with echocardiography. *P < 0.01 vs. the corresponding sham + V group, #P < 0.01 vs. MI + V group (n = 8 per group), V, vehicle (dimethyl sulfoxide). For all panels, two-way ANOVA analysis was used.
4. Discussion
Since the generation of periostin deficient mice, many studies examined the roles of this factor in the regeneration of various tissues including bone, heart and skin as well as in tumor growth.11 Based on results of studies using MI models in adult rodents, the role of periostin in myocardial regeneration has been controversial. Kühn et al. reported that periostin released from patches placed over the infarcted area of the adult rat heart induced proliferation of differentiated cardiomyocytes and improved cardiac function, while suppressing myocardial fibrosis and hypertrophy.16 Cho et al.17 demonstrated that injection of mesenchymal stem cells overexpressing periostin into the infarcted regions of rat hearts attenuated post-MI remodelling. These findings supported involvement of periostin in promoting myocardial regeneration in the adult heart. However, Lorts et al.13 showed no significant difference in post-infarction myocardial regeneration between mice with modulated periostin expression (transgenic and knockout mice) and their corresponding strain-matched controls. Taniyama et al.18 reported that inhibition of periostin-exon 17 attenuated post-MI fibrosis in adult rats but did not affect cardiomyocyte proliferation. These reports suggested, in contrast to other findings, that periostin is not involved in post-infarction myocardial regeneration in the adult heart. Similarly, based on results obtained using neonatal rodent cardiomyocytes, the regeneration promoting ability of periostin was disputed.13,16 A recent study by White et al.19,20 indicated that the capacity of the neonatal mammalian heart for regeneration required sympathetic innervation,19,20 which might explain why periostin exerted no regenerative effect in cultured cardiomyocytes, where sympathetic activity was not a factor.13 It is generally believed that the capacity for cardiac regeneration is absent in the adult mammalian heart, while recent studies confirmed its existence in the neonatal mammalian heart.2,4,6,19 Accumulated evidence demonstrated that a variety of injuries could induce heart regeneration, through cardiomyocyte proliferation, in newt, zebrafish and newborn mice (review by Leone et al.21). Based on such evidence, taken together, we postulated that using an in vivo neonatal heart injury model could help resolve controversies regarding the role of periostin in myocardial regeneration. Therefore we designed this study.
The regenerative model of the murine heart is controversial. Andersen et al.22 found no evidence of complete regeneration and questioned the utility of the apical resection model, whereas Konfino et al.23 observed significantly greater scar formation following left coronary artery ligation associated with a lack of induction of cardiomyocyte proliferation. These findings contradicted substantial reports from various laboratories, demonstrating that neonatal mice have the capacity for heart regeneration in response to injuries, including resection of the apex and occlusion of the left coronary artery.2,4,6,20,21,24–28 There is also controversy regarding the capacity for heart regeneration in response to cryo-injury. Strungs et al.29 demonstrated that the apex of the heart ventricle, cryoinjured at 1 days after birth, had no visible scar and could fully regenerate myocardium, but at least two groups reported that cryo-injury30 or cryo-transmural infarction31 led to scar formation. The technical difficulties of inducing neonatal heart injury and various choices in anesthesia may have contributed to the variations in results on regeneration reported by different laboratories.23,32 Blom et al.33 recently characterized this model clearly with video recording and demonstrated that the MI-induced scars completely disappeared by 21 days post-injury.
Periostin overexpression was proposed to promote re-entry of differentiated cardiomyocytes into the cell cycle and, consequently, contribute to myocardial repair following MI.16 Negative results in some studies13 were, therefore, not surprising because periostin may be necessary but not sufficient to induce cardiomyocyte regeneration. In neonatal mice, O'Meara et al.4 demonstrated that interleukin 13 induced entry of cardiomyocytes into the cell cycle and identified STAT3 and periostin as critical mediators of interleukin 13 signalling. This supports the concept that periostin can contribute to inducing regeneration but, alone, is not sufficient. In our study, we verified the regeneration-promoting potential of periostin using periostin knockout mice. We found that, in this strain of neonatal mice subjected to MI, periostin deficiency impaired the regenerative capacity of cardiomyocytes. Similar results were described by others using non-cardiovascular disease models, with recent studies showing that periostin promoted pancreatic exocrine regeneration and neural stem cell proliferation.34,35
It was reported that plasma or myocardial ANP and BNP were elevated shortly after MI in humans and adult rodents.36,37 However, no reports described myocardial ANP expression in neonatal mice with MI. We found that, in neonatal mice, myocardial ANP was downregulated at 7 days after MI but was upregulated at 21 d. Schoensiegel et al.38 reported that plasma levels of the N-terminal propeptide of ANP were not increased in adult mice subjected to non-ischaemic MI for 4 weeks. In a recent report by Bielmann et al.,35 BNP stimulated cardiac progenitor cell proliferation and differentiation in murine hearts after birth and BNP administration induced heart regeneration. Becker et al.39 also demonstrated in vitro that ANP induced proliferation of neonatal murine cardiomyocytes. The potentially interesting association between periostin and natriuretic peptides should be further investigated in the future.
With regard to regeneration mechanisms of periostin, it was previously reported that PI3K, extracellular-signal-regulated kinases and STAT3/STAT6 were involved.4,40 Emerging evidence has shown that the GSK3β-cyclin D1 signalling pathway is closely associated with cell proliferation and cardiovascular diseases,41–44 but whether periostin is also involved in this pathway is unknown. In our study, we found that periostin ablation led to upregulation of GSK3β and downregulation of cyclin D1, while a GSK3 inhibitor partially rescued the regeneration capacity of the heart after MI in the neonatal periostin knockout mice.
In adult mice with MI, whether GSK3β is beneficial or detrimental for cardiac remodelling has been controversial.42,45–47 However, it is generally believed that GSK-3β is critical for embryonic cardiomyocyte proliferation and differentiation. GSK3β deletion induced embryonic lethality, caused by near obliteration of the ventricular cavities by proliferating cardiomyocytes. In addition, terminal cardiomyocyte differentiation was substantially blunted in embryoid bodies with GSK3β deficiency.44,45 Ahmad et al. reported that cardiomyocyte-specific GSK3α deletion attenuated post-infarction cardiac remodelling and heart failure.48 These results were consistent with our observations that increased GSK3β in periostin knockout mice impaired post-MI regeneration of the myocardium, while SB216763, a pan inhibitor of both GSK3α and GSK3β, improved myocyte regeneration and attenuated cardiac remodelling in post infarcted periostin knockout mice.
The role of periostin in myocardial fibrosis in adult animals is also unclear.12,16,49 In our study, we focused on the role of periostin in cardiomyocyte regeneration in neonatal mice with MI. Unlike adult mammalian hearts, that respond to injury with scar formation, neonatal mouse hearts respond to MI with cardiomyocyte proliferation. We demonstrated that, in wildtype mice, myocardial fibrosis was significantly formed at 7 days after MI but was completely replaced by myocardium at 21 d, in agreement with previous studies.2,6 In periostin knockout mice, myocardial fibrosis in the infarcted area was still present at 21 days after MI, possibly a net result of impaired cardiomyocyte regeneration capacity, counterbalancing the anti-fibrotic effects of periostin deficiency on cardiac fibroblasts.50,51 In addition, other mechanisms may have also contributed to the impaired cardiomyocyte regenerative capacity in the periostin knockout mice. Periostin can affect collagen formation and recruitment of macrophages.52,53 Schwanekamp et al. showed that loss of periostin decreased macrophage recruitment to atherosclerotic lesions.54 Although periostin deficiency induced a large set of differentially expressed genes related to fibroblast function and contributed to post-MI rupture by attenuating scar (fibrosis) formation in adult mice,52 it was also likely to reduce macrophage recruitment. This would, in turn, inhibit myocardial regeneration and eventually lead to replacement of the infarcted myocardium with fibrotic tissue in newborn mice.24 Therefore, it would be worthwhile to, in future studies, investigate the contribution of macrophages to impairment of myocardial regeneration associated with periostin deficiency.
We further found that periostin ablation impaired post-MI angiogenesis, results supported by previous studies in adult animals. Kühn et al. reported that periostin improved post-MI ventricular remodelling, reduced fibrosis and increased angiogenesis.16 Hakuno et al.55 demonstrated that periostin induced angiogenesis and promoted tube formation by mobilization of endothelial cells. We noted a significant decrease of CD31 positive endothelial cells, indicating capillaries, in periostin knockout mice with MI, suggesting that periostin affected angiogenesis-associated endothelial cells. Our findings were consistent with previous reports showing that periostin promoted tube formation by mobilization of endothelial cells.55,56 In the postnatal heart, endocardial cells contributed to postnatal vascular development in the heart, an effect that was enhanced in response to hypoxia.57 This was consistent with our finding that, in wildtype mice, capillary density was higher in the infarcted areas of hearts from the MI group than in the corresponding areas of hearts from the sham group. However, it remains unclear whether periostin can affect the response of endocardial cells to ischaemia.
Although it was believed that the majority of newly formed cardiomyocytes are derived from pre-existing cardiomyocytes,2 there is evidence that resident non-myocytes can also be reprogrammed into cardiomyocyte-like cells by addition of Gata4, Mef2c and Tbx5, an affect confirmed by using genetic lineage tracing and periostin-Cre R26R-lacZ mice in a murine MI model.58,59 It would interesting to investigate the contributions of periostin on non-myocyte derived cardiomyocyte regeneration using genetic lineage tracing.
In conclusion, our findings indicate that a lack of periostin impairs post-MI regeneration of cardiomyocytes and angiogenesis, effects mediated by the PI3K/GSK3β/cyclin D1 signalling pathway.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31271513 to Y.L.), the Municipal Planning Projects of Scientific Technology of Guangzhou (20150401001), and the Provincial Natural Science Foundation of Guangdong (2014A030313342, 2015A030313301, and 2015A030313298).
Supplementary Material
References
- 1. Milasinovic D, Mohl W.. Contemporary perspective on endogenous myocardial regeneration. World J Stem Cells 2015;7:793–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA.. Transient regenerative potential of the neonatal mouse heart. Science 2011;331:1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang Y, Cheng HW, Qiu Y, Dupee D, Noonan M, Lin YD, Fisch S, Unno K, Sereti KI, Liao R.. MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res 2015;117:450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Meara CC, Wamstad JA, Gladstone RA, Fomovsky GM, Butty VL, Shrikumar A, Gannon JB, Boyer LA, Lee RT.. Transcriptional reversion of cardiac myocyte fate during mammalian cardiac regeneration. Circ Res 2015;116:804–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J, Gao E, Vite A, Yi R, Gomez L, Goossens S, van Roy F, Radice GL.. Alpha-catenins control cardiomyocyte proliferation by regulating yap activity. Circ Res 2014;116:70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA.. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci USA 2012;110:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA.. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014;157:565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Segers VF, Lee RT.. Protein therapeutics for cardiac regeneration after myocardial infarction. J Cardiovasc Transl Res 2010;3:469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katsuragi N, Morishita R, Nakamura N, Ochiai T, Taniyama Y, Hasegawa Y, Kawashima K, Kaneda Y, Ogihara T, Sugimura K.. Periostin as a novel factor responsible for ventricular dilation. Circulation 2004;110:1806–1813. [DOI] [PubMed] [Google Scholar]
- 10. Chen B, Lu D, Fu Y, Zhang J, Huang X, Cao S, Xu D, Bin J, Kitakaze M, Huang Q, Liao Y.. Olmesartan prevents cardiac rupture in mice with myocardial infarction by modulating growth differentiation factor 15 and p53. Br J Pharmacol 2014;171:3741–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kudo A. Periostin in fibrillogenesis for tissue regeneration: periostin actions inside and outside the cell. Cell Mol Life Sci 2011;68:3201–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A.. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med 2008;205:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lorts A, Schwanekamp JA, Elrod JW, Sargent MA, Molkentin JD.. Genetic manipulation of periostin expression in the heart does not affect myocyte content, cell cycle activity, or cardiac repair. Circ Res 2009;104:e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mahmoud AI, Porrello ER, Kimura W, Olson EN, Sadek HA.. Surgical models for cardiac regeneration in neonatal mice. Nat Protoc 2014;9:305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ehler E, Moore-Morris T, Lange S.. Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kühn B, del Monte F, Hajjar RJ, Chang Y-S, Lebeche D, Arab S, Keating MT.. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med 2007;13:962–969. [DOI] [PubMed] [Google Scholar]
- 17. Cho Y-H, Cha M-J, Song B-W, Kim I-K, Song H, Chang W, Lim S, Ham O, Lee S-Y, Choi E, Kwon HM, Hwang K-C.. Enhancement of MSC adhesion and therapeutic efficiency in ischemic heart using lentivirus delivery with periostin. Biomaterials 2012;33:1376–1385. [DOI] [PubMed] [Google Scholar]
- 18. Taniyama Y, Katsuragi N, Sanada F, Azuma J, Iekushi K, Koibuchi N, Okayama K, Ikeda-Iwabu Y, Muratsu J, Otsu R, Rakugi H, Morishita R.. Selective blockade of periostin exon 17 preserves cardiac performance in acute myocardial infarction. Hypertension 2016;67:356–361. [DOI] [PubMed] [Google Scholar]
- 19. White IA, Gordon J, Balkan W, Hare JM.. Sympathetic reinnervation is required for mammalian cardiac regeneration. Circ Res 2015;117:990–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mahmoud AI, O'Meara CC, Gemberling M, Zhao L, Bryant DM, Zheng R, Gannon JB, Cai L, Choi WY, Egnaczyk GF, Burns CE, Burns CG, MacRae CA, Poss KD, Lee RT.. Nerves regulate cardiomyocyte proliferation and heart regeneration. Dev Cell 2015;34:387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leone M, Magadum A, Engel FB.. Cardiomyocyte proliferation in cardiac development and regeneration: a guide to methodologies and interpretations. Am J Physiol Heart Circ Physiol 2015;309:H1237–1250. [DOI] [PubMed] [Google Scholar]
- 22. Andersen DC, Ganesalingam S, Jensen CH, Sheikh SP.. Do neonatal mouse hearts regenerate following heart apex resection? Stem Cell Reports 2014;2:406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konfino T, Landa N, Ben-Mordechai T, Leor J.. The type of injury dictates the mode of repair in neonatal and adult heart. J Am Heart Assoc 2015;4:e001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN.. Macrophages are required for neonatal heart regeneration. J Clin Invest 2014;124:1382–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haubner BJ, Adamowicz-Brice M, Khadayate S, Tiefenthaler V, Metzler B, Aitman T, Penninger JM.. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging (Albany NY) 2012;4:966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bryant DM, O'Meara CC, Ho NN, Gannon J, Cai L, Lee RT.. A systematic analysis of neonatal mouse heart regeneration after apical resection. J Mol Cell Cardiol 2015;79:315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haubner BJ, Schuetz T, Penninger JM.. A reproducible protocol for neonatal ischemic injury and cardiac regeneration in neonatal mice. Basic Res Cardiol 2016;111:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, Li L, Sun Z, Olson EN, Amendt BA, Martin JF.. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 2016;534:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strungs EG, Ongstad EL, O'Quinn MP, Palatinus JA, Jourdan LJ, Gourdie RG.. Cryoinjury models of the adult and neonatal mouse heart for studies of scarring and regeneration. Methods Mol Biol 2013;1037:343–353. [DOI] [PubMed] [Google Scholar]
- 30. Polizzotti BD, Ganapathy B, Haubner BJ, Penninger JM, Kuhn B.. A cryoinjury model in neonatal mice for cardiac translational and regeneration research. Nat Protoc 2016;11:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darehzereshki A, Rubin N, Gamba L, Kim J, Fraser J, Huang Y, Billings J, Mohammadzadeh R, Wood J, Warburton D, Kaartinen V, Lien CL.. Differential regenerative capacity of neonatal mouse hearts after cryoinjury. Dev Biol 2015;399:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sen S, Sadek HA.. Neonatal heart regeneration: mounting support and need for technical standards. J Am Heart Assoc 2015;4:e001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blom JN, Lu X, Arnold P, Feng Q.. Myocardial Infarction in Neonatal Mice, A Model of Cardiac Regeneration. J Vis Exp 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smid JK, Faulkes S, Rudnicki MA.. Periostin induces pancreatic regeneration. Endocrinology 2015;156:824–836. [DOI] [PubMed] [Google Scholar]
- 35. Bielmann C, Rignault-Clerc S, Liaudet L, Li F, Kunieda T, Sogawa C, Zehnder T, Waeber B, Feihl F, Rosenblatt-Velin N.. Brain natriuretic peptide is able to stimulate cardiac progenitor cell proliferation and differentiation in murine hearts after birth. Basic Res Cardiol 2015;110:455. [DOI] [PubMed] [Google Scholar]
- 36. Morita E, Yasue H, Yoshimura M, Ogawa H, Jougasaki M, Matsumura T, Mukoyama M, Nakao K.. Increased plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation 1993;88:82–91. [DOI] [PubMed] [Google Scholar]
- 37. Nakanishi M, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, Kawakami R, Nakagawa Y, Tanimoto K, Yasuno S, Usami S, Li Y, Adachi Y, Fukamizu A, Garbers DL, Nakao K.. Role of natriuretic peptide receptor guanylyl cyclase-A in myocardial infarction evaluated using genetically engineered mice. Hypertension 2005;46:441–447. [DOI] [PubMed] [Google Scholar]
- 38. Schoensiegel F, Bekeredjian R, Schrewe A, Weichenhan D, Frey N, Katus HA, Ivandic BT.. Atrial natriuretic peptide and osteopontin are useful markers of cardiac disorders in mice. Comp Med 2007;57:546–553. [PubMed] [Google Scholar]
- 39. Becker JR, Chatterjee S, Robinson TY, Bennett JS, Panakova D, Galindo CL, Zhong L, Shin JT, Coy SM, Kelly AE, Roden DM, Lim CC, MacRae CA.. Differential activation of natriuretic peptide receptors modulates cardiomyocyte proliferation during development. Development 2014;141:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rojas-Munoz A, Rajadhyksha S, Gilmour D, van Bebber F, Antos C, Rodriguez Esteban C, Nusslein-Volhard C, Izpisua Belmonte JC.. ErbB2 and ErbB3 regulate amputation-induced proliferation and migration during vertebrate regeneration. Dev Biol 2009;327:177–190. [DOI] [PubMed] [Google Scholar]
- 41. Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E, Force T.. Cardiac fibroblast glycogen synthase kinase-3 regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014;130:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, DeCaul M, Shang X, Patel S, Woodgett JR, Force T, Zhou J.. Glycogen synthase kinase-3 regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res 2010;106:1635–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dimova N, Wysoczynski M, Rokosh G.. Stromal cell derived factor-1alpha promotes C-Kit+ cardiac stem/progenitor cell quiescence through casein kinase 1alpha and GSK3beta. Stem Cells 2014;32:487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS.. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 2008;118:3609–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lal H, Ahmad F, Woodgett J, Force T.. The GSK-3 family as therapeutic target for myocardial diseases. Circ Res 2014;116:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Webb IG, Sicard P, Clark JE, Redwood S, Marber MS.. Myocardial stress remodelling after regional infarction is independent of glycogen synthase kinase-3 inactivation. J Mol Cell Cardiol 2010;49:897–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J.. Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ Res 2011;109:502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahmad F, Lal H, Zhou J, Vagnozzi RJ, Yu JE, Shang X, Woodgett JR, Gao E, Force T.. Cardiomyocyte-specific deletion of Gsk3alpha mitigates post-myocardial infarction remodeling, contractile dysfunction, and heart failure. J Am Coll Cardiol 2014;64:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ladage D, Yaniz-Galende E, Rapti K, Ishikawa K, Tilemann L, Shapiro S, Takewa Y, Muller-Ehmsen J, Schwarz M, Garcia MJ, Sanz J, Hajjar RJ, Kawase Y.. Stimulating myocardial regeneration with periostin Peptide in large mammals improves function post-myocardial infarction but increases myocardial fibrosis. PLoS One 2013;8:e59656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S, Wettschureck N.. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ Res 2016;118:1906–1917. [DOI] [PubMed] [Google Scholar]
- 51. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD.. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW 2nd, Conway SJ, Aronow BJ, Robbins J, Molkentin JD.. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res 2007;101:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, Rich JN, Bao S.. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 2015;17:170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schwanekamp JA, Lorts A, Vagnozzi RJ, Vanhoutte D, Molkentin JD.. Deletion of periostin protects against atherosclerosis in mice by altering inflammation and extracellular matrix remodeling. Arterioscler Thromb Vasc Biol 2016; 36:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hakuno D, Kimura N, Yoshioka M, Mukai M, Kimura T, Okada Y, Yozu R, Shukunami C, Hiraki Y, Kudo A, Ogawa S, Fukuda K.. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J Clin Invest 2010;120: 2292–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim BR, Jang IH, Shin SH, Kwon YW, Heo SC, Choi EJ, Lee JS, Kim JH.. Therapeutic angiogenesis in a murine model of limb ischemia by recombinant periostin and its fasciclin I domain. Biochim Biophys Acta 2014;1842:1324–1332. [DOI] [PubMed] [Google Scholar]
- 57. Tian X, Hu T, Zhang H, He L, Huang X, Liu Q, Yu W, He L, Yang Z, Yan Y, Yang X, Zhong TP, Pu WT, Zhou B.. Vessel formation. De novo formation of a distinct coronary vascular population in neonatal heart. Science 2014;345:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D.. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012;485:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ma H, Wang L, Yin C, Liu J, Qian L.. In vivo cardiac reprogramming using an optimal single polycistronic construct. Cardiovasc Res 2015;108:217–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.